Molecular and Cellular Substrates for the Friedreich Ataxia. Significance of Contactin Expression and of Antioxidant Administration




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
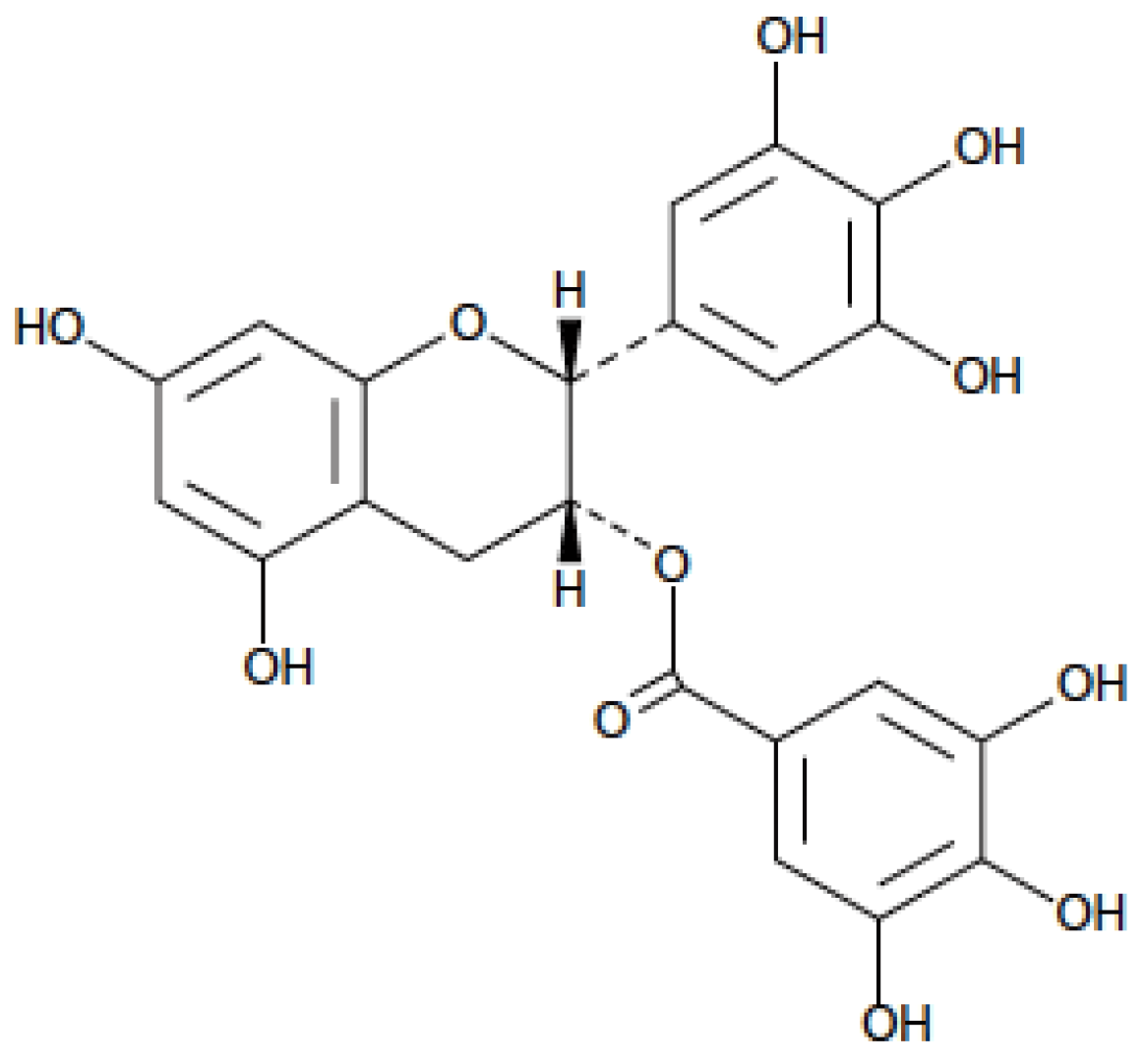
1. Introduction
2. Results
2.1. Rodent Models for the Friedreich Ataxia: Cerebellar and Spinal Cord Phenotypes and Effects of Antioxidant Treatment
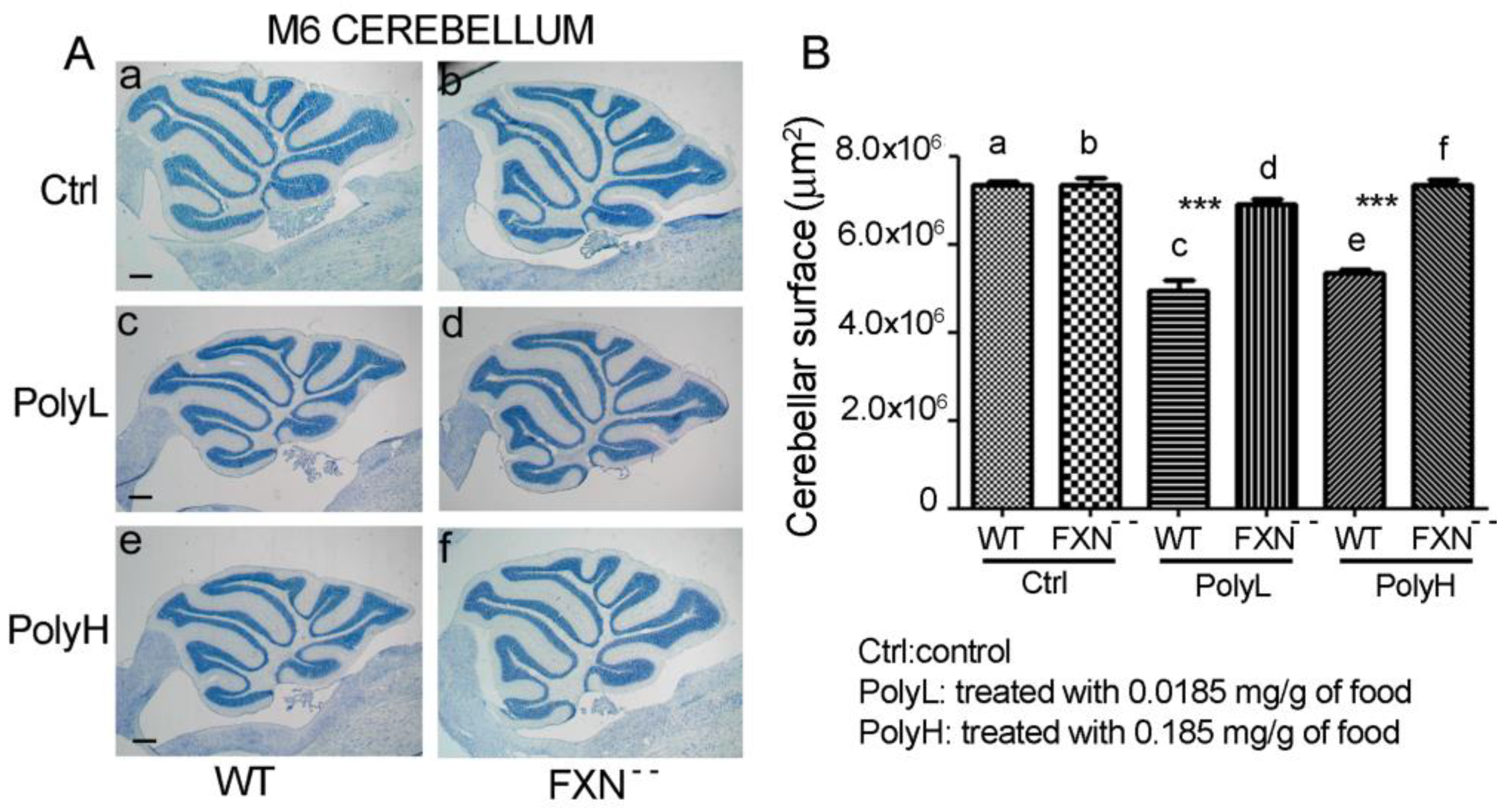
2.1.1. Effects of Frataxin Gene Mutation on Cerebellar Development
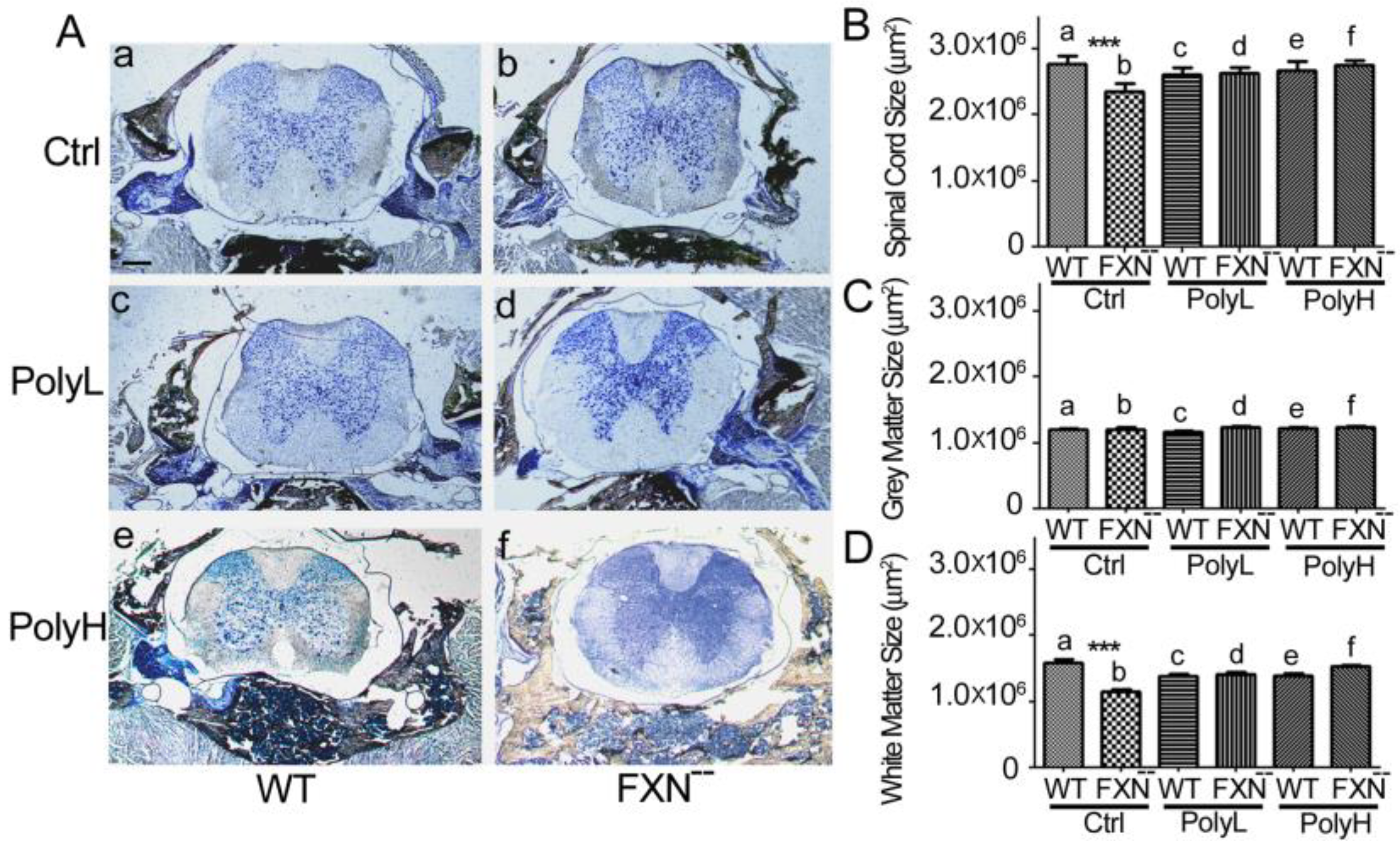
2.1.2. Effects of Frataxin Gene Mutation on the Spinal Cord Development
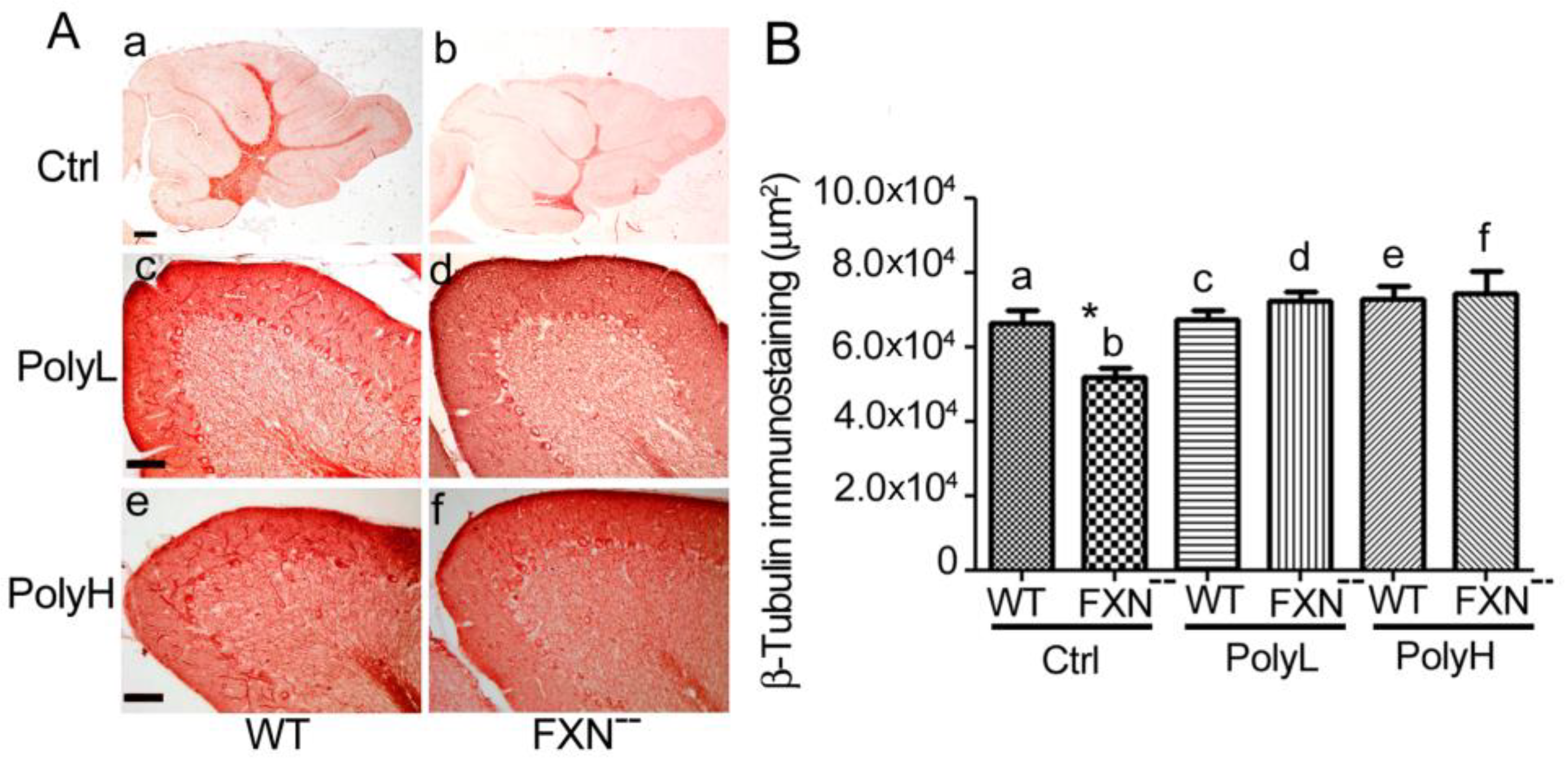
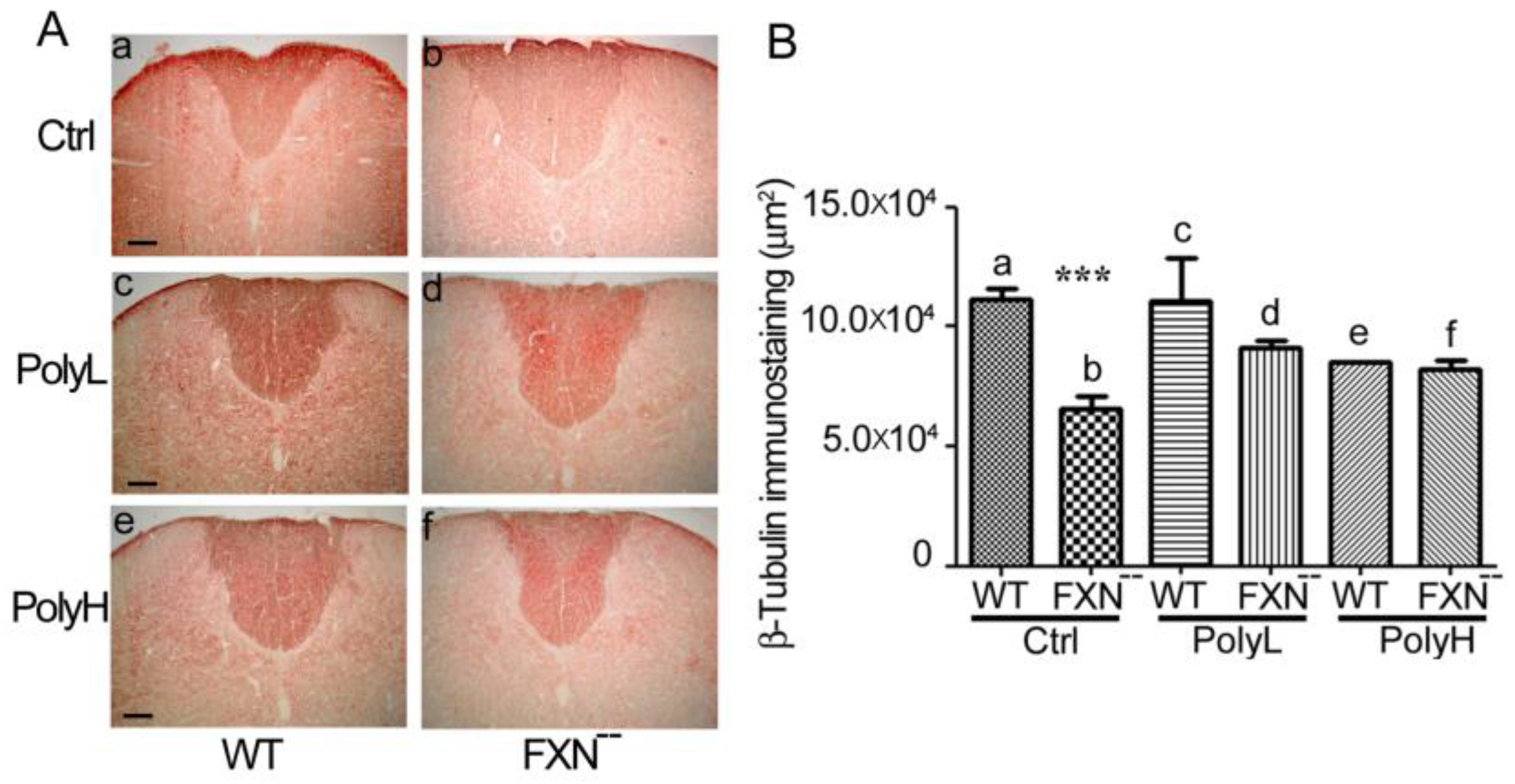
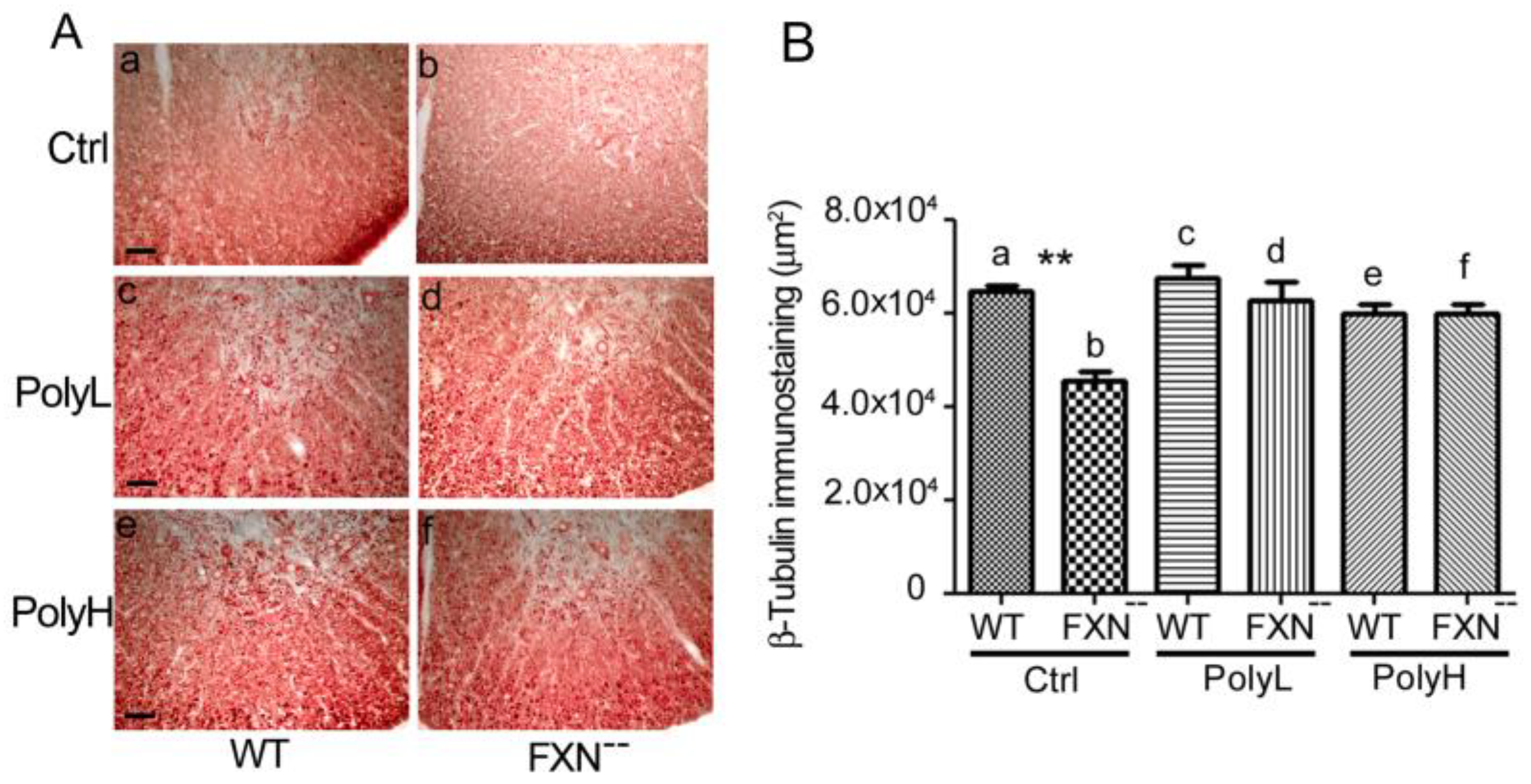
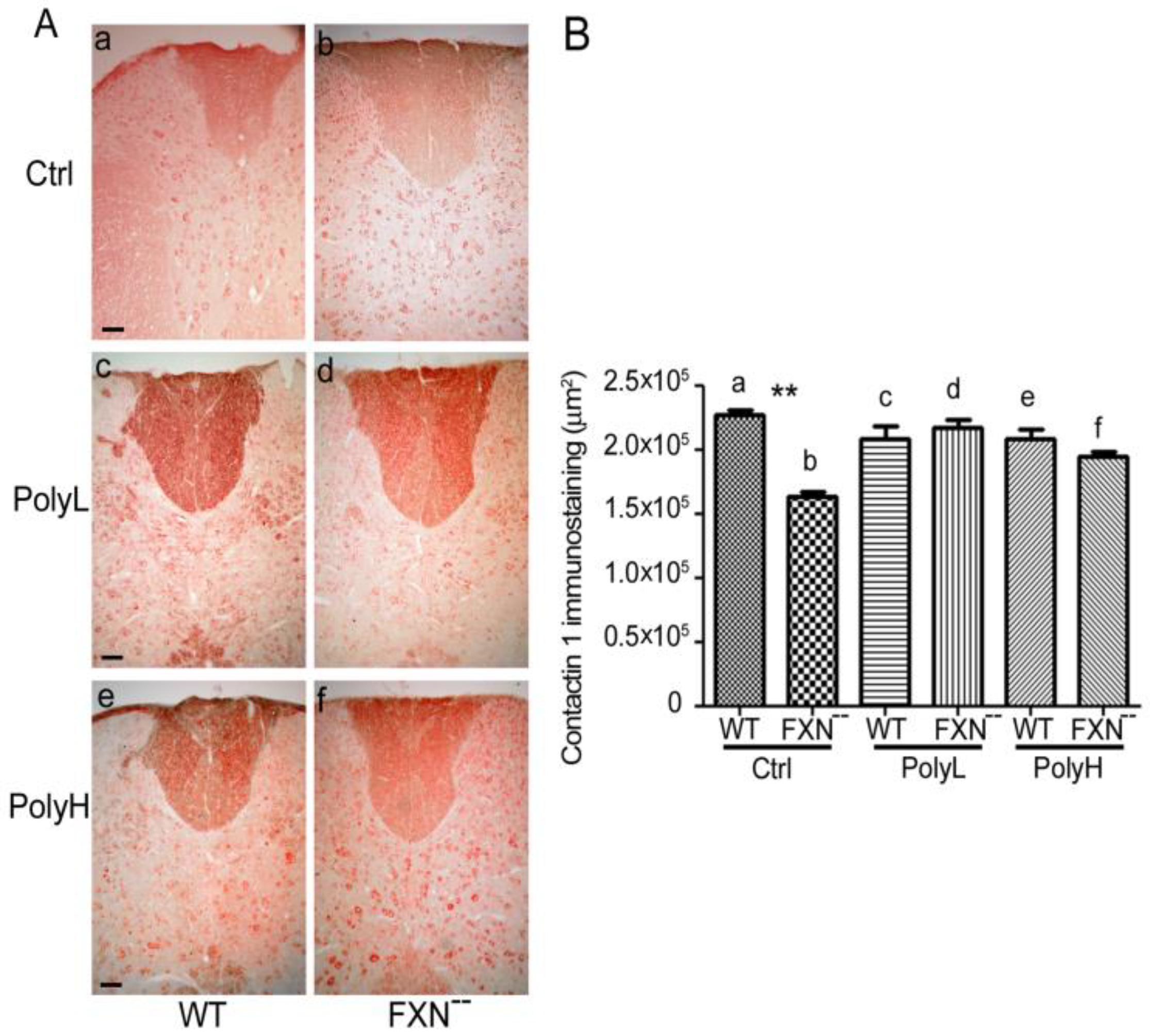
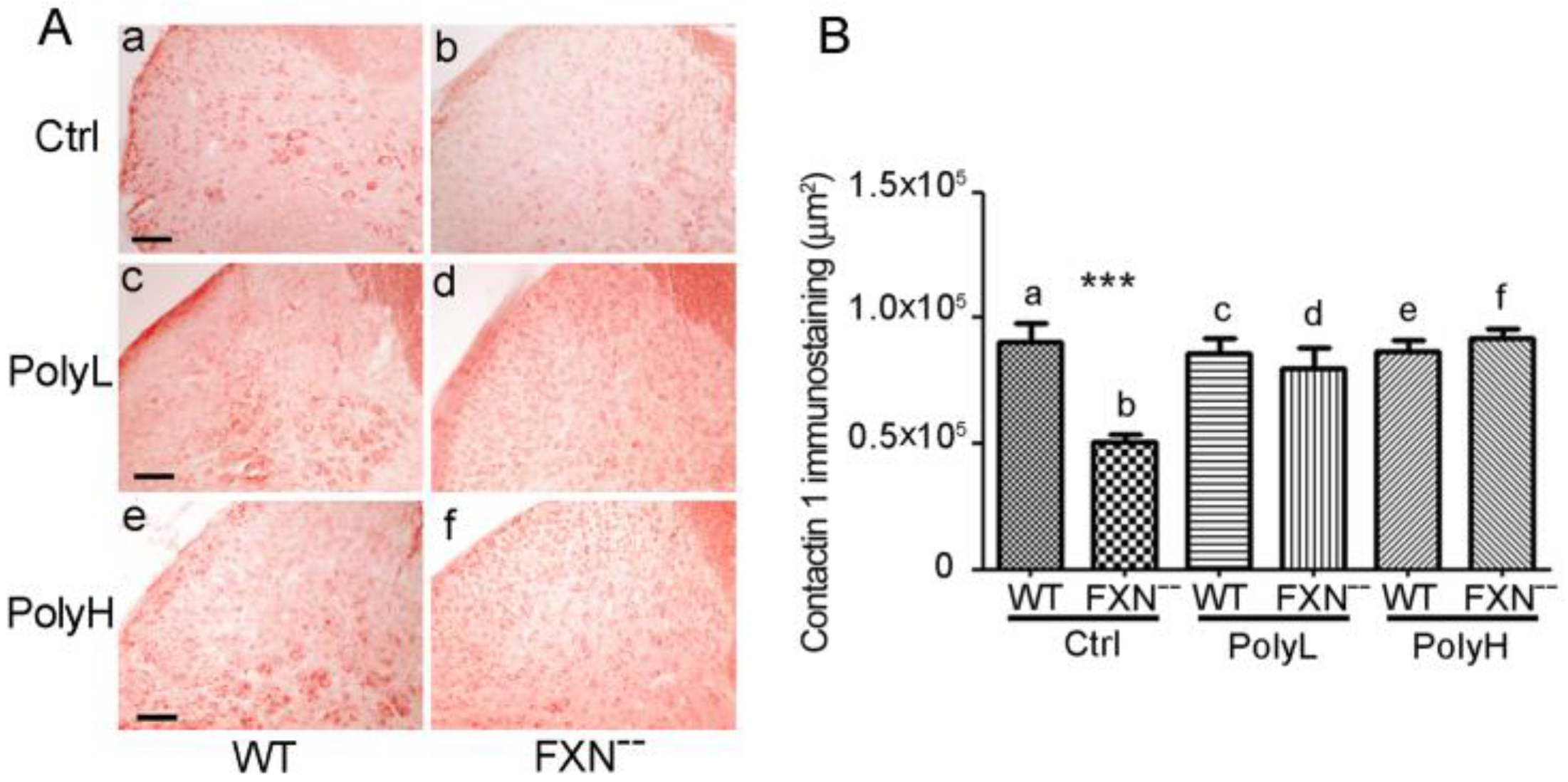
2.1.3. Contactin 1 Expression Profile
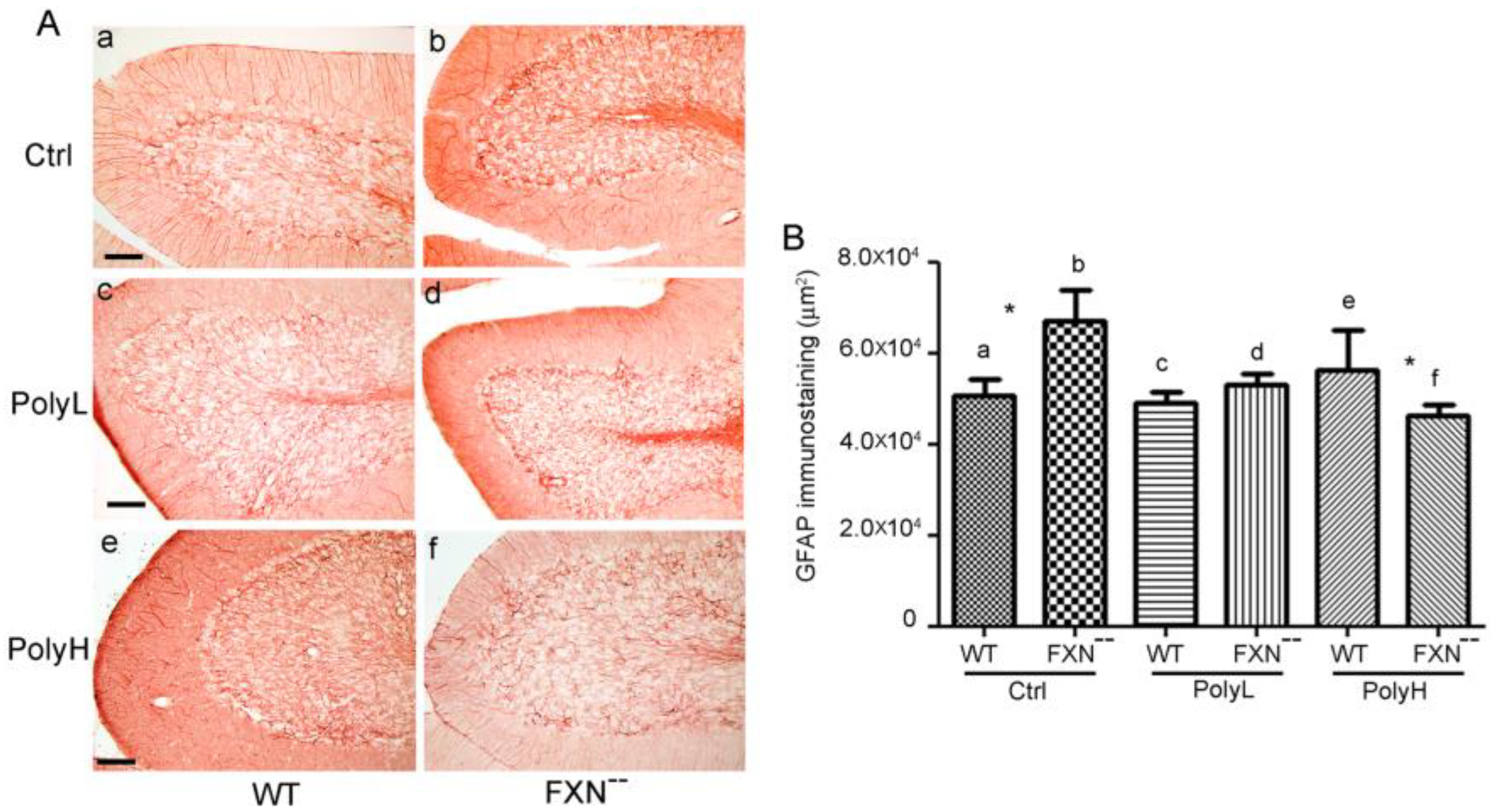
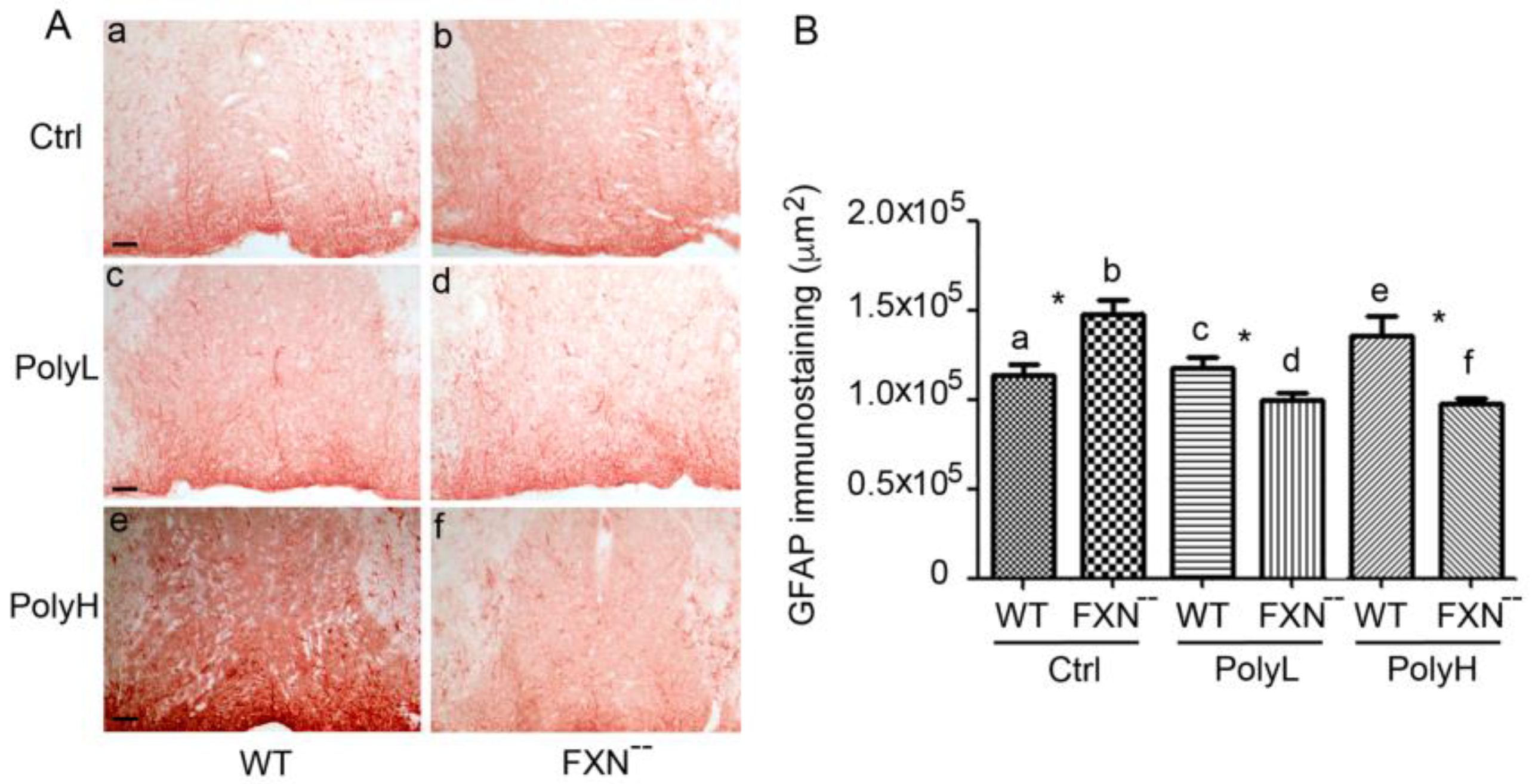
2.2. Glial Cells Phenotype
3. Discussion
4. Materials and Methods
4.1. Transgenic Mice Lines
4.2. Phenotype Analysis
4.3. Morphological and Morphometric Analysis
4.4. Statistical Analyses
4.5. Animal Breeding
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cook, A.; Giunti, P. Friedreich’s ataxia: Clinical features, pathogenesis and management. Br. Med. Bull. 2017, 124, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H. The neuropathology of the adult cerebellum. Handb. Clin. Neurol. 2018, 154, 129–149. [Google Scholar] [PubMed]
- Delatycki, M.B.; Bidichandani, S.I. Friedreich ataxia-pathogenesis and implications for therapies. Neurobiol. Dis. 2019, 132. [Google Scholar] [CrossRef]
- Pandolfo, M. Friedreich ataxia. Semin. Pediatr. Neurol. 2003, 10, 163–172. [Google Scholar] [CrossRef]
- Pandolfo, M. Friedreich ataxia. Arch Neurol. 2008, 65, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M.; Pastore, A. The pathogenesis of Friedreich ataxia and the structure and function of Frataxin. J. Neurol. 2009, 256, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H. Friedreich’s ataxia: Pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011, 303, 1–12. [Google Scholar] [CrossRef]
- Koeppen, A.H. The Neuropathology of Spinocerebellar Ataxia Type 3/Machado-Joseph Disease. Adv. Exp. Med. Biol. 2018, 1049, 233–241. [Google Scholar]
- Parkinson, M.H.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Giunti, P. Clinical features of Friedreich’s ataxia: Classical and atypical phenotypes. J. Neurochem. 2013, 126, 103–117. [Google Scholar] [CrossRef]
- Wolf, N.I.; Koenig, M. Progressive cerebellar atrophy: Hereditary ataxias and disorders with spinocerebellar degeneration. Handb. Clin. Neurol. 2013, 113, 1869–1878. [Google Scholar]
- Chiang, S.; Kovacevic, Z.; Sahni, S.; Lane, D.J.; Merlot, A.M.; Kalinowski, D.S.; Huang, M.L.; Richardson, D.R. Frataxin and the molecular mechanism of mitochondrial iron-loading in Friedreich’s ataxia. Clin. Sci. (Lond.) 2016, 130, 853–870. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.; Kalinowski, D.S.; Jansson, P.J.; Richardson, D.R.; Huang, M.L. Mitochondrial dysfunction in the neuro-degenerative and cardio-degenerative disease, Friedreich’s ataxia. Neurochem. Int. 2018, 117, 35–48. [Google Scholar] [CrossRef]
- Serrano, M. Epigenetic cerebellar diseases. In Handbook Clinical of Neurology; Aminoff, M.J., Boller, F., Swaab, D.F., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 155, pp. 227–244. [Google Scholar]
- Simon, D.; Seznec, H.; Gansmuller, A.; Carelle, N.; Weber, P.; Metzger, D.; Rustin, P.; Koenig, M.; Puccio, H. Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J. Neurosci. 2004, 24, 1987–1995. [Google Scholar] [CrossRef] [PubMed]
- Jayadev, S.; Bird, T.D. Hereditary ataxias: Overview. Genet. Med. 2013, 15, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Bereznyakova, O.; Dupré, N. Spastic ataxias. Handb. Clin. Neurol. 2018, 155, 191–203. [Google Scholar] [PubMed]
- Weidemann, F.; Störk, S.; Liu, D.; Hu, K.; Herrmann, S.; Ertl, G.; Niemann, M. Cardiomyopathy of Friedreich ataxia. J. Neurochem. 2013, 126, 88–93. [Google Scholar] [CrossRef]
- Sommerville, R.B.; Vincenti, M.G.; Winborn, K.; Casey, A.; Stitziel, N.O.; Connolly, A.M.; Mann, D.L. Diagnosis and management of adult hereditary cardio-neuromuscular disorders: A model for the multidisciplinary care of complex genetic disorders. Trends Cardiovasc. Med. 2017, 27, 51–58. [Google Scholar] [CrossRef]
- Hanson, E.; Sheldon, M.; Pacheco, B. Alkubeysi, M.; Raizada, V. Heart disease in Friedreich’s ataxia. World J. Cardiol. 2019, 11, 1–12. [Google Scholar] [CrossRef]
- González-Cabo, P.; Palau, F. Mitochondrial pathophysiology in Friedreich’s ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 53–64. [Google Scholar] [CrossRef]
- Gomes, C.M.; Santos, R. Neurodegeneration in Friedreich’s ataxia: From defective to oxidative stress. Oxid. Med. Cell Longev. 2013, 487534. [Google Scholar]
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The role of oxidative stress in Friedreich’s. ataxia. FEBS Lett. 2018, 592, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Li, K. Iron Pathophysiology in Friedreich’s Ataxia. Adv. Exp. Med. Biol. 2019, 1173, 125–143. [Google Scholar] [PubMed]
- Pastore, A.; Puccio, H. Frataxin: A protein in search for a function. J. Neurochem. 2013, 126, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Alfadhel, M.; Nashabat, M.; Abu Ali, Q.; Hundallah, K. Mitochondrial iron-sulfur cluster biogenesis from molecular understanding to clinical disease. Neurosciences 2017, 22, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Fox, N.G.; Yu, X.; Feng, X.; Bailey, H.J.; Martelli, A.; Nabhan, J.F.; Strain-Damerell, C.; Bulawa, C.; Yue, W.W.; Han, S. Structure of the human Frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef]
- Sandi, C.; Sandi, M.; Anjomani Virmouni, S.; Al-Mahdawi, S.; Pook, M.A. Epigenetic-based therapies for Friedreich ataxia. Front. Genet. 2014, 5, 165. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Becker, A.B.; Qian, J.; Feustel, P.J. Friedreich Ataxia: Hypoplasia of Spinal Cord and Dorsal Root Ganglia. J. Neuropathol. Exp. Neurol. 2017, 76, 101–108. [Google Scholar] [CrossRef][Green Version]
- Proske, U. What is the role of muscle receptors in proprioception? Muscle Nerve. 2005, 31, 780–787. [Google Scholar] [CrossRef]
- Imai, F.; Yoshida, Y. Molecular mechanisms underlying monosynaptic sensory-motor circuit development in the spinal cord. Dev. Dyn. 2018, 247, 581–587. [Google Scholar] [CrossRef]
- Selvadurai, L.P.; Harding, I.H.; Corben, L.A.; Stagnitti, M.R.; Storey, E.; Egan, G.F.; Delatycki, M.B.; Georgiou-Karistianis, N. Cerebral and cerebellar grey matter atrophy in Friedreich ataxia: The IMAGE-FRDA study. J. Neurol. 2016, 263, 2215–2223. [Google Scholar] [CrossRef]
- Selvadurai, L.P.; Harding, I.H.; Corben, L.A.; Georgiou-Karistianis, N. Cerebral abnormalities in Friedreich ataxia: A review. Neurosci. Biobehav. Rev. 2018, 84, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Nazıroğlu, M.; Braidy, N. Thermo-Sensitive TRP Channels: Novel Targets for Treating Chemotherapy-Induced Peripheral Pain. Front. Physiol. 2017, 8, 1040. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Lodi, R.; Tonon, C.; D’Agata, V.; Sapienza, M.; Scapagnini, G.; Mangiameli, A.; Pennisi, G.; Stella, A.M.; Butterfield, D.A. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J. Neurol. Sci. 2005, 233, 145–162. [Google Scholar] [CrossRef]
- Richardson, T.E.; Kelly, H.N.; Yu, A.E.; Simpkins, J.W. Therapeutic strategies in Friedreich’s ataxia. Brain Res. 2013, 1514, 91–97. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aranca, T.V.; Jones, T.M.; Shaw, J.D.; Staffetti, J.S.; Ashizawa, T.; Kuo, S.H.; Fogel, B.L.; Wilmot, G.R.; Perlman, S.L.; Onyik, C.U.; et al. Emerging therapies in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2016, 6, 49–65. [Google Scholar] [CrossRef]
- Kearney, M.; Orrell, R.W.; Fahey, M.; Pandolfo, M. Pharmacological treatments for Friedreich ataxia. Cochrane Database Syst. Rev. 2016, 2016, CD007791. [Google Scholar] [CrossRef]
- Strawser, C.; Schadt, K.; Hauser, L.; McCormick, A.; Wells, M.; Larkindale, J.; Lin, H.; Lynch, D.R. Pharmacological therapeutics in Friedreich ataxia: The present state. Expert Rev. Neurother. 2017, 17, 895–907. [Google Scholar] [CrossRef]
- Gottesfeld, J.M. Molecular Mechanisms and Therapeutics for the GAATTC Expansion Disease Friedreich Ataxia. Neurotherapeutics 2019, 16, 1032–1049. [Google Scholar] [CrossRef]
- Schirinzi, T.; Sancesario, A.; Bertini, E.; Castelli, E.; Vasco, G. Speech and Language Disorders in Friedreich Ataxia: Highlights on Phenomenology, Assessment, and Therapy. Cerebellum 2020, 19, 126–130. [Google Scholar] [CrossRef]
- Zhang, S.; Napierala, M.; Napierala, J.S. Therapeutic Prospects for Friedreich’s Ataxia. Trends Pharmacol. Sci. 2019, 40, 229–233. [Google Scholar] [CrossRef]
- Jones, J.; Estirado, A.; Redondo, C.; Pacheco-Torres, J.; Sirerol-Piquer, M.S.; Garcia-Verdugo, J.M.; Martinez, S. Mesenchymal stem cells improve motor functions and decrease neurodegeneration in ataxic mice. Mol. Ther. 2015, 23, 130–138. [Google Scholar] [CrossRef]
- Long, A.; Napierala, J.S.; Polak, U.; Hauser, L.; Koeppen, A.H.; Lynch, D.R.; Napierala, M. Somatic instability of the expanded GAA repeats in Friedreich’s ataxia. PLoS ONE 2017, 12, e0189990. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Magrone, T.; Magrone, M.; Russo, M.A.; Jirillo, E. Recent advances on the anti-inflammatory and anti-oxidant properties of red grape polyphenols: In vitro and in vivo studies. Antioxidants 2020, 9, 35. [Google Scholar] [CrossRef]
- Vauzour, D. Dietary polyphenols as modulators of brain functions: Biological actions and molecular mechanisms underpinning their beneficial effects. Oxid. Med. Cell. Longev. 2012, 2012, 914273. [Google Scholar] [CrossRef]
- Magrone, T.; Russo, M.A.; Jirillo, E. Cocoa and dark chocolate polyphenols: From Biology to Clinical Applications. Front. Immunol. 2017, 8, 677. [Google Scholar] [CrossRef] [PubMed]
- Marzulli, G.; Magrone, T.; Kawaguchi, K.; Kumazawa, Y.; Jirillo, E. Fermented grape marc (FGM): Immunomodulating properties and its potential exploitation in the treatment of neurodegenerative diseases. Curr. Pharm. Des. 2012, 18, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.C.; Xu, H.; Yuan, Y.; Chen, J.Y.; Zhang, Y.J.; Lin, Y.; Yuan, S.Y. Delayed Treatment with Green Tea Polyphenol EGCG Promotes Neurogenesis After Ischemic Stroke in Adult Mice. Mol. Neurobiol. 2017, 54, 3652–3664. [Google Scholar] [CrossRef]
- Singh, N.A.; Mandal, A.K.; Khan, Z.A. Potential neuroprotective properties of epigallocatechin-3-gallate (EGCG). Nutr. J. 2015, 15, 60. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhao, Y.; Nan, Y.; Wang, X.; Chen, Y.; Wang, S. (-)-Epigallocatechin-3-gallate ameliorates memory impairment and rescues the abnormal synaptic protein levels in the frontal cortex and hippocampus in a mouse model of Alzheimer’s disease. Neuroreport 2017, 28, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Ayyalasomayajula, N.; Ajumeera, R.; Chellu, C.S.; Challa, S. Mitigative effects of epigallocatechin gallate in terms of diminishing apoptosis and oxidative stress generated by the combination of lead and amyloid peptides in human neuronal cells. J. Biochem. Mol. Toxicol. 2019, 33, e22393. [Google Scholar] [CrossRef] [PubMed]
- Park, D.J.; Kang, J.B.; Koh, P.O. Epigallocatechin gallate alleviates neuronal cell damage against focal cerebral ischemia in rats. J. Vet. Med. Sci. 2020, 82, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Raposo, D.; Morgado, C.; Pereira-Terra, P.; Tavares, I. Nociceptive spinal cord neurons of laminae I-III exhibit oxidative stress damage during diabetic neuropathy which is prevented by early antioxidant treatment with epigallocatechin-gallate (EGCG). Brain Res. Bull. 2015, 110, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Gennarini, G.; Bizzoca, A.; Picocci, S.; Puzzo, D.; Corsi, P.; Furley, A.J.W. The role of Gpi-anchored axonal glycoproteins in neural development and neurological disorders. Mol. Cell Neurosci. 2017, 81, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Pekna, M. Astrocyte reactivity and reactive astrogliosis: Costs and benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M. Reactive gliosis in the pathogenesis of CNS diseases. Biochim. Biophys. Acta 2016, 1862, 483–491. [Google Scholar] [CrossRef]
- Perdomini, M.; Hick, A.; Puccio, H.; Pook, M.A. Animal and cellular models of Friedreich ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 65–79. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, K.K. Glial fibrillary acidic protein: From intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef]
- Storey, E. Genetic cerebellar ataxias. Semin. Neurol. 2014, 34, 280–292. [Google Scholar] [CrossRef]
- Morral, J.A.; Davis, A.N.; Qian, J.; Gelman, B.B.; Koeppen, A.H. Pathology and pathogenesis of sensory neuropathy in Friedreich’s ataxia. Acta Neuropathol. 2010, 120, 97–108. [Google Scholar] [CrossRef]
- Piermarini, E.; Cartelli, D.; Pastore, A.; Tozzi, G.; Compagnucci, C.; Giorda, E.; D’Amico, J.; Petrini, S.; Bertini, E.; Cappelletti, G.; et al. Frataxin silencing alters microtubule stability in motor neurons: Implications for Friedreich’s ataxia. Hum. Mol. Genet. 2016, 25, 4288–4301. [Google Scholar] [CrossRef] [PubMed]
- Tamarit, J.; Obis, E.; Ros, J. Oxidative stress and altered lipid metabolism in Friedreich ataxia. Free Radic Biol. Med. 2016, 100, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Ramirez, R.L.; Becker, A.B.; Bjork, S.T.; Levi, S.; Santambrogio, P.; Parsons, P.J.; Kruger, P.C.; Yang, K.X.; Feustel, P.J.; et al. The pathogenesis of cardiomyopathy in Friedreich ataxia. PLoS ONE 2015, 10, e0116396. [Google Scholar] [CrossRef] [PubMed]
- Middeldorp, J.; Hol, E.M. GFAP in health and disease. Prog. Neurobiol. 2011, 93, 421–443. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A central element in neurological diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Avci, H.X.; Leist, M.; Kobolák, J.; Dinnyés, A. Astrocyte Differentiation of Human Pluripotent Stem Cells: New Tools for Neurological Disorder Research. Front. Cell. Neurosci. 2016, 10, 215. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Hara, M.; Kobayakawa, K.; Matsumoto, Y.; Nakashima, Y. Astrocyte reactivity and astrogliosis after spinal cord injury. Neurosci. Res. 2018, 126, 39–43. [Google Scholar] [CrossRef]
- Franco, C.; Genis, L.; Navarro, J.A.; Perez-Domper, P.; Fernandez, A.M.; Schneuwly, S.; Torres Alemán, I. A role for astrocytes in cerebellar deficits in frataxin deficiency: Protection by insulin-like growth factor I. Mol. Cell Neurosci. 2017, 80, 100–110. [Google Scholar] [CrossRef]
- Hang, L.; Basil, A.H.; Lim, K.L. Nutraceuticals in Parkinson’s Disease. Neuromolecular Med. 2016, 18, 306–321. [Google Scholar] [CrossRef]
- Xu, Q.; Langley, M.; Kanthasamy, A.G.; Reddy, M.B. Epigallocatechin Gallate Has a Neurorescue Effect in a Mouse Model of Parkinson Disease. J. Nutr. 2017, 147, 1926–1931. [Google Scholar] [CrossRef]
- Syarifah-Noratiqah, S.B.; Naina-Mohamed, I.; Zulfarina, M.S.; Qodriyah, H.M.S. Natural Polyphenols in the Treatment of Alzheimer’s Disease. Curr. Drug Targets 2018, 19, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Molino, S.; Dossena, M.; Buonocore, D.; Ferrari, F.; Venturini, L.; Ricevuti, G.; Verri, M. Polyphenols in dementia: From molecular basis to clinical trials. Life Sci. 2016, 161, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Pervin, M.; Unno, K.; Ohishi, T.; Tanabe, H.; Miyoshi, N.; Nakamura, Y. Beneficial Effects of Green Tea Catechins on Neurodegenerative Diseases. Molecules 2018, 23, 1297. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, R.; Mousseau, N. Mitigating Alzheimer’s Disease with Natural Polyphenols: A Review. Curr. Alzheimer Res. 2019, 16, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Koyama, N.; Tan, J.; Segawa, T.; Maeda, M.; Town, T. Combined treatment with the phenolics (-)-epigallocatechin-3-gallate and ferulic acid improves cognition and reduces Alzheimer-like pathology in mice. J. Biol. Chem. 2019, 294, 2714–2731. [Google Scholar] [CrossRef]
- Chatterjee, M.; Schild, D.; Teunissen, C.E. Contactins in the central nervous system: Role in health and disease. Neural. Regen. Res. 2020, 14, 206–216. [Google Scholar]
- Gennarini, G.; Durbec, P.; Boned, A.; Rougon, G.; Goridis, C. Transfected F3/F11 neuronal cell surface protein mediates intercellular adhesion and promotes neurite outgrowth. Neuron 1991, 6, 595–606. [Google Scholar] [CrossRef]
- Faivre-Sarrailh, C.; Devaux, J.J. Neuro-glial interactions at the nodes of Ranvier: Implication in health and diseases. Front. Cell. Neurosci. 2013, 7, 196. [Google Scholar] [CrossRef]
- Colakoğlu, G.; Bergstrom-Tyrberg, U.; Berglund, E.O.; Ranscht, B. Contactin1 regulates myelination and nodal/paranodal domain organization in the central nervous system. Proc. Natl. Acad. Sci. USA 2014, 111, E394–E403. [Google Scholar] [CrossRef]
- Manso, C.; Querol, L.; Mekaouche, M.; Illa, I.; Devaux, J.J. Contactin-1 IgG4 antibodies cause paranode dismantling and conduction defects. Brain 2016, 139, 1700–1712. [Google Scholar] [CrossRef]
- Zoupi, L.; Savvaki, M.; Kalemaki, K.; Kalafatakis, I.; Sidiropoulou, K.; Karagogeos, D. The function of contactin-2/TAG-1 in oligodendrocytes in health and demyelinating pathology. Glia 2018, 66, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Ranscht, B. Sequence of Contactin, a 130-kD glycoprotein concentrated in areas of interneuronal contact, defines a new member of the immunoglobulin supergene family in the nervous system. J. Cell Biol. 1988, 107, 1561–1573. [Google Scholar] [CrossRef]
- Gennarini, G.; Cibelli, G.; Rougon, G.; Mattei, M.G.; Goridis, C. The mouse neuronal cell surface protein F3: A phosphatidylinositol-anchored member of the immunoglobulin superfamily related to chicken contactin. J. Cell Biol. 1989, 109, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Oguro-Ando, A.; Zuko, A.; Kleijer, K.T.E.; Burbach, J.P.H. A current view on contactin-4, -5 and -6: Implications in neurodevelopmental disorders. Mol. Cell Neurosci. 2017, 81, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Fehlmi, J.; Scherer, S.S.; Willison, H.J.; Rinaldi, S. Nodes, paranodes and neuropathies. J. Neurol. Neurosurg. Psychiatry 2018, 89, 61–71. [Google Scholar] [CrossRef]
- Zou, Y.; Zhang, W.F.; Liu, H.Y.; Li, X.; Zhang, X.; Ma, X.F.; Sun, Y.; Jiang, S.Y.; Ma, Q.H.; Xu, D.E. Structure and function of the contactin-associated protein family in myelinated axons and their relationship with nerve diseases. Neural Regen. Res. 2017, 12, 1551–1558. [Google Scholar]
- Scott, R.; Sánchez-Aguilera, A.; van Elst, K.; Lim, L.; Dehorter, N.; Bae, S.E.; Bartolini, G.; Peles, E.; Kas, M.J.H.; Bruining, H.; et al. Loss of Cntnap2 causes axonal excitability deficits, developmental delay in cortical myelination, and abnormal stereotyped motor behavior. Cereb. Cortex 2019, 29, 586–597. [Google Scholar] [CrossRef]
- Dubessy, A.L.; Mazuir, E.; Rappeneau, Q.; Ou, S.; Abi Ghanem, C.; Piquand, K.; Aigrot, M.S.; Thétiot, M.; Desmazières, A.; Chan, E.; et al. Role of a Contactin multi-molecular complex secreted by oligodendrocytes in nodal protein clustering in the CNS. Glia 2019, 67, 2248–2263. [Google Scholar] [CrossRef]
- Leshchyns’ka, I.; Sytnyk, V. Synaptic Cell Adhesion Molecules in Alzheimer’s Disease. Neural Plast. 2016, 6427537. [Google Scholar] [CrossRef]
- Torres, V.I.; Vallejo, D.; Inestrosa, N.C. Emerging Synaptic Molecules as Candidates in the Etiology of Neurological Disorders. Neural Plast. 2017, 8081758. [Google Scholar] [CrossRef]
- Chatterjee, M.; Nöding, B.; Willemse, E.A.J.; Koel-Simmelink, M.J.A.; van der Flier, W.M.; Schild, D.; Teunissen, C. Detection of contactin-2 in cerebrospinal fluid (CSF) of patients with Alzheimer’s disease using Fluorescence Correlation Spectroscopy (FCS). Clin. Biochem. 2017, 50, 1061–1066. [Google Scholar] [CrossRef]
- Chatterjee, M.; Teunissen, C. Contactins & Alzheimer’s Disease: Synaptic Proteins, Contactins may contribute to the pathology of Alzheimer’s disease. Neuroscience 2020, 424, 182–183. [Google Scholar] [PubMed]
- Zhang, R.; Engler, A.; Taylor, V. Notch: An interactive player in neurogenesis and disease. Cell Tissue Res. 2018, 371, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.; Zhang, R.; Taylor, V. Notch and Neurogenesis. Adv. Exp. Med. Biol. 2018, 1066, 223–234. [Google Scholar]
- Hu, Q.D.; Ang, B.T.; Karsak, M.; Hu, W.P.; Cui, X.Y.; Duka, T.; Takeda, Y.; Chia, W.; Sankar, N.; Ng, Y.K.; et al. F3/contactin acts as a functional ligand for Notch during oligodendrocyte maturation. Cell 2003, 115, 163–175. [Google Scholar] [CrossRef]
- Bizzoca, A.; Corsi, P.; Polizzi, A.; Pinto, M.F.; Xenaki, D.; Furley, A.J.; Gennarini, G. F3/Contactin acts as a modulator of neurogenesis during cerebral cortex development. Dev. Biol. 2012, 365, 133–151. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Picocci, S.; Bizzoca, A.; Corsi, P.; Magrone, T.; Jirillo, E.; Gennarini, G. Modulation of Nerve Cell Differentiation: Role of Polyphenols and of Contactin Family Components. Front. Cell Dev. Biol. 2019, 7, 119. [Google Scholar] [CrossRef] [PubMed]
- Magrone, T.; Spagnoletta, A.; Bizzoca, A.; Russo, M.A.; Jirillo, E.; Gennarini, G. Polyphenol effects on splenic cytokine response in post-weaning Contactin 1 overexpressing transgenic mice. Molecules 2019, 24, 2205. [Google Scholar] [CrossRef]
- Gu, L.T.; Yang, J.; Su, S.Z.; Liu, W.W.; Shi, Z.G.; Wang, Q.R. Green Tea Polyphenols Protects Cochlear Hair Cells from Ototoxicity by Inhibiting Notch Signalling. Neurochem. Res. 2015, 40, 1211–1219. [Google Scholar] [CrossRef]
- Virgintino, D.; Ambrosini, M.; D’Errico, P.; Bertossi, M.; Papadaki, C.; Karagogeos, D.; Gennarini, G. Regional distribution and cell type-specific expression of the mouse F3 axonal glycoprotein: A developmental study. J. Comp. Neurol. 1999, 413, 357–372. [Google Scholar] [CrossRef]
- Compton, A.G.; Albrecht, D.E.; Seto, J.T.; Cooper, S.T.; Ilkovski, B.; Jones, K.J.; Challis, D.; Mowat, D.; Ranscht, B.; Bahlo, M.; et al. Mutations in contactin-1, a neural adhesion and neuromuscular junction protein, cause a familial form of lethal congenital myopathy. Am. J. Hum. Genet. 2008, 83, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.A.; Lu, I.L.; Tsai, J.W. Contactin-1/F3 Regulates Neuronal Migration and Morphogenesis Through Modulating RhoA Activity. Front. Mol. Neurosci. 2018, 11, 422. [Google Scholar] [CrossRef] [PubMed]
- Ocklenburg, S.; Gerding, W.M.; Arning, L.; Genç, E.; Epplen, J.T.; Güntürkün, O.; Beste, C. Myelin Genes and the Corpus Callosum: Proteolipid Protein 1 (PLP1) and Contactin 1 (CNTN1) GeneVariation Modulates Interhemispheric Integration. Mol. Neurobiol. 2017, 54, 7908–7916. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.Y.; Takagishi, Y.; Okabe, E.; Chishima, Y.; Kanou, Y.; Murase, S.; Mizumura, K.; Inaba, M.; Komatsu, Y.; Hayashi, Y.; et al. A novel Caspr mutation causes the shambling mouse phenotype by disrupting axoglial interactions of myelinated nerves. J. Neuropathol. Exp. Neurol. 2009, 68, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Zarei-Kheirabadi, M.; Vaccaro, A.R.; Rahimi-Movaghar, V.; Kiani, S.; Baharvand, H. An Overview of Extrinsic and Intrinsic Mechanisms Involved in Astrocyte Development in the Central Nervous System. Stem Cells Dev. 2019, 29, 266–280. [Google Scholar] [CrossRef]
- Anjomani Virmouni, S.; Sandi, C.; Al-Mahdawi, S.; Pook, M.A. Cellular, molecular and functional characterisation of YAC transgenic mouse models of Friedreich ataxia. PLoS ONE 2014, 9, e107416. [Google Scholar] [CrossRef]
- Anjomani Virmouni, S.; Ezzatizadeh, V.; Sandi, C.; Sandi, M.; Al-Mahdawi, S.; Chutake, Y.; Pook, M.A. A novel GAA-repeat-expansion-based mouse model of Friedreich’s ataxia. Dis. Model. Mech. 2015, 8, 225–235. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bizzoca, A.; Caracciolo, M.; Corsi, P.; Magrone, T.; Jirillo, E.; Gennarini, G. Molecular and Cellular Substrates for the Friedreich Ataxia. Significance of Contactin Expression and of Antioxidant Administration. Molecules 2020, 25, 4085. https://doi.org/10.3390/molecules25184085
Bizzoca A, Caracciolo M, Corsi P, Magrone T, Jirillo E, Gennarini G. Molecular and Cellular Substrates for the Friedreich Ataxia. Significance of Contactin Expression and of Antioxidant Administration. Molecules. 2020; 25(18):4085. https://doi.org/10.3390/molecules25184085
Chicago/Turabian StyleBizzoca, Antonella, Martina Caracciolo, Patrizia Corsi, Thea Magrone, Emilio Jirillo, and Gianfranco Gennarini. 2020. "Molecular and Cellular Substrates for the Friedreich Ataxia. Significance of Contactin Expression and of Antioxidant Administration" Molecules 25, no. 18: 4085. https://doi.org/10.3390/molecules25184085
APA StyleBizzoca, A., Caracciolo, M., Corsi, P., Magrone, T., Jirillo, E., & Gennarini, G. (2020). Molecular and Cellular Substrates for the Friedreich Ataxia. Significance of Contactin Expression and of Antioxidant Administration. Molecules, 25(18), 4085. https://doi.org/10.3390/molecules25184085