
Unravelling the Photoprotective Mechanisms of Nature-Inspired Ultraviolet Filters Using Ultrafast Spectroscopy


Abstract
1. Introduction
1.1. Ultraviolet Radiation and Biological Systems
1.1.1. Previous Attempt to Address Sun Protection
1.1.2. Challenges with Existing Sunscreens
1.1.3. Nature-Inspired Sunscreens
1.1.4. Plant Ultraviolet Filters
1.1.5. Microbial Ultraviolet Filters
1.2. Experimental Technique
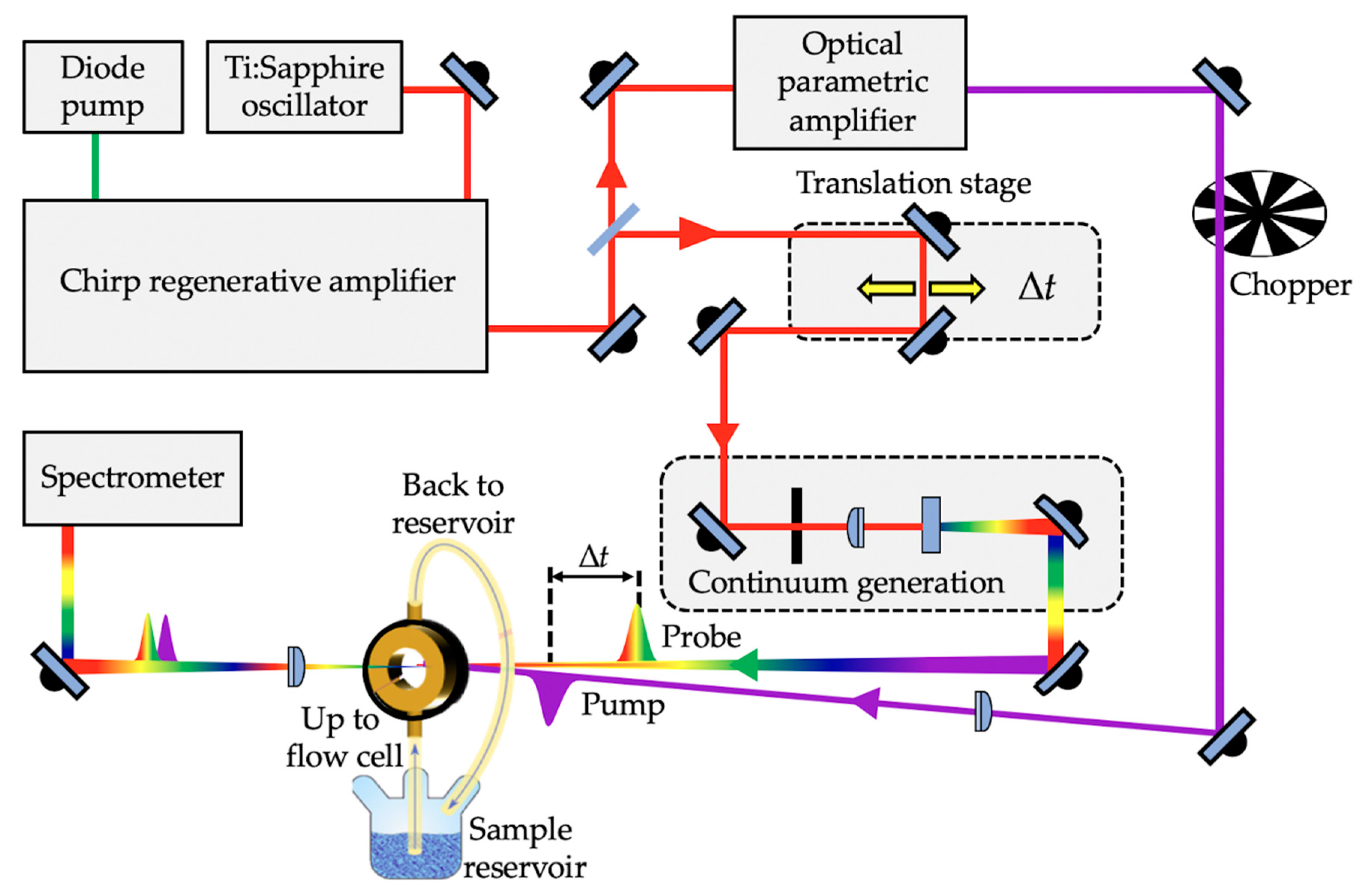
1.2.1. Femtosecond Pump–Probe Spectroscopy
1.2.2. Solution-Phase Transient Electronic Absorption Spectroscopy
1.2.3. Photophysical and Photochemical Processes
2. Case Studies
2.1. Plant-Inspired Ultraviolet Filters
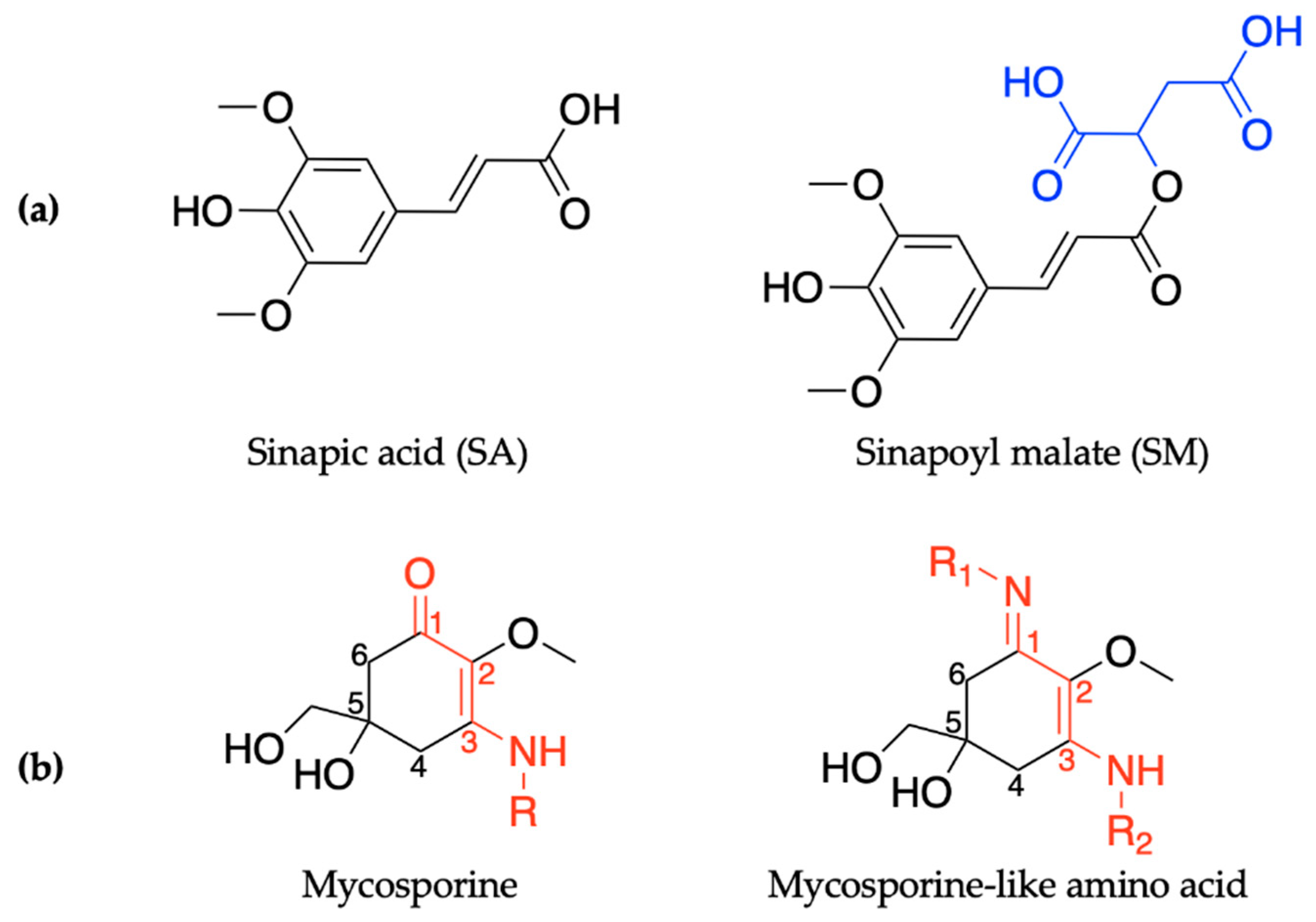
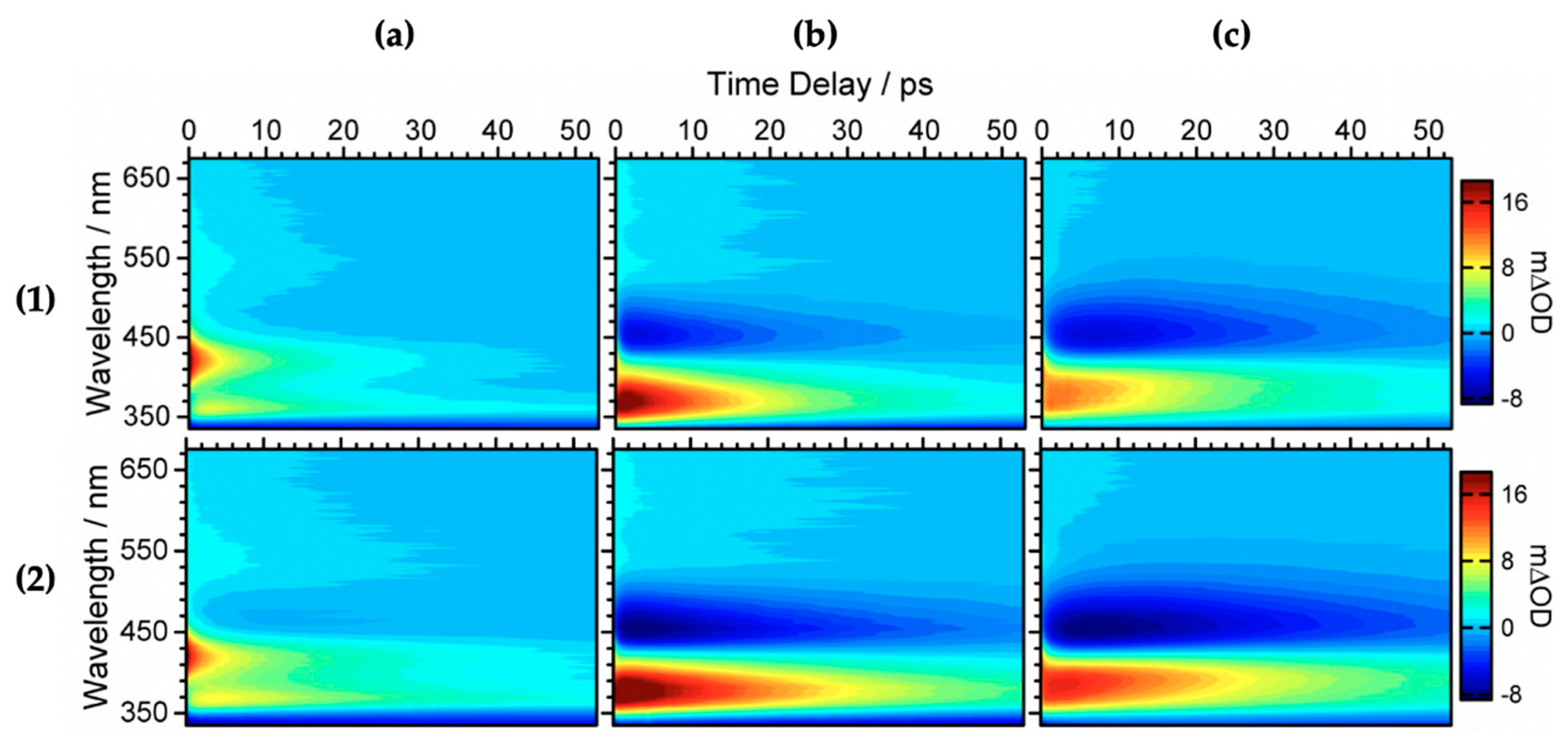
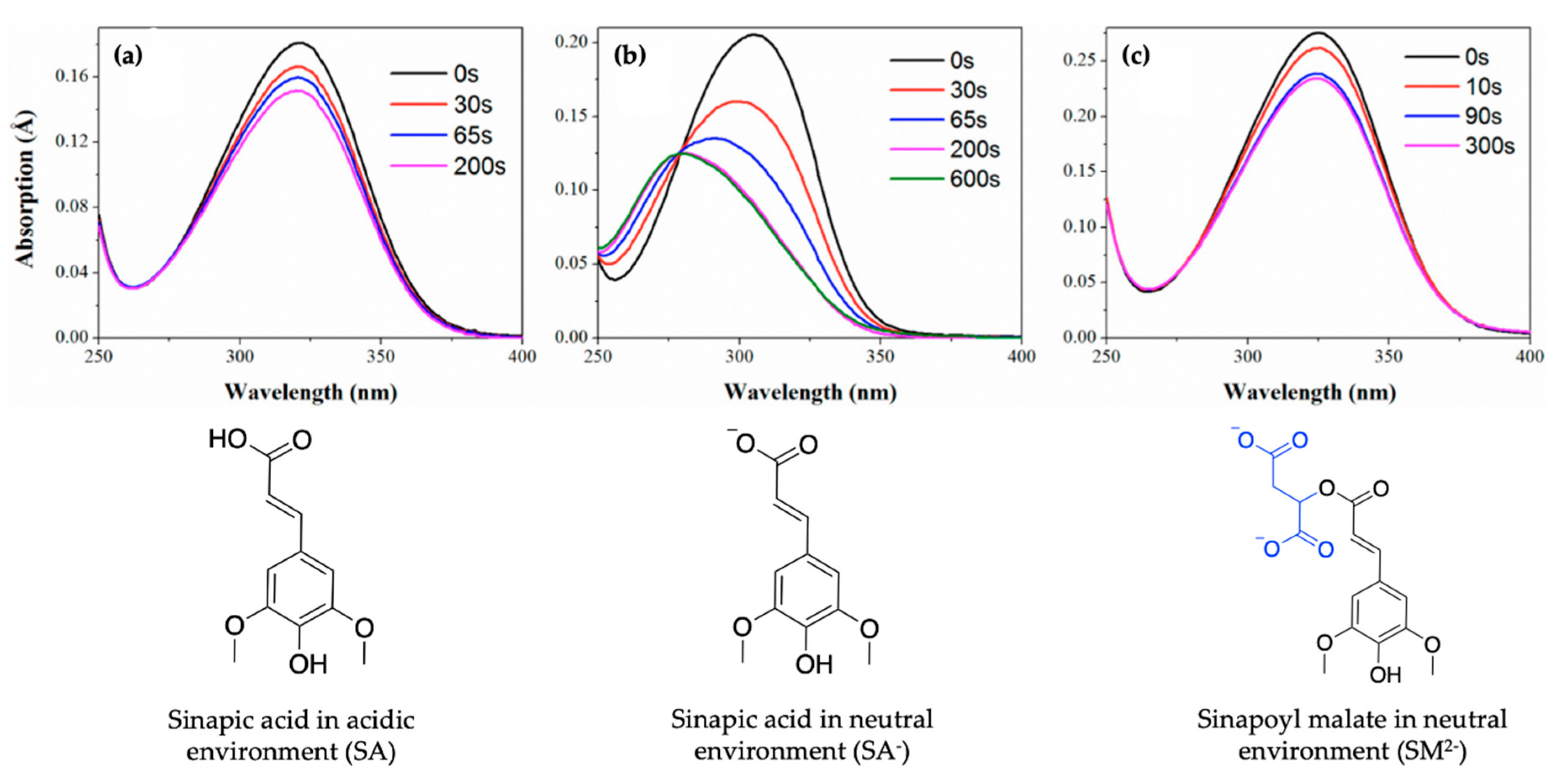
2.1.1. Sinapoyl Malate and Sinapic Acid
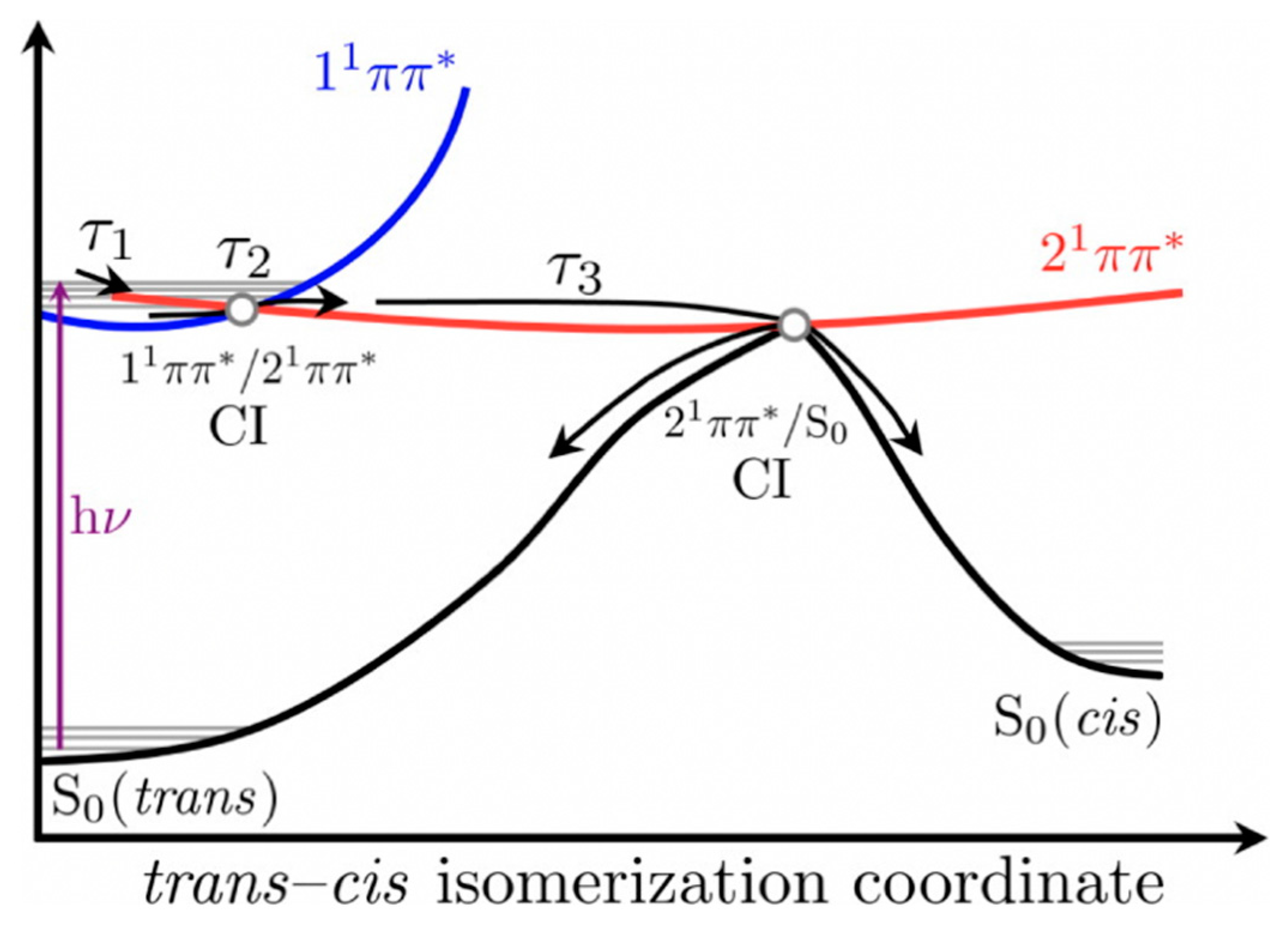
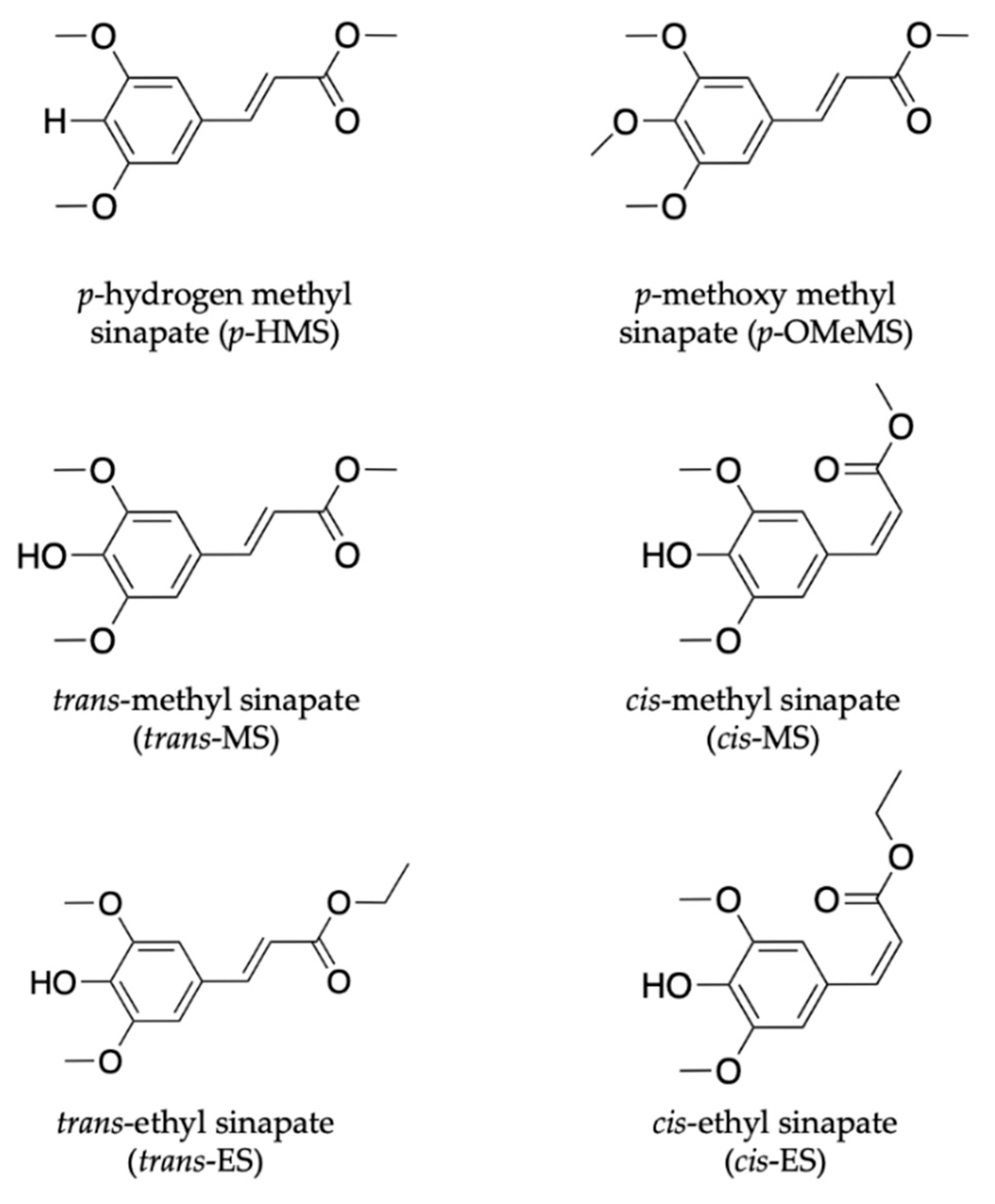
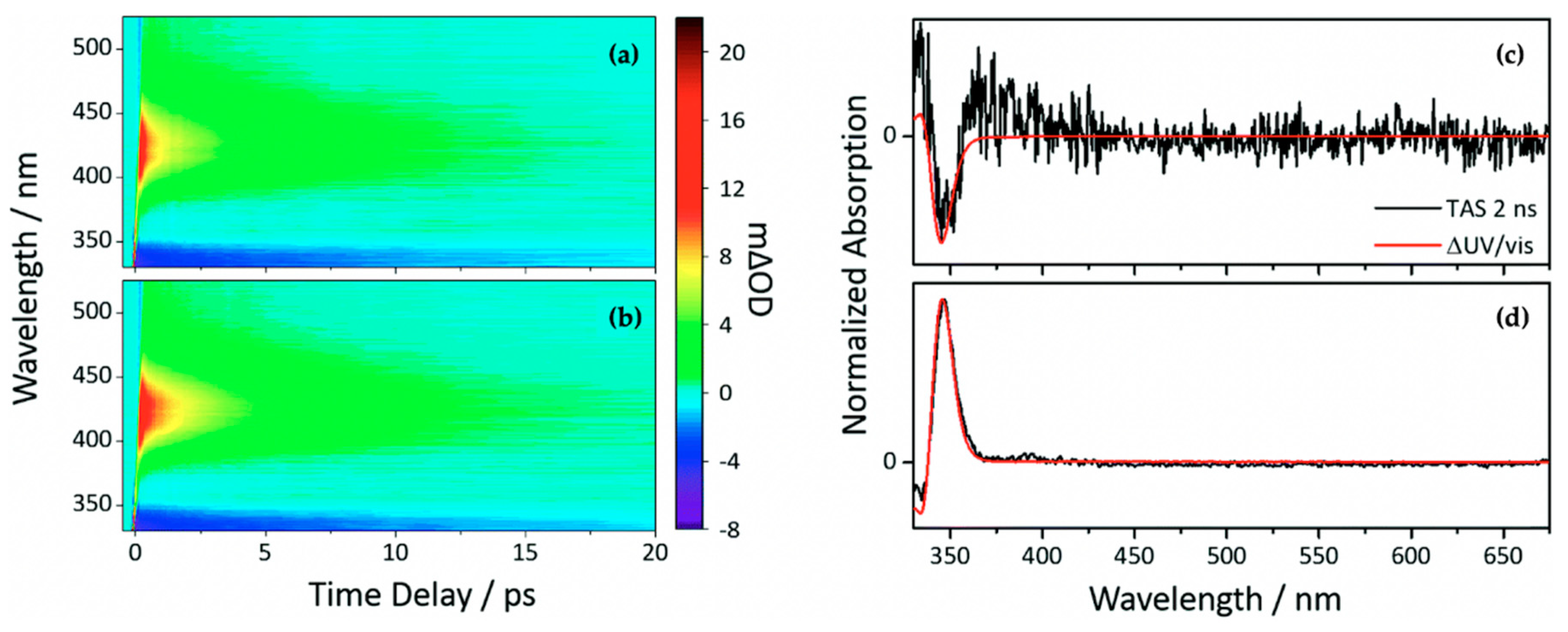
2.1.2. Sinapate Ester Derivatives
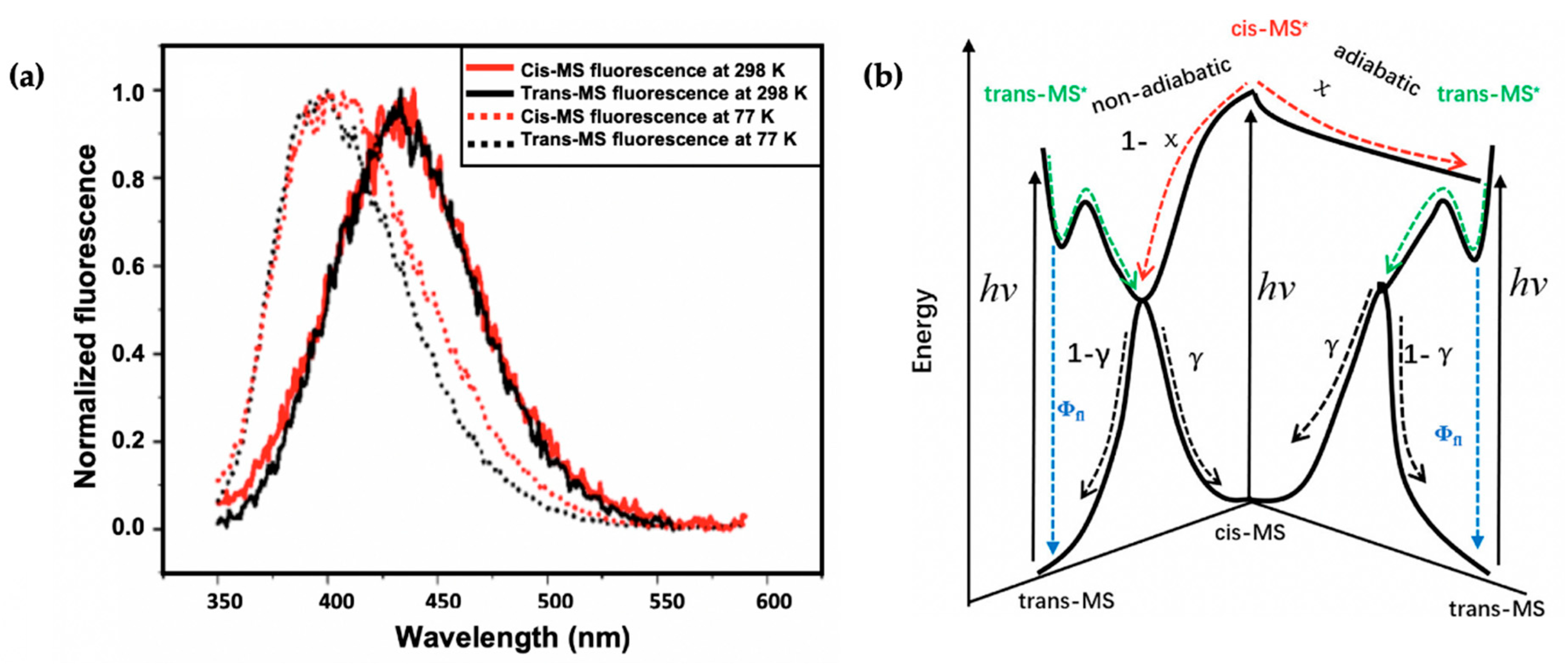
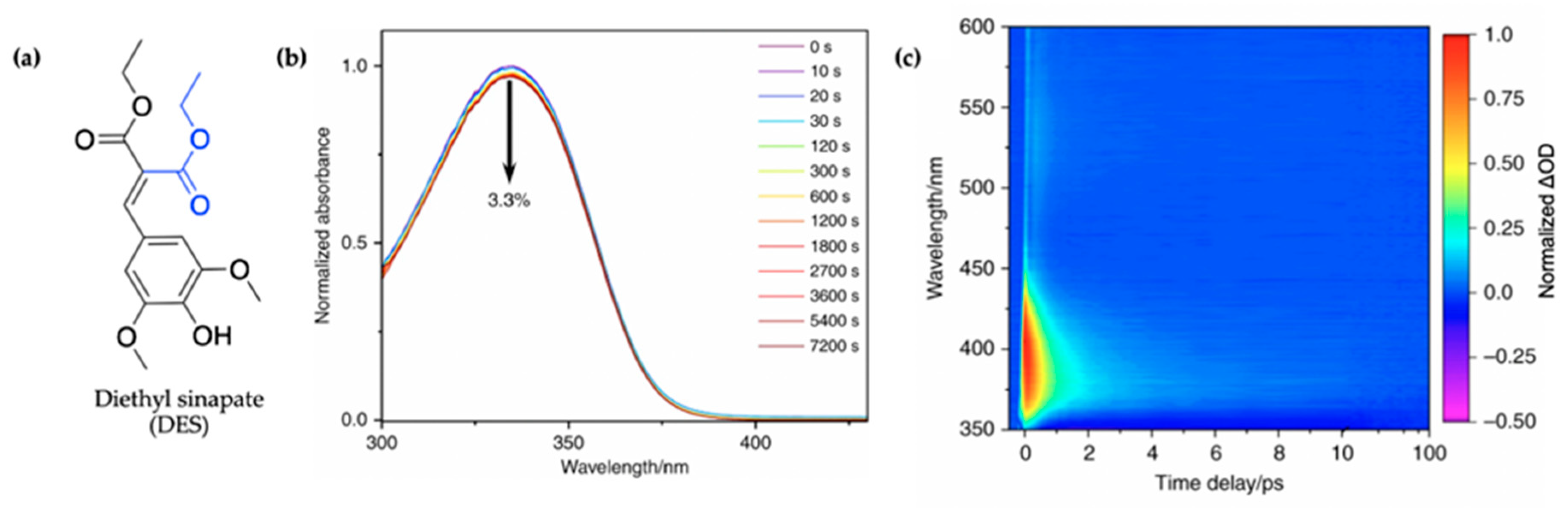
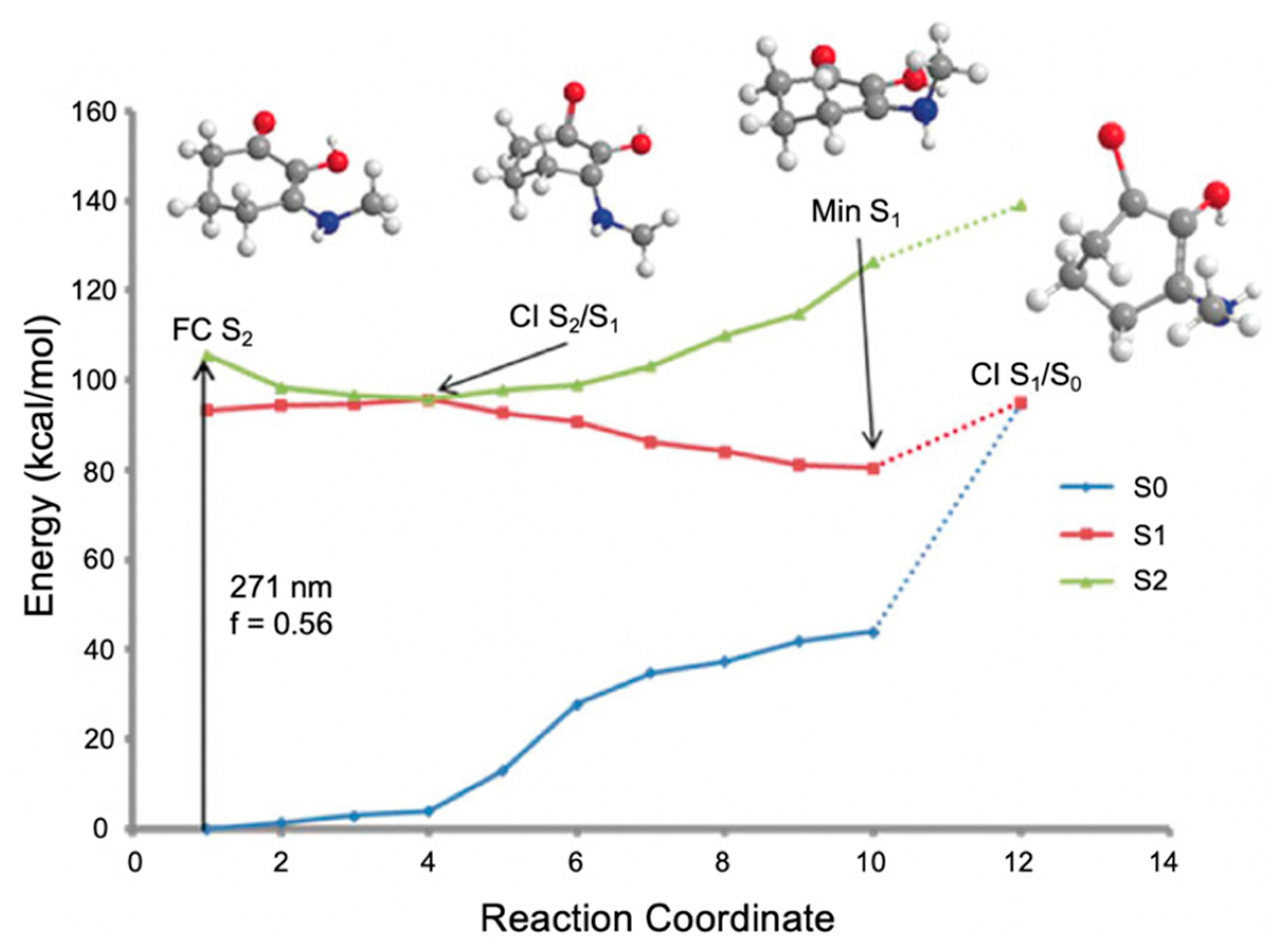
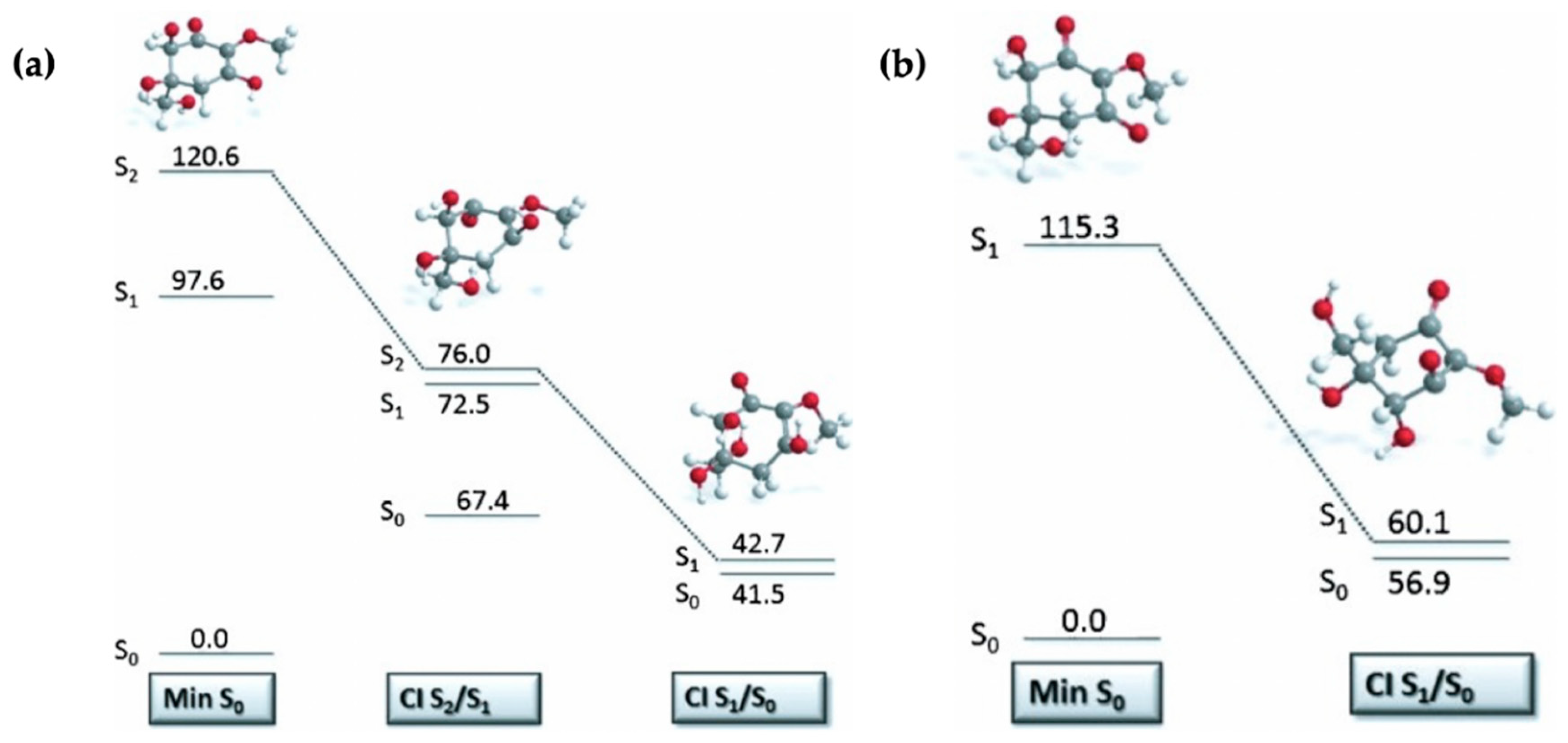
2.1.3. Symmetrically Functionalized Sinapate Ester
2.2. Microbial-Inspired Ultraviolet Filters
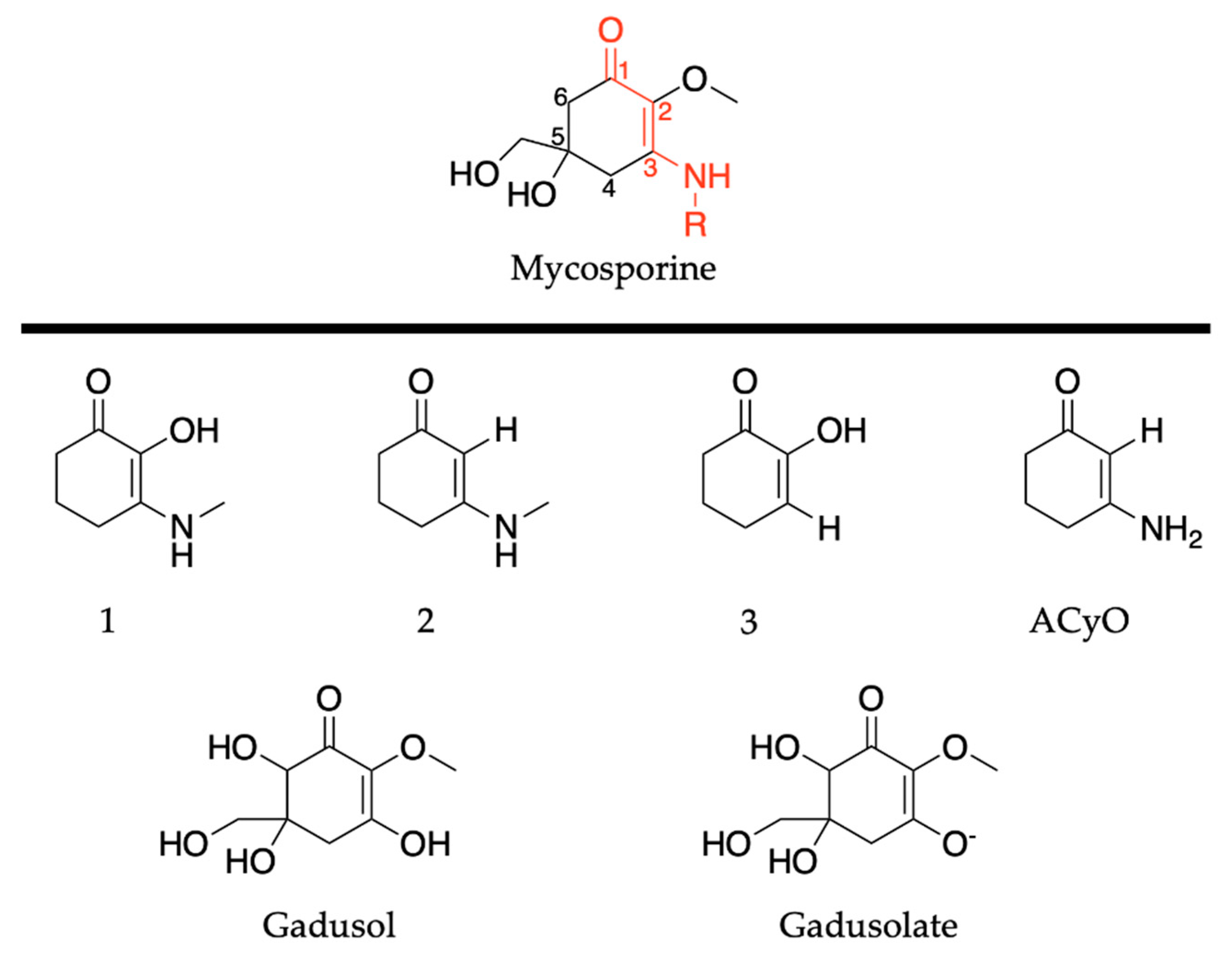
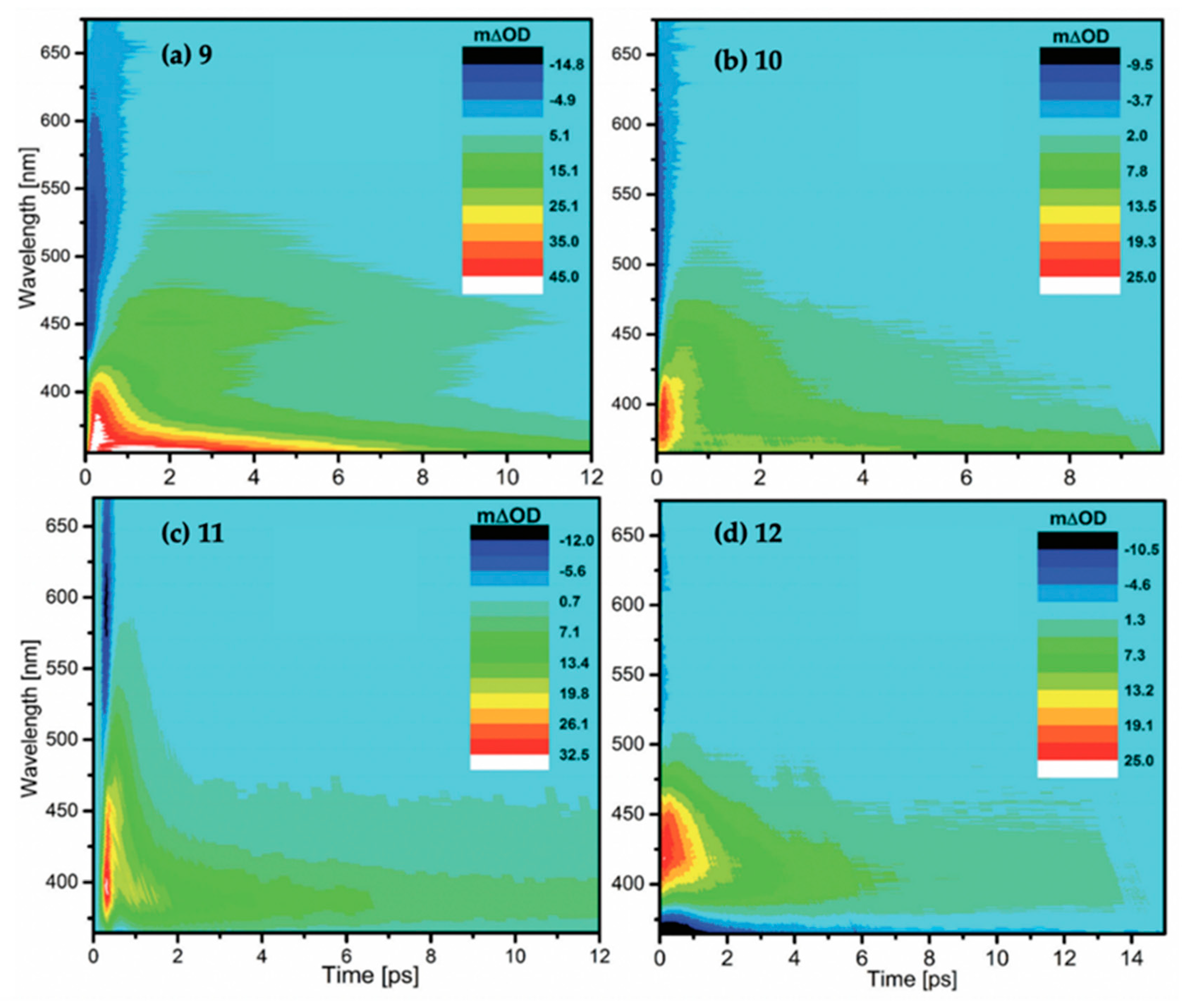
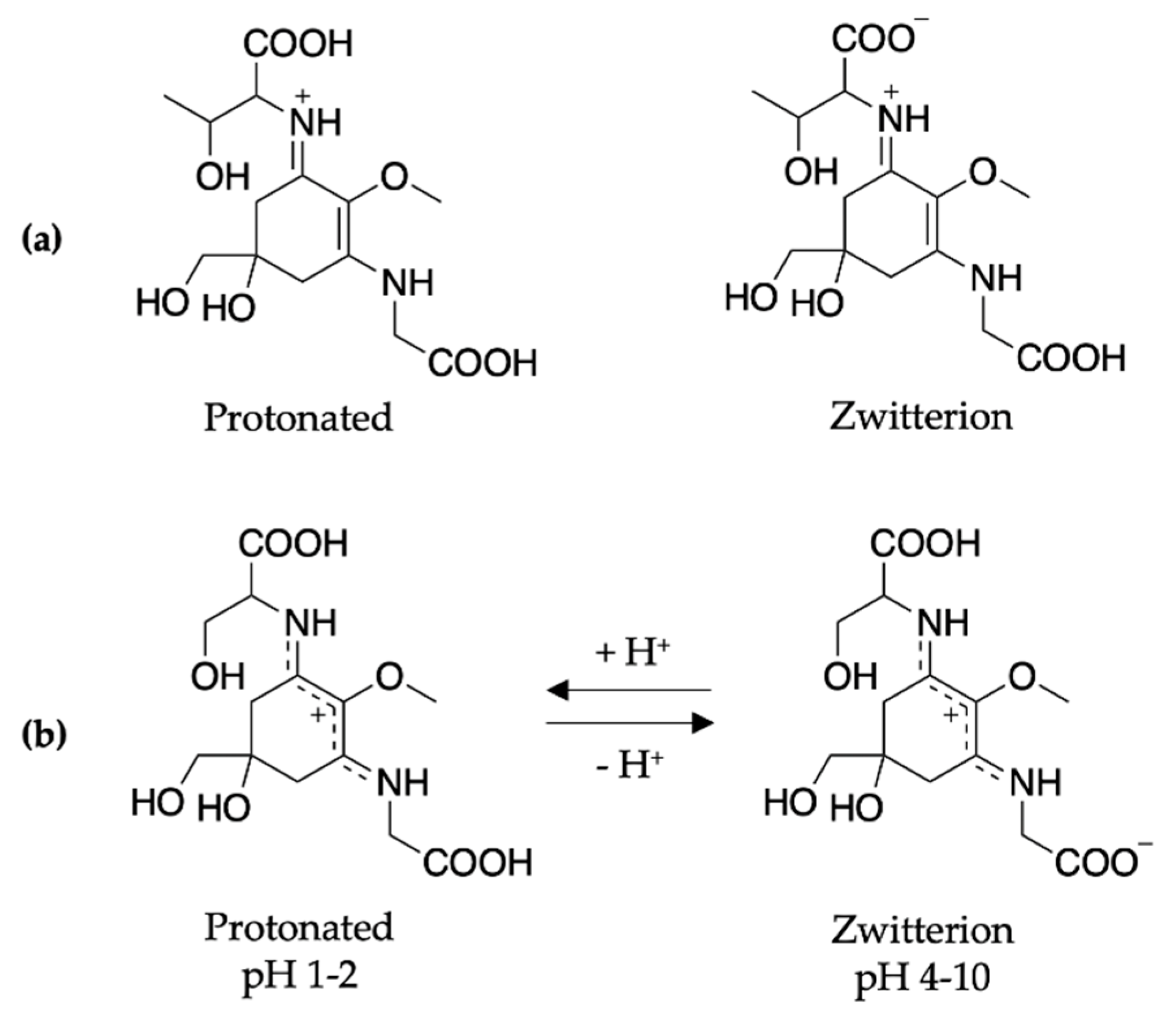
2.2.1. Mycosporines
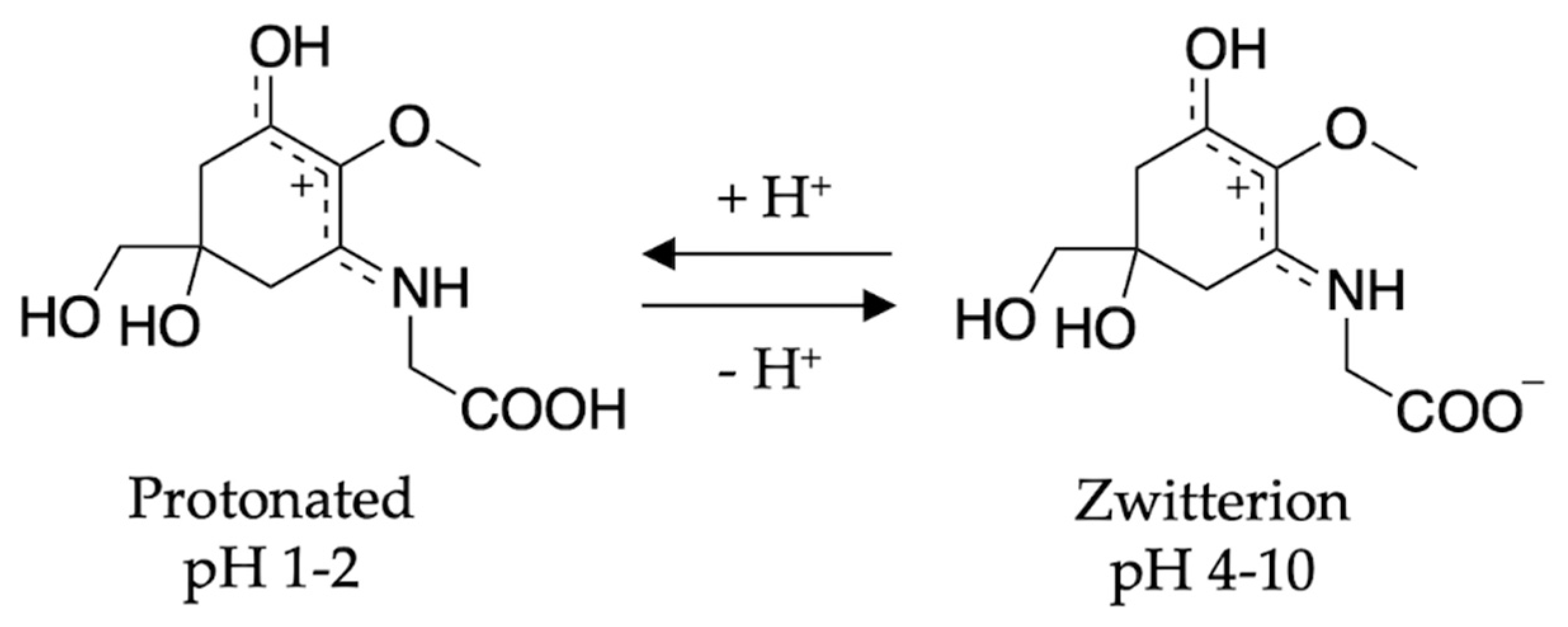
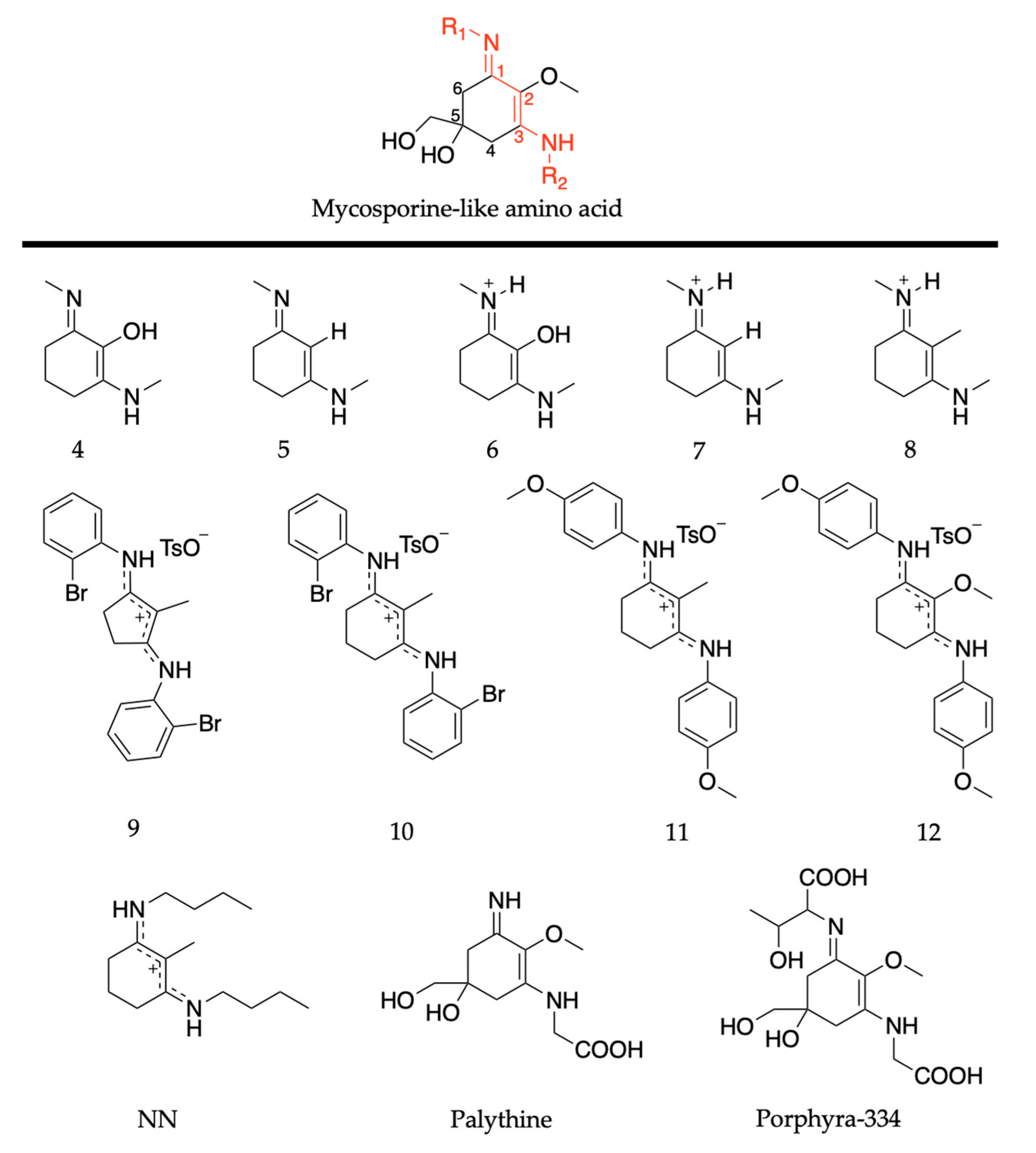
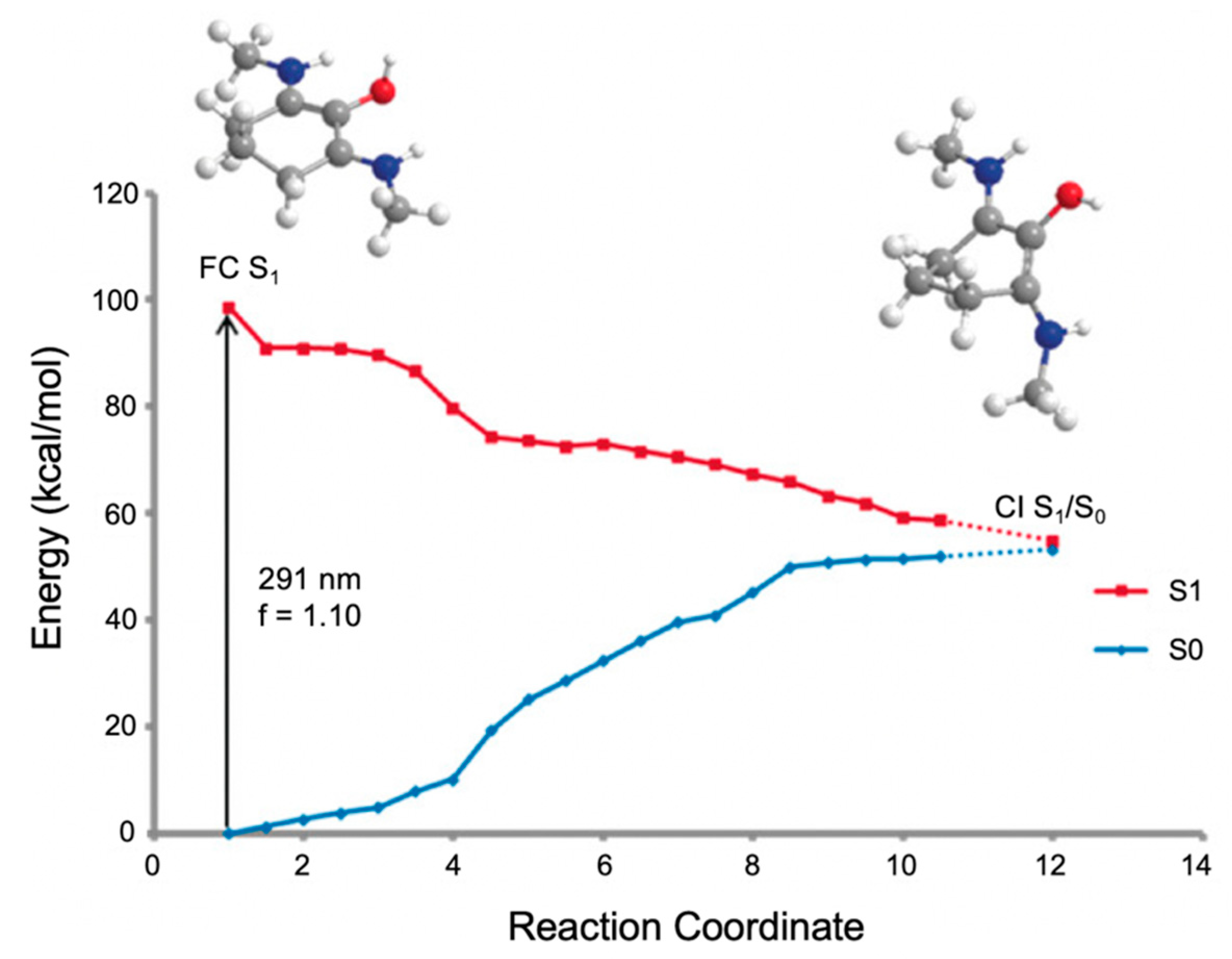
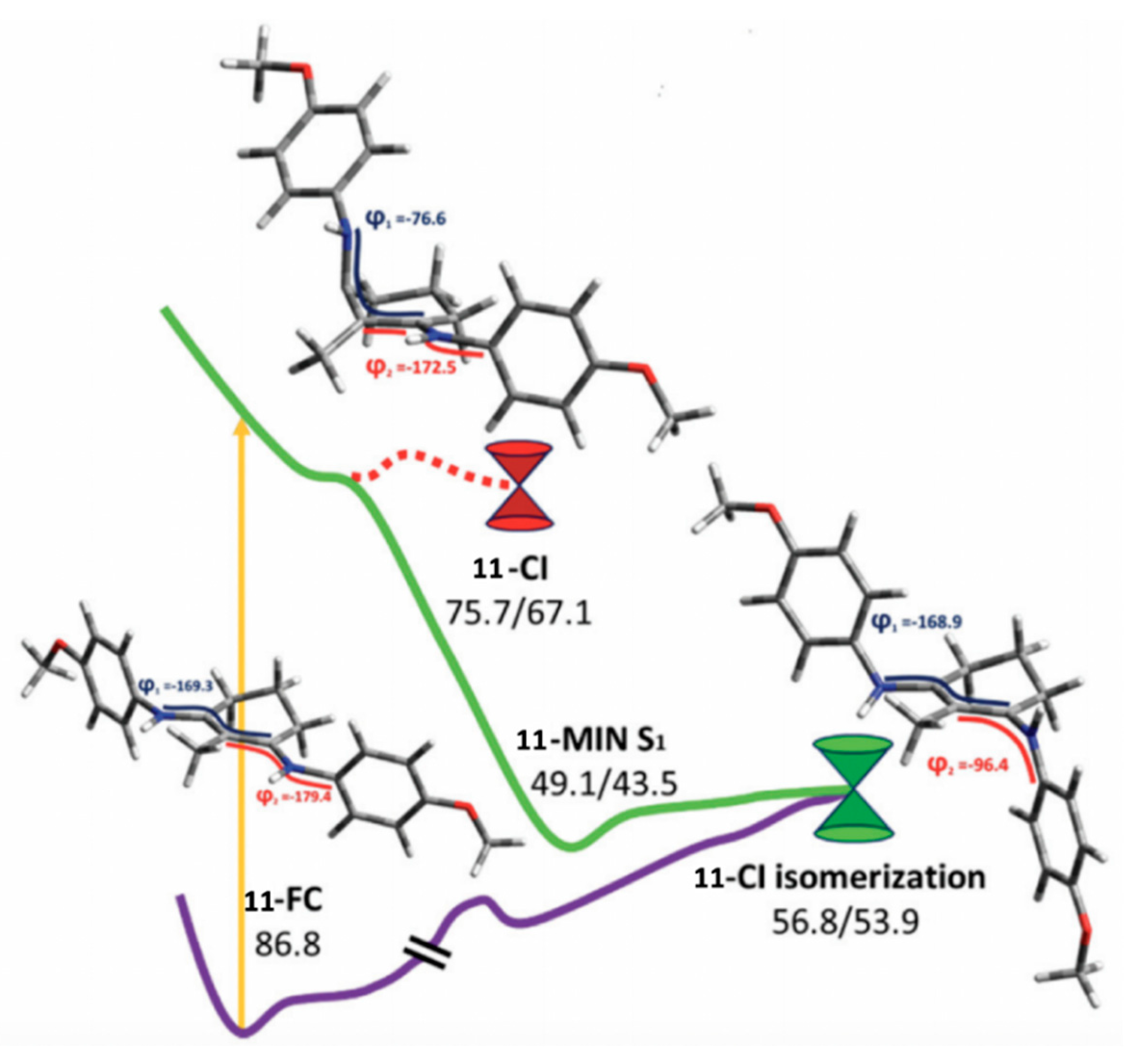
2.2.2. Mycosporine-Like Amino Acids
3. Future Direction for Nature-Inspired Ultraviolet Filters
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lacis, A.A.; Hansen, J. A parameterization for the absorption of solar radiation in the Earth’s atmosphere. J. Atmos. Sci. 1974, 31, 118–133. [Google Scholar] [CrossRef]
- Frederick, J.E.; Snell, H.E.; Haywood, E.K. Solar ultraviolet radiation at the Earth’s surface. Photochem. Photobiol. 1989, 50, 443–450. [Google Scholar] [CrossRef]
- Matsumi, Y.; Kawasaki, M. Photolysis of atmospheric ozone in the ultraviolet region. Chem. Rev. 2003, 103, 4767–4782. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Claveau, J.; Rossi, A.B. Ultraviolet radiation and the skin: Photobiology and sunscreen photoprotection. J. Am. Acad. Dermatol. 2017, 76, S100–S109. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am. J. Clin. Nutr. 2004, 80, 1678S–1688S. [Google Scholar] [CrossRef]
- Mithal, A.; Wahl, D.A.; Bonjour, J.-P.; Burckhardt, P.; Dawson-Hughes, B.; Eisman, J.A.; Fuleihan, G.E.-H.; Josse, R.G.; Lips, P.; Morales-Torres, J. Global vitamin D status and determinants of hypovitaminosis D. Osteoporos. Int. 2009, 20, 1807–1820. [Google Scholar] [CrossRef]
- Lips, P.; Van Schoor, N.M. The effect of vitamin D on bone and osteoporosis. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 585–591. [Google Scholar] [CrossRef]
- Wirz-Justice, A.; Graw, P.; Kräuchi, K.; Sarrafzadeh, A.; English, J.; Arendt, J.; Sand, L. ‘Natural’ light treatment of seasonal affective disorder. J. Affect. Disord. 1996, 37, 109–120. [Google Scholar] [CrossRef]
- Humble, M.B. Vitamin D, light and mental health. J. Photochem. Photobiol. B Biol. 2010, 101, 142–149. [Google Scholar] [CrossRef]
- Dahle, J.; Kvam, E. Induction of delayed mutations and chromosomal instability in fibroblasts after UVA-, UVB-, and X-radiation. Cancer Res. 2003, 63, 1464–1469. [Google Scholar]
- Baker, L.A.; Marchetti, B.; Karsili, T.N.V.; Stavros, V.G.; Ashfold, M.N.R. Photoprotection: Extending lessons learned from studying natural sunscreens to the design of artificial sunscreen constituents. Chem. Soc. Rev. 2017, 46, 3770–3791. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.D.N.; Staniforth, M.; Stavros, V.G. Photophysics of sunscreen molecules in the gas phase: A stepwise approach towards understanding and developing next-generation sunscreens. Proc. R. Soc. A 2016, 472, 20160677. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Srinivas, C.R. Photoprotection. Indian J. Dermatol. Venereol. 2007, 73, 73–79. [Google Scholar]
- Gallagher, R.P.; Lee, T.K. Adverse effects of ultraviolet radiation: A brief review. Prog. Biophys. Mol. Biol. 2006, 92, 119–131. [Google Scholar] [CrossRef]
- Wlaschek, M.; Tantcheva-Poór, I.; Naderi, L.; Ma, W.; Schneider, L.A.; Razi-Wolf, Z.; Schüller, J.; Scharffetter-Kochanek, K. Solar UV irradiation and dermal photoaging. J. Photochem. Photobiol. B Biol. 2001, 63, 41–51. [Google Scholar] [CrossRef]
- Taylor, H.R.; West, S.K.; Rosenthal, F.S.; Muñoz, B.; Newland, H.S.; Abbey, H.; Emmett, E.A. Effect of ultraviolet radiation on cataract formation. N. Engl. J. Med. 1988, 319, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, D.L.; Saladi, R.N.; Fox, J.L. Ultraviolet radiation and skin cancer. Int. J. Dermatol. 2010, 49, 978–986. [Google Scholar] [CrossRef]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef]
- Fedorow, H.; Tribl, F.; Halliday, G.; Gerlach, M.; Riederer, P.; Double, K.L. Neuromelanin in human dopamine neurons: Comparison with peripheral melanins and relevance to Parkinson’s disease. Prog. Neurobiol. 2005, 75, 109–124. [Google Scholar] [CrossRef]
- Eller, M.S.; Gilchrest, B.A. Tanning as part of the eukaryotic SOS response. Pigment Cell Res. 2000, 13, 94–97. [Google Scholar] [CrossRef]
- Mekhaldi, F.; Muscheler, R.; Adolphi, F.; Aldahan, A.; Beer, J.; McConnell, J.R.; Possnert, G.; Sigl, M.; Svensson, A.; Synal, H.-A.; et al. Multiradionuclide evidence for the solar origin of the cosmic-ray events of ᴀᴅ 774/5 and 993/4. Nat. Commun. 2015, 6, 8611. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.D.; Saraf, S. In vitro sun protection factor determination of herbal oils used in cosmetics. Pharmacogn. Res. 2010, 2, 22–25. [Google Scholar]
- Aldahan, A.S.; Shah, V.V.; Mlacker, S.; Nouri, K. The history of sunscreen. JAMA Dermatol. 2015, 151, 1316. [Google Scholar] [CrossRef]
- Urbach, F. The historical aspects of sunscreens. J. Photochem. Photobiol. B Biol. 2001, 64, 99–104. [Google Scholar] [CrossRef]
- Hockberger, P.E. A History of Ultraviolet Photobiology for Humans, Animals and Microorganisms. Photochem. Photobiol. 2002, 76, 561–579. [Google Scholar] [CrossRef]
- Gasparro, F.P. Epilogue: New perspectives in sunscreen photobiology. In Sunscreen Photobiology: Molecular, Cellular and Physiological Aspects; Gasparro, F.P., Ed.; Springer: Berlin/Heidelberg, Germany, 1997; pp. 177–186. [Google Scholar]
- Boehm, F.; Clarke, K.; Edge, R.; Fernandez, E.; Navaratnam, S.; Quilhot, W.; Rancan, F.; Truscott, T.G. Lichens–Photophysical studies of potential new sunscreens. J. Photochem. Photobiol. B Biol. 2009, 95, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Schaap, I.; Slijkerman, D.M.E. An environmental risk assessment of three organic UV-filters at Lac Bay, Bonaire, Southern Caribbean. Mar. Pollut. Bull. 2018, 135, 490–495. [Google Scholar] [CrossRef]
- Downs, C.A.; Kramarsky-Winter, E.; Segal, R.; Fauth, J.; Knutson, S.; Bronstein, O.; Ciner, F.R.; Jeger, R.; Lichtenfeld, Y.; Woodley, C.M.; et al. Toxicopathological effects of the sunscreen UV filter, oxybenzone (benzophenone-3), on coral planulae and cultured primary cells and its environmental contamination in Hawaii and the U.S. Virgin Islands. Arch. Environ. Contam. Toxicol. 2016, 70, 265–288. [Google Scholar] [CrossRef]
- Kockler, J.; Oelgemöller, M.; Robertson, S.; Glass, B.D. Photostability of sunscreens. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 91–110. [Google Scholar] [CrossRef]
- Gonzalez, H.; Tarras-Wahlberg, N.; Strömdahl, B.; Juzeniene, A.; Moan, J.; Larkö, O.; Rosén, A.; Wennberg, A.-M. Photostability of commercial sunscreens upon sun exposure and irradiation by ultraviolet lamps. BMC Dermatol. 2007, 7, 1. [Google Scholar] [CrossRef]
- Battie, C.; Jitsukawa, S.; Bernerd, F.; Del Bino, S.; Marionnet, C.; Verschoore, M. New insights in photoaging, UVA induced damage and skin types. Exp. Dermatol. 2014, 23, 7–12. [Google Scholar] [CrossRef]
- Brenner, M.; Hearing, V.J. The protective role of melanin against UV damage in human skin. Photochem. Photobiol. 2008, 84, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Afonso, S.; Horita, K.; Sousa e Silva, J.P.; Almeida, I.F.; Amaral, M.; Lobão, P.A.; Costa, P.C.; Miranda, M.S.; Esteves da Silva, J.C.G.; Sousa Lobo, J.M. Photodegradation of avobenzone: Stabilization effect of antioxidants. J. Photochem. Photobiol. B Biol. 2014, 140, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Mturi, G.J.; Martincigh, B.S. Photostability of the sunscreening agent 4-tert-butyl-4′-methoxydibenzoylmethane (avobenzone) in solvents of different polarity and proticity. J. Photochem. Photobiol. A Chem. 2008, 200, 410–420. [Google Scholar] [CrossRef]
- Fourtanier, A.; Moyal, D.; Seite, S. UVA filters in sun-protection products: Regulatory and biological aspects. Photochem. Photobiol. Sci. 2012, 11, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Hanson, K.M.; Cutuli, M.; Rivas, T.; Antuna, M.; Saoub, J.; Tierce, N.T.; Bardeen, C.J. Effects of solvent and micellar encapsulation on the photostability of avobenzone. Photochem. Photobiol. Sci. 2020, 19, 390–398. [Google Scholar] [CrossRef]
- Downs, C.A.; Kramarsky-Winter, E.; Fauth, J.E.; Segal, R.; Bronstein, O.; Jeger, R.; Lichtenfeld, Y.; Woodley, C.M.; Pennington, P.; Kushmaro, A.; et al. Toxicological effects of the sunscreen UV filter, benzophenone-2, on planulae and in vitro cells of the coral, Stylophora pistillata. Ecotoxicology 2013, 23, 175–191. [Google Scholar] [CrossRef]
- Danovaro, R.; Bongiorni, L.; Corinaldesi, C.; Giovannelli, D.; Damiani, E.; Astolfi, P.; Greci, L.; Pusceddu, A. Sunscreens cause coral bleaching by promoting viral infections. Environ. Health Perspect. 2008, 116, 441–447. [Google Scholar] [CrossRef]
- Ramos, S.; Homem, V.; Alves, A.; Santos, L. Advances in analytical methods and occurrence of organic UV-filters in the environment—A review. Sci. Total Environ. 2015, 526, 278–311. [Google Scholar] [CrossRef]
- Alonso, M.B.; Feo, M.L.; Corcellas, C.; Gago-Ferrero, P.; Bertozzi, C.P.; Marigo, J.; Flach, L.; Meirelles, A.C.O.; Carvalho, V.L.; Azevedo, A.F.; et al. Toxic heritage: Maternal transfer of pyrethroid insecticides and sunscreen agents in dolphins from Brazil. Environ. Pollut. 2015, 207, 391–402. [Google Scholar] [CrossRef]
- Gago-Ferrero, P.; Díaz-Cruz, M.S.; Barceló, D. UV filters bioaccumulation in fish from Iberian river basins. Sci. Total Environ. 2015, 518–519, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Warshaw, E.M.; Wang, M.Z.; Maibach, H.I.; Belsito, D.V.; Zug, K.A.; Taylor, J.S.; Mathias, C.G.; Sasseville, D.; Zirwas, M.J.; Fowler, J.F., Jr.; et al. Patch test reactions associated with sunscreen products and the importance of testing to an expanded series: Retrospective analysis of North American Contact Dermatitis Group data, 2001 to 2010. Dermatitis 2013, 24, 176–182. [Google Scholar] [CrossRef] [PubMed]
- SCCP (Scientific Committee on Consumer Products). Opinion on Benzophenone-3. Available online: https://ec.europa.eu/health/archive/ph_risk/committees/04_sccp/docs/sccp_o_078.pdf (accessed on 19 December 2006).
- Heurung, A.R.; Raju, S.I.; Warshaw, E.M. Benzophenones. Dermatitis 2014, 25, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Matta, M.K.; Zusterzeel, R.; Pilli, N.R.; Patel, V.; Volpe, D.A.; Florian, J.; Oh, L.; Bashaw, E.; Zineh, I.; Sanabria, C.; et al. Effect of sunscreen application under maximal use conditions on plasma concentration of sunscreen active ingredients: A randomized clinical trial. JAMA 2019, 321, 2082–2091. [Google Scholar] [CrossRef]
- Matta, M.K.; Florian, J.; Zusterzeel, R.; Pilli, N.R.; Patel, V.; Volpe, D.A.; Yang, Y.; Oh, L.; Bashaw, E.; Zineh, I.; et al. Effect of sunscreen application on plasma concentration of sunscreen active ingredients: A randomized clinical trial. JAMA 2020, 323, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; McMicheal, T.; Smith, W.; Armstrong, B. Solar ultraviolet radiation. In Environmental Burden of Disease Series, No. 13; World Health Organization: Geneva, Switzerland, 2006. [Google Scholar]
- Jenkins, G.I. Signal transduction in responses to UV-B radiation. Annu. Rev. Plant Biol. 2009, 60, 407–431. [Google Scholar] [CrossRef]
- Frohnmeyer, H.; Staiger, D. Ultraviolet-B radiation-mediated responses in plants. Balancing damage and protection. Plant Physiol. 2003, 133, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Ballare, C.L.; Scopel, A.L.; Stapleton, A.E.; Yanovsky, M.J. Solar Ultraviolet-B Radiation Affects Seedling Emergence, DNA Integrity, Plant Morphology, Growth Rate, and Attractiveness to Herbivore Insects in Datura ferox. Plant Physiol. 1996, 112, 161–170. [Google Scholar] [CrossRef]
- Fraser, C.M.; Chapple, C. The Phenylpropanoid Pathway in Arabidopsis. Arab. Book 2011, 9, e0152. [Google Scholar] [CrossRef]
- Dean, J.C.; Kusaka, R.; Walsh, P.S.; Allais, F.; Zwier, T.S. Plant Sunscreens in the UV-B: Ultraviolet Spectroscopy of Jet-Cooled Sinapoyl Malate, Sinapic Acid, and Sinapate Ester Derivatives. J. Am. Chem. Soc. 2014, 136, 14780–14795. [Google Scholar] [CrossRef]
- Baker, L.A.; Stavros, V.G. Observing and understanding the ultrafast photochemistry in small molecules: Applications to sunscreens. Sci. Prog. 2016, 99, 282–311. [Google Scholar] [CrossRef] [PubMed]
- Holt, E.L.; Stavros, V.G. Applications of ultrafast spectroscopy to sunscreen development, from first principles to complex mixtures. Int. Rev. Phys. Chem. 2019, 38, 243–285. [Google Scholar] [CrossRef]
- Shick, J.M.; Dunlap, W.C. Mycosporine-like amino acids and related Gadusols: Biosynthesis, acumulation, and UV-protective functions in aquatic organisms. Annu. Rev. Physiol. 2002, 64, 223–262. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, S.Z.; Van Dijken, G.L.; Gonzalez-Rodas, G. Effects of ultraviolet radiation on marine ecosystems. Int. J. Environ. Stud. 1996, 51, 199–216. [Google Scholar] [CrossRef]
- Balskus, E.P.; Walsh, C.T. The genetic and molecular basis for sunscreen biosynthesis in cyanobacteria. Science 2010, 329, 1653–1656. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.P.; Singh, S.P.; Hader, D.P. Database on mycosporines and mycosporine-like amino acids (MAAs) in fungi, cyanobacteria, macroalgae, phytoplankton and animals. J. Photochem. Photobiol. B Biol. 2007, 89, 29–35. [Google Scholar] [CrossRef]
- Gao, Q.; Garcia-Pichel, F. Microbial ultraviolet sunscreens. Nat. Rev. Microbiol. 2011, 9, 791–802. [Google Scholar] [CrossRef]
- Oren, A.; Gunde-Cimerman, N. Mycosporines and mycosporine-like amino acids: UV protectants or multipurpose secondary metabolites? FEMS Microbiol. Lett. 2007, 269, 1–10. [Google Scholar] [CrossRef]
- Losantos, R.; Sampedro, D.; Churio, M.S. Photochemistry and photophysics of mycosporine-like amino acids and gadusols, nature’s ultraviolet screens. Pure Appl. Chem. 2015, 87, 979–996. [Google Scholar] [CrossRef]
- Bandaranayake, W.M. Mycosporines: Are they nature’s sunscreens? Nat. Prod. Rep. 1998, 15, 159–172. [Google Scholar] [CrossRef]
- Carreto, J.I.; Carignan, M.O. Mycosporine-Like Amino Acids: Relevant Secondary Metabolites. Chemical and Ecological Aspects. Mar. Drugs 2011, 9, 387–446. [Google Scholar] [CrossRef] [PubMed]
- Dunlap, W.C.; Chalker, B.E.; Bandaranayake, W.M.; Wu Won, J.J. Nature’s sunscreen from the Great Barrier Reef, Australia. Int. J. Cosmet. Sci. 1998, 20, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Adams, N.L.; Shick, J.M. Mycosporine-like Amino Acids Provide Protection Against Ultraviolet Radiation in Eggs of the Green Sea Urchin Strongylocentrotus droebachiensis. Photochem. Photobiol. 1996, 64, 149–158. [Google Scholar] [CrossRef]
- Rastogi, R.P.; Sinha, R.P.; Singh, S.P.; Häder, D.-P. Photoprotective compounds from marine organisms. J. Ind. Microbiol. Biotechnol. 2010, 37, 537–558. [Google Scholar] [CrossRef]
- Rosic, N.N. Mycosporine-Like Amino Acids: Making the Foundation for Organic Personalised Sunscreens. Mar. Drugs 2019, 17, 638. [Google Scholar] [CrossRef]
- Shaath, N.A. Ultraviolet filters. Photochem. Photobiol. Sci. 2010, 9, 464–469. [Google Scholar] [CrossRef]
- Chrapusta, E.; Kaminski, A.; Duchnik, K.; Bober, B.; Adamski, M.; Bialczyk, J. Mycosporine-Like Amino Acids: Potential Health and Beauty Ingredients. Mar. Drugs 2017, 15, 326. [Google Scholar] [CrossRef]
- Yakovleva, I.; Bhagooli, R.; Takemura, A.; Hidaka, M. Differential susceptibility to oxidative stress of two scleractinian corals: Antioxidant functioning of mycosporine-glycine. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2004, 139, 721–730. [Google Scholar] [CrossRef]
- Ito, S.; Hirata, Y. Isolation and structure of a mycosporine from the zoanthid Palythoa tuberculosa. Tetrahedron Lett. 1977, 18, 2429–2430. [Google Scholar] [CrossRef]
- Takano, S.; Uemura, D.; Hirata, Y. Isolation and structure of a new amino acid, palythine, from the zoanthid Palythoa tuberculosa. Tetrahedron Lett. 1978, 26, 2299–2300. [Google Scholar] [CrossRef]
- Hirata, Y.; Uemura, D.; Ueda, K.; Takano, S. Several compounds from Palyfhoa tuberculosa (Coelenterata). Pure Appl. Chem. 1979, 51, 1875–1883. [Google Scholar] [CrossRef]
- Conde, F.R.; Churio, M.S.; Previtali, C.M. The photoprotector mechanism of mycosporine-like amino acids. Excited-state properties and photostability of porphyra-334 in aqueous solution. J. Photochem. Photobiol. B Biol. 2000, 56, 139–144. [Google Scholar] [CrossRef]
- Conde, F.R.; Churio, M.S.; Previtali, C.M. The deactivation pathways of the excited-states of the mycosporine-like amino acids shinorine and porphyra-334 in aqueous solution. Photochem. Photobiol. Sci. 2004, 3, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Conde, F.R.; Churio, M.S.; Previtali, C.M. Experimental study of the excited-state properties and photostability of the mycosporine-like amino acid palythine in aqueous solution. Photochem. Photobiol. Sci. 2007, 6, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.P.; Klisch, M.; Gröniger, A.; Häder, D.-P. Mycosporine-like amino acids in the marine red alga Gracilaria cornea—Effects of UV and heat. Environ. Exp. Bot. 2000, 43, 33–43. [Google Scholar] [CrossRef]
- Rastogi, R.P.; Incharoensakdi, A. Characterization of UV-screening compounds, mycosporine-like amino acids, and scytonemin in the cyanobacterium Lyngbya sp. CU2555. FEMS Microbiol. Ecol. 2014, 87, 244–256. [Google Scholar] [CrossRef]
- Rastogi, R.P.; Incharoensakdi, A. UV radiation-induced biosynthesis, stability and antioxidant activity of mycosporine-like amino acids (MAAs) in a unicellular cyanobacterium Gloeocapsa sp. CU2556. J. Photochem. Photobiol. B Biol. 2014, 130, 287–292. [Google Scholar] [CrossRef]
- Moliné, M.; Arbeloa, E.M.; Flores, M.R.; Libkind, D.; Farías, M.E.; Bertolotti, S.G.; Churio, M.S.; Van Broock, M.R. UVB Photoprotective Role of Mycosporines in Yeast: Photostability and Antioxidant Activity of Mycosporine-Glutaminol-Glucoside. Radiat. Res. 2011, 175, 44–50. [Google Scholar] [CrossRef]
- Arbeloa, E.M.; Bertolotti, S.G.; Churio, M.S. Photophysics and reductive quenching reactivity of gadusol in solution. Photochem. Photobiol. Sci. 2011, 10, 133–142. [Google Scholar] [CrossRef]
- Woolley, J.M.; Stavros, V.G. Unravelling photoprotection in microbial natural products. Sci. Prog. 2019, 102, 287–303. [Google Scholar] [CrossRef]
- Losantos, R.; Lamas, I.; Montero, R.; Longarte, A.; Sampedro, D. Photophysical characterization of new and efficient synthetic sunscreens. Phys. Chem. Chem. Phys. 2019, 21, 11376–11384. [Google Scholar] [CrossRef] [PubMed]
- Woolley, J.M.; Staniforth, M.; Horbury, M.D.; Richings, G.W.; Wills, M.; Stavros, V.G. Unravelling the Photoprotection Properties of Mycosporine Amino Acid Motifs. J. Phys. Chem. Lett. 2018, 9, 3043–3048. [Google Scholar] [CrossRef] [PubMed]
- Sampedro, D. Computational exploration of natural sunscreens. Phys. Chem. Chem. Phys. 2011, 13, 5584–5586. [Google Scholar] [CrossRef]
- Losantos, R.; Churio, M.S.; Sampedro, D. Computational Exploration of the Photoprotective Potential of Gadusol. ChemistryOpen 2015, 4, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, K.; Hatakeyama, M.; Boero, M.; Nobusada, K.; Hori, H.; Misonou, T.; Nakamura, S. How seaweeds release the excess energy from sunlight to surrounding sea water. Phys. Chem. Chem. Phys. 2017, 19, 15745–15753. [Google Scholar] [CrossRef]
- Losantos, R.; Funes-Ardoiz, I.; Aguilera, J.; Herrera-Ceballos, E.; Garcia-Iriepa, C.; Campos, P.J.; Sampedro, D. Rational Design and Synthesis of Efficient Sunscreens to Boost the Solar Protection Factor. Angew. Chem. Int. Ed. 2017, 56, 2632–2635. [Google Scholar] [CrossRef]
- Hatakeyama, M.; Koizumi, K.; Boero, M.; Nobusada, K.; Hori, H.; Misonou, T.; Kobayashi, T.; Nakamura, S. Unique Structural Relaxations and Molecular Conformations of Porphyra-334 at the Excited State. J. Phys. Chem. B 2019, 123, 7649–7656. [Google Scholar] [CrossRef]
- Zewail, A.H. Laser Femtochemistry. Science 1988, 242, 1645–1653. [Google Scholar] [CrossRef]
- Zewail, A.H. Femtochemistry: Atomic-scale dynamics of the chemical bond. J. Phy. Chem. A 2000, 104, 5660–5694. [Google Scholar] [CrossRef]
- Berera, R.; Van Grondelle, R.; Kennis, J.T.M. Ultrafast transient absorption spectroscopy: Principles and application to photosynthetic systems. Photosynth. Res. 2009, 101, 105–118. [Google Scholar] [CrossRef]
- Gupta, V.P. Molecular and Laser Spectroscopy: Advances and Applications; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Strickland, D.; Mourou, G. Compression of amplified chirped optical pulses. Opt. Commun. 1985, 56, 219–221. [Google Scholar] [CrossRef]
- Bradler, M.; Baum, P.; Riedle, E. Femtosecond continuum generation in bulk laser host materials with sub-μJ pump pulses. Appl. Phys. B 2009, 97, 561–574. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Plasmonics in biology and plasmon-controlled fluorescence. Plasmonics 2006, 1, 5–33. [Google Scholar] [CrossRef] [PubMed]
- Chatterley, A.S.; West, C.W.; Stavros, V.G.; Verlet, J.R.R. Time-resolved photoelectron imaging of the isolated deprotonated nucleotides. Chem. Sci. 2014, 5, 3963–3975. [Google Scholar] [CrossRef]
- Snellenburg, J.J.; Laptenok, S.; Seger, R.; Mullen, K.M.; Van Stokkum, I.H.M. Glotaran: A Java-Based Graphical User Interface for the R Package TIMP. J. Stat. Softw. 2012, 49, 1–22. [Google Scholar] [CrossRef]
- Mullen, K.M.; Van Stokkum, I.H.M. TIMP: An R Package for Modeling Multi-way Spectroscopic Measurements. J. Stat. Softw. 2007, 18, 1–46. [Google Scholar] [CrossRef]
- Beckwith, J.S.; Rumble, C.A.; Vauthey, E. Data analysis in transient electronic spectroscopy—An experimentalist’s view. Int. Rev. Phys. Chem. 2020, 39, 135–216. [Google Scholar] [CrossRef]
- Morabito, K.; Shapley, N.C.; Steeley, K.G.; Tripathi, A. Review of sunscreen and the emergence of non-conventional absorbers and their applications in ultraviolet protection. Int. J. Cosmet. Sci. 2011, 33, 385–390. [Google Scholar] [CrossRef]
- Saewan, N.; Jimtaisong, A. Natural products as photoprotection. J. Cosmet. Dermatol. 2015, 14, 47–63. [Google Scholar] [CrossRef]
- Baker, L.A.; Horbury, M.D.; Greenough, S.E.; Allais, F.; Walsh, P.S.; Habershon, S.; Stavros, V.G. Ultrafast photoprotecting sunscreens in natural plants. J. Phys. Chem. Lett. 2016, 7, 56–61. [Google Scholar] [CrossRef]
- Baker, L.A.; Horbury, M.D.; Greenough, S.E.; Coulter, P.M.; Karsili, T.N.V.; Roberts, G.M.; Orr-Ewing, A.J.; Ashfold, M.N.R.; Stavros, V.G. Probing the ultrafast energy dissipation mechanism of the sunscreen oxybenzone after UVA irradiation. J. Phys. Chem. Lett. 2015, 6, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Liu, Y.; Yang, S.; Flourat, A.L.; Allais, F.; Han, K. Ultrafast Barrierless Photoisomerization and Strong Ultraviolet Absorption of Photoproducts in Plant Sunscreens. J. Phys. Chem. Lett. 2017, 8, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Horbury, M.D.; Quan, W.-D.; Flourat, A.L.; Allais, F.; Stavros, V.G. Elucidating nuclear motions in a plant sunscreen during photoisomerization through solvent viscosity effects. Phys. Chem. Chem. Phys. 2017, 19, 21127–21131. [Google Scholar] [CrossRef]
- Rodrigues, N.D.N.; Stavros, V.G. From fundamental science to product: A bottom-up approach to sunscreen development. Sci. Prog. 2018, 101, 8–31. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, X.; Luo, J.; Yang, S. Excited-state dynamics of sinapate esters in aqueous solution and polyvinyl alcohol film. J. Lumin. 2019, 206, 469–473. [Google Scholar] [CrossRef]
- Baker, L.A.; Staniforth, M.; Flourat, A.L.; Allais, F.; Stavros, V.G. Gas-Solution Phase Transient Absorption Study of the Plant Sunscreen Derivative Methyl Sinapate. ChemPhotoChem 2018, 2, 743–748. [Google Scholar] [CrossRef]
- Zhao, X.; Luo, J.; Liu, Y.; Pandey, P.; Yang, S.; Wei, D.; Han, K. Substitution Dependent Ultrafast Ultraviolet Energy Dissipation Mechanisms of Plant Sunscreens. J. Phys. Chem. Lett. 2019, 10, 5244–5249. [Google Scholar] [CrossRef]
- Sharma, A.; Bányiová, K.; Babica, P.; Yamani, N.E.; Collins, A.R.; Čupr, P. Different DNA damage response of cis and trans isomers of commonly used UV filter after the exposure on adult human liver stem cells and human lymphoblastoid cells. Sci. Total Environ. 2017, 593–594, 18–26. [Google Scholar] [CrossRef]
- Zhao, X.; Luo, J.; Yang, S.; Han, K. New Insight into the Photoprotection Mechanism of Plant Sunscreens: Adiabatic Relaxation Competing with Nonadiabatic Relaxation in the cis → trans Photoisomerization of Methyl Sinapate. J. Phys. Chem. Lett. 2019, 10, 4197–4202. [Google Scholar] [CrossRef]
- Horbury, M.D.; Flourat, A.L.; Greenough, S.E.; Allais, F.; Stavros, V.G. Investigating isomer specific photoprotection in a model plant sunscreen. Chem. Commun. 2018, 54, 936–939. [Google Scholar] [CrossRef]
- Horbury, M.D.; Holt, E.L.; Mouterde, L.M.M.; Balaguer, P.; Cebrián, J.; Blasco, L.; Allais, F.; Stavros, V.G. Towards symmetry driven and nature inspired UV filter design. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Crews, D.; McLachlan, J.A. Epigenetics, evolution, endocrine disruption, health, and disease. Endocrinology 2006, 147, s4–s10. [Google Scholar] [CrossRef] [PubMed]
- Andersson, K.; Malmqvist, P.Å.; Roos, B.O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar] [CrossRef]
- Roos, B.O.; Taylor, P.R.; Siegbahn, P.E.M. A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach. Chem. Phys. 1980, 48, 157–173. [Google Scholar] [CrossRef]
- Olivucci, M. (Ed.) Computational Photochemistry; Elsevier: Amsterdam, The Netherlands, 2005; Volume 16. [Google Scholar]
- Sampedro, D. UV/Vis Spectroscopy, Photochemical Reactions and Photosynthesis. In Photochemistry; Maes, K.J., Willems, J.M., Eds.; Nova Science Publishers: New York, NY, USA, 2011. [Google Scholar]
- Avenel-Audran, M.; Dutartre, H.; Goossens, A.; Jeanmougin, M.; Comte, C.; Bernier, C.; Benkalfate, L.; Michel, M.; Ferrier-Lebouëdec, M.C.; Vigan, M.; et al. Octocrylene, an Emerging Photoallergen. Arch. Dermatol. 2010, 146, 753–757. [Google Scholar] [CrossRef]
- Karlsson, I.; Vanden Broecke, K.; Martensson, J.; Goossens, A.; Borje, A. Clinical and experimental studies of octocrylene’s allergenic potency. Contact Dermat. 2011, 64, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, R.P.; Incharoensakdi, A. Analysis of UV-absorbing photoprotectant mycosporine-like amino acid (MAA) in the cyanobacterium Arthrospira sp. CU2556. Photochem. Photobiol. Sci. 2014, 13, 1016–1024. [Google Scholar] [CrossRef]
- Plack, P.A.; Fraser, N.W.; Grant, P.T.; Middleton, C.; Mitchell, A.I.; Thomson, R.H. Gadusol, an enolic derivative of cyclohexane-1,3-dione present in the roes of cod and other marine fish. Isolation, properties and occurrence compared with ascorbic acid. Biochem. J. 1981, 199, 741–747. [Google Scholar] [CrossRef]
- Braslavsky, S.E.; Heibel, G.E. Time-resolved photothermal and photoacoustic methods applied to photoinduced processes in solution. Chem. Rev. 1992, 92, 1381–1410. [Google Scholar] [CrossRef]
- Arnaut, L.G.; Caldwell, R.A.; Elbert, J.E.; Melton, L.A. Recent advances in photoacoustic calorimetry: Theoretical basis and improvements in experimental design. Rev. Sci. Instrum. 1992, 63, 5381–5389. [Google Scholar] [CrossRef]
- Gensch, T.; Viappiani, C. Time-resolved photothermal methods: Accessing time-resolved thermodynamics of photoinduced processes in chemistry and biology. Photochem. Photobiol. Sci. 2003, 2, 699–721. [Google Scholar] [CrossRef] [PubMed]
- Abbruzzetti, S.; Viappiani, C.; Murgida, D.H.; Erra-Balsells, R.; Bilmes, G.M. Non-toxic, water-soluble photocalorimetric reference compounds for UV and visible excitation. Chem. Phys. Lett. 1999, 304, 167–172. [Google Scholar] [CrossRef]
- Matsuyama, K.; Matsumoto, J.; Yamamoto, S.; Nagasaki, K.; Inoue, Y.; Nishijima, M.; Mori, T. pH-Independent Charge Resonance Mechanism for UV Protective Functions of Shinorine and Related Mycosporine-like Amino Acids. J. Phys. Chem. A 2015, 119, 12722–12729. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.D.N.; Cole-Filipiak, N.C.; Horbury, M.D.; Staniforth, M.; Karsili, T.N.V.; Peperstraete, Y.; Stavros, V.G. Photophysics of the sunscreen ingredient menthyl anthranilate and its precursor methyl anthranilate: A bottom-up approach to photoprotection. J. Photochem. Photobiol. A Chem. 2018, 353, 376–384. [Google Scholar] [CrossRef]
- Dunkelberger, A.D.; Kieda, R.D.; Marsh, B.M.; Crim, F.F. Picosecond Dynamics of Avobenzone in Solution. J. Phys. Chem. A 2015, 119, 6155–6161. [Google Scholar] [CrossRef]
- ISO 24443:2012. Determination of Sunscreen UVA Photoprotection In Vitro; International Organization for Standardization: Geneva, Switzerland, 2012. [Google Scholar]
- Marx, D.; Hutter, J. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Klisch, M.; Richter, P.; Puchta, R.; Häder, D.P.; Bauer, W. The Stereostructure of Porphyra-334: An Experimental and Calculational NMR Investigation. Evidence for an Efficient ‘Proton Sponge’. Helv. Chim. Acta 2007, 90, 488–511. [Google Scholar] [CrossRef]
- Zhang, Z.; Tashiro, Y.; Matsukawa, S.; Ogawa, H. Influence of pH and temperature on the ultraviolet-absorbing properties of porphyra-334. Fish. Sci. 2005, 71, 1382–1384. [Google Scholar] [CrossRef]
- Orallo, D.E.; Bertolotti, S.G.; Churio, M.S. Photophysicochemical characterization of mycosporine-like amino acids in micellar solutions. Photochem. Photobiol. Sci. 2017, 16, 1117–1125. [Google Scholar] [CrossRef]
- Orallo, D.E.; Fangio, M.F.; Poblet, M.; Carignan, M.O.; Bernal, L.; Carreto, J.I.; Bertolotti, S.G.; Churio, M.S. Photochemistry and Photophysics of Shinorine Dimethyl Ester. Photochem. Photobiol. 2018, 94, 829–833. [Google Scholar] [CrossRef]
- Rini, M.; Holm, A.-K.; Nibbering, E.T.J.; Fidder, H. Ultrafast UV-mid-IR Investigation of the Ring Opening Reaction of a Photochromic Spiropyran. J. Am. Chem. Soc. 2003, 125, 3028–3034. [Google Scholar] [CrossRef]
- Holm, A.-K.; Rini, M.; Nibbering, E.T.J.; Fidder, H. Femtosecond UV/mid-IR study of photochromism of the spiropyran 1′,3′-dihydro-1′,3′,3′-trimethyl-6-nitrospiro[2 H-1-benzopyran-2,2′-(2H)-indole] in solution. Chem. Phys. Lett. 2003, 376, 214–219. [Google Scholar] [CrossRef]
- Murdock, D.; Harris, S.J.; Luke, J.; Grubb, M.P.; Orr-Ewing, A.J.; Ashfold, M.N.R. Transient UV pump–IR probe investigation of heterocyclic ring-opening dynamics in the solution phase: The role played by nσ* states in the photoinduced reactions of thiophenone and furanone. Phys. Chem. Chem. Phys. 2014, 16, 21271–21279. [Google Scholar] [CrossRef] [PubMed]
- Murdock, D.; Harris, S.J.; Clark, I.P.; Greetham, G.M.; Towrie, M.; Orr-Ewing, A.J.; Ashfold, M.N.R. UV-Induced Isomerization Dynamics of N-Methyl-2-pyridone in Solution. J. Phys. Chem. A 2015, 119, 88–94. [Google Scholar] [CrossRef]
- Gelzinis, A.; Augulis, R.; Butkus, V.; Robert, B.; Valkunas, L. Two-dimensional spectroscopy for non-specialists. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Hybl, J.D.; Albrecht, A.W.; Faeder, S.M.; Jonas, D.M. Two-dimensional electronic spectroscopy. Chem. Phys. Lett. 1998, 297, 307–313. [Google Scholar] [CrossRef]
- Schlau-Cohen, G.S.; De Re, E.; Cogdell, R.J.; Fleming, G.R. Determination of Excited-State Energies and Dynamics in the B Band of the Bacterial Reaction Center with 2D Electronic Spectroscopy. J. Phys. Chem. Lett. 2012, 3, 2487–2492. [Google Scholar] [CrossRef]
- Huxter, V.M.; Oliver, T.A.A.; Budker, D.; Fleming, G.R. Vibrational and electronic dynamics of nitrogen-vacancy centres in diamond revealed by two-dimensional ultrafast spectroscopy. Nat. Phys. 2013, 9, 744–749. [Google Scholar] [CrossRef]
- Taylor, V.C.A.; Tiwari, D.; Duchi, M.; Donaldson, P.M.; Clark, I.P.; Fermin, D.J.; Oliver, T.A.A. Investigating Electron-Phonon Coupling in Formamidinium Lead Iodide Perovskite Using Ultrafast Laser Spectroscopy. In 18th International Conference on Nanotechnology, NANO 2018; (IEEE International Conference on Nanotechnology (IEEE-NANO)); IEEE Computer Society: Cork, Ireland, 2019; Volume 2018. [Google Scholar]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| τn | Dioxane | Acetonitrile | Methanol | |
|---|---|---|---|---|
| SA | τ1/fs | 93 ± 17 | 52 ± 5 | 572 ± 87 |
| τ2/ps | 0.90 ± 0.19 | 0.57 ± 0.04 | 3.79 ± 0.72 | |
| τ3/ps | 12.2 ± 1.1 | 17.0 ± 0.66 | 25.5 ± 1.6 | |
| SM | τ1/fs | 119 ± 28 | 51 ± 4 | 619 ± 101 |
| τ2/ps | 1.62 ± 0.15 | 0.63 ± 0.04 | 4.81 ± 0.77 | |
| τ3/ps | 22.4 ± 1.9 | 27.3 ± 0.77 | 33.5 ± 1.7 |
| τivr (fs) | τiso (ps) | τpp (ns) | |
|---|---|---|---|
| cis-ES | 330 ± 40 | 5.05 ± 0.06 | >>2 |
| trans-ES | 290 ± 40 | 4.60 ± 0.04 | >>2 |
| k1/s−1 (×1013) | k2/s−1 (×1012) | k3/s−1 (×1011) | k4/s−1 (×1010) | k5/s−1 (×108) | |
|---|---|---|---|---|---|
| VC/AB | 0.7 ± 0.2 | 3.0 ± 0.3 | 4.24 ± 0.07 | 1.02 ± 0.06 | <<5 |
| AB | 2.5 ± 2.5 | 2.1 ± 0.2 | 5.3 ± 0.1 | 2.6 ± 0.1 | <<5 |
| Ethanol | 1.1 ± 0.5 | 11 ± 5 | 12.6 ± 0.6 | 15.5 ± 0.1 | <<5 |
| Cyclohexane | 0.7 ± 0.2 | n/a | 16 ± 1 | 6.3 ± 0.2 | n/a |
| τ0 | τ1 | τ2 | τ3 | |
|---|---|---|---|---|
| 9 | 70 ± 21 fs | 415 ± 43 fs | 5 ± 0.22 ps | - |
| 10 | - | 320 ± 110 fs | 1.7 ± 0.48 ps | 9.7 ± 1.04 ps |
| 11 | - | 206 ± 65 fs | 397 ± 190 fs | 6 ± 0.4 ps |
| 12 | - | 872 ± 201 fs | 1.8 ± 0.15 ps | 10.2 ± 0.22 ps |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abiola, T.T.; Whittock, A.L.; Stavros, V.G. Unravelling the Photoprotective Mechanisms of Nature-Inspired Ultraviolet Filters Using Ultrafast Spectroscopy. Molecules 2020, 25, 3945. https://doi.org/10.3390/molecules25173945
Abiola TT, Whittock AL, Stavros VG. Unravelling the Photoprotective Mechanisms of Nature-Inspired Ultraviolet Filters Using Ultrafast Spectroscopy. Molecules. 2020; 25(17):3945. https://doi.org/10.3390/molecules25173945
Chicago/Turabian StyleAbiola, Temitope T., Abigail L. Whittock, and Vasilios G. Stavros. 2020. "Unravelling the Photoprotective Mechanisms of Nature-Inspired Ultraviolet Filters Using Ultrafast Spectroscopy" Molecules 25, no. 17: 3945. https://doi.org/10.3390/molecules25173945
APA StyleAbiola, T. T., Whittock, A. L., & Stavros, V. G. (2020). Unravelling the Photoprotective Mechanisms of Nature-Inspired Ultraviolet Filters Using Ultrafast Spectroscopy. Molecules, 25(17), 3945. https://doi.org/10.3390/molecules25173945