1. Introduction
The knowledge of a suitable molecular target and its specificity for a given pathology is a necessary condition in a targeted radionuclide therapy approach. Many malignant tumors possess an infiltrating character with no defined margins or spread out metastases around the whole body. Only the selective binding of a radiopharmaceutical to a molecular target allows for the reliable imaging or safe ablation of cancer lesions with minimal side effects.
Neurokinin 1 receptor (NK1R; tachykinin 1 receptor) is a well-known G protein-coupled receptor for neuropeptide Substance P (SP) and a promising system for an anticancer therapeutic molecular target [
1,
2]. The activation of the NK1R by its endogenous ligand creates significant proliferative impulses for tumor cells promoting growth and development, including angiogenesis and metastasis. At the same time, the frequent formation of SP-NK1R complexes stimulate the cellular up-regulation of NK1R on tumor cell surfaces [
3], thus providing an even greater cell sensitivity for the mitogen action of SP. On the other hand, the blockage of SP action by using antagonists of NK1R on SP-sensitive tumor cells can selectively induce an anti-tumor effect through the mechanism of cell apoptosis [
4,
5].
Antagonists of NK1R are a very diverse and numerous group of compounds, though clinical applications have only been found for four compounds. They are applied to the prevention of nausea and vomiting induced by chemotherapy or surgical complications [
2,
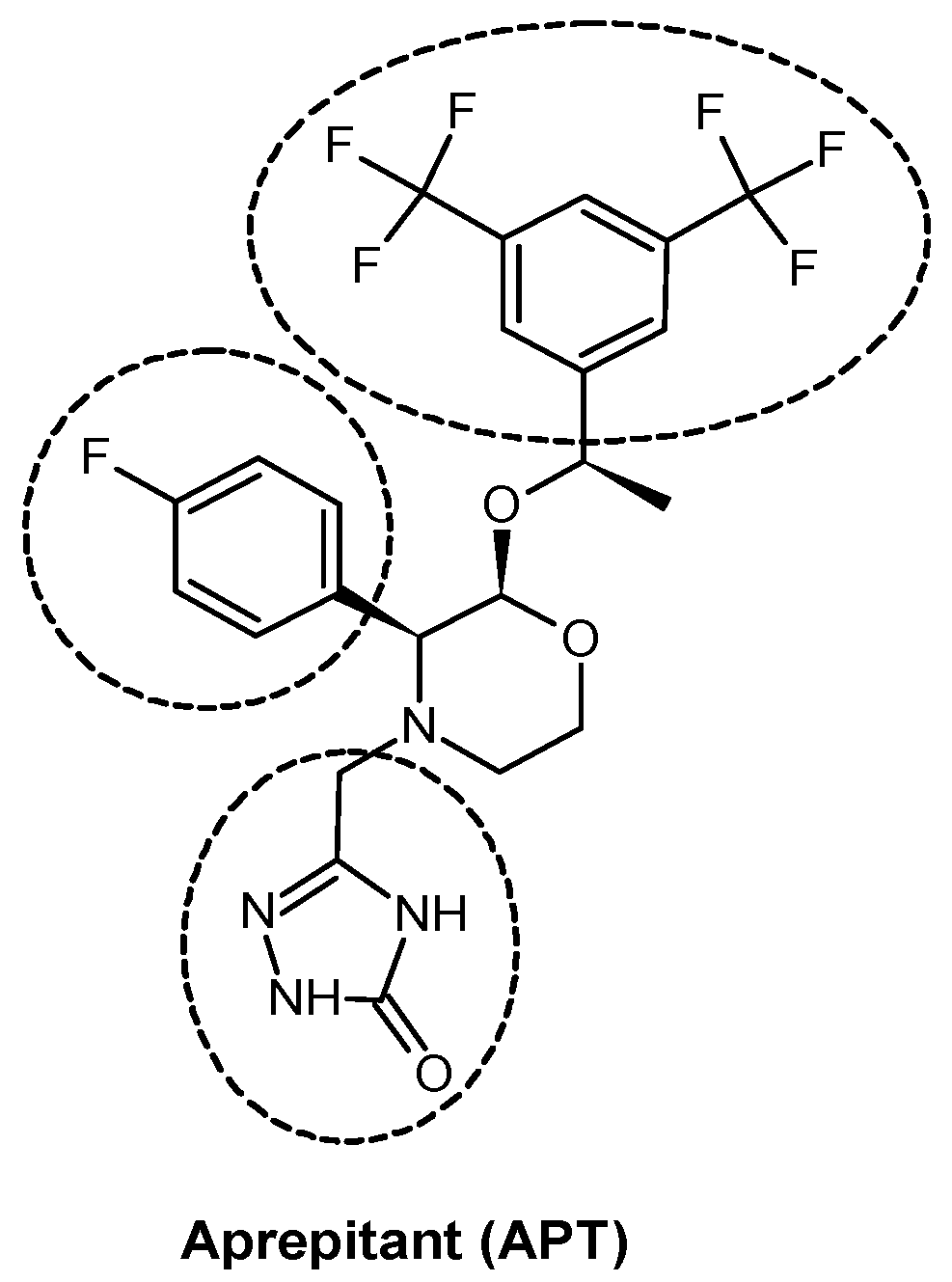
6]. One of the best known and widely studied compounds in this group is aprepitant (APT;
Figure 1)—a lipophilic and low molecular weight morpholine derivative with a high and selective affinity for NK1R. APT possesses anti-tumor activity, as has been determined in many cancer cell lines [
5,
7,
8,
9,
10,
11,
12]. Moreover, the phenomenon of the synergism of the anti-tumor activity of NK1R antagonists with an inhibitory effect on the cancer cell growth of other agents has been confirmed [
4]. It has been shown in vitro that the application of microtubule destabilizing agents in combination with antagonists of NK1R possess synergism in apoptotic effect in human glioblastoma, bladder, cervical and breast cancer cells [
13]. More remarkable cytotoxic synergism has been proven in a combination of aprepitant and ritonavir (an antiretroviral agent) in the human glioblastoma GAMG cell line [
14]. The application of these two drugs with temozolomide, an alkylating chemotherapeutic used clinically to treat glioblastoma, gives an even stronger synergistic effect.
What is most relevant is that APT is a fairly safe drug with a known pharmacological profile, with tolerability similar to placebo- and dose- related action. This could be shown by the fact that aprepitant’s half-maximal inhibitory concentration (IC
50) value determined for the human embryonic kidney (HEK) 293 cell line (a low expression NK1R control) is higher than the aprepitant IC
100 values determined for numerous tumor cell lines overexpressing NK1R [
15]. For the reasons described above, APT’s structure is an interesting scaffold for creating conjugates for carrying radionuclides to NK1R-positive tumors.
By looking at aprepitant in terms of molecular structure, it can be seen that the compound (
Figure 1) consists of a morpholine core decorated by three ‘arms,’ which are:
- (i)
p-fluorophenyl,
- (ii)
3,5-bis-trifluoromethylphenyl suspended at an ether linker, and
- (iii)
a triazolinone moiety suspended at a methylene linker.
In the course of extensive structure-activity studies on NK1R antagonists [
16,
17,
18], it has been established that the first two features (in particular: the distance and mutual positioning of two aromatic rings) are critical for high affinity and, therefore, for NK1R antagonism. On the other hand, the third element, triazolinone ring, can be, at least in some cases, safely modified without a significant loss of affinity [
18]. This was exploited in attempts to improve the solubility of aprepitant derivatives, resulting in the derivative L-760,735.
That this site tolerates some modifications is now well-understood in terms of protein-ligand interactions. A recently reported X-ray structure of an NK1R-aprepitant complex [
19] revealed that the triazolinone ring is located relatively close to the extracellular end of the receptor binding pocket, where it participates in hydrogen bonding to E193 and W184. However, E193A mutation has virtually no effect on aprepitant’s affinity, thus suggesting that the interactions in this area are of less importance to high affinity binding. Therefore, it seemed the most rational that a convenient site for functionalizing the APT structure is at this very ring. Nevertheless, the performed functionalization of the APT molecule required confirmation that the obtained conjugate still had a sufficiently high affinity for the receptor.
Based on that knowledge, we focused our efforts on the syntheses and in vitro evaluation of newly designed radioconjugates of aprepitant with gallium-68 or lutetium-177 radionuclides. For this purpose, we have proposed two functionalization routes of the APT molecule, followed by conjugation of different macrocyclic chelators DOTA, Bn-DOTA, and Bn-DOTAGA, as well as acrylic chelator DTPA dedicated to 68Ga and 177Lu. For conjugates showing full stability in human serum, molecular modelling studies for human NK1R and preliminary in vitro examination were performed. These reported findings indicate new perspectives of aprepitant applications in the form of selective theranostic-like concept radiopharmaceuticals for NK1R-positive tumors.
2. Results and Discussion
2.1. Syntheses of Aprepitant-Based Radioconjugates
2.1.1. Syntheses of Aprepitant Derivatives
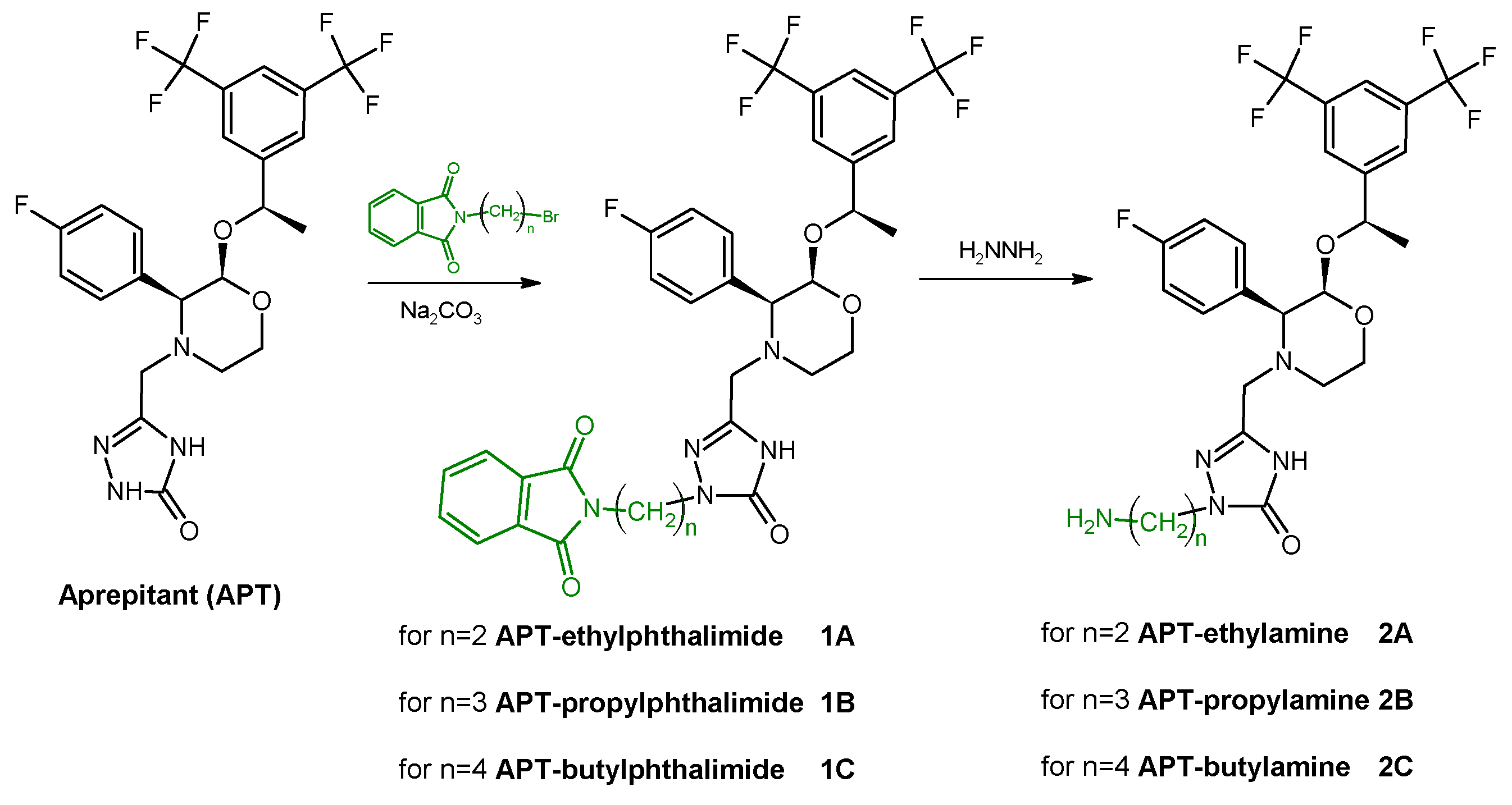
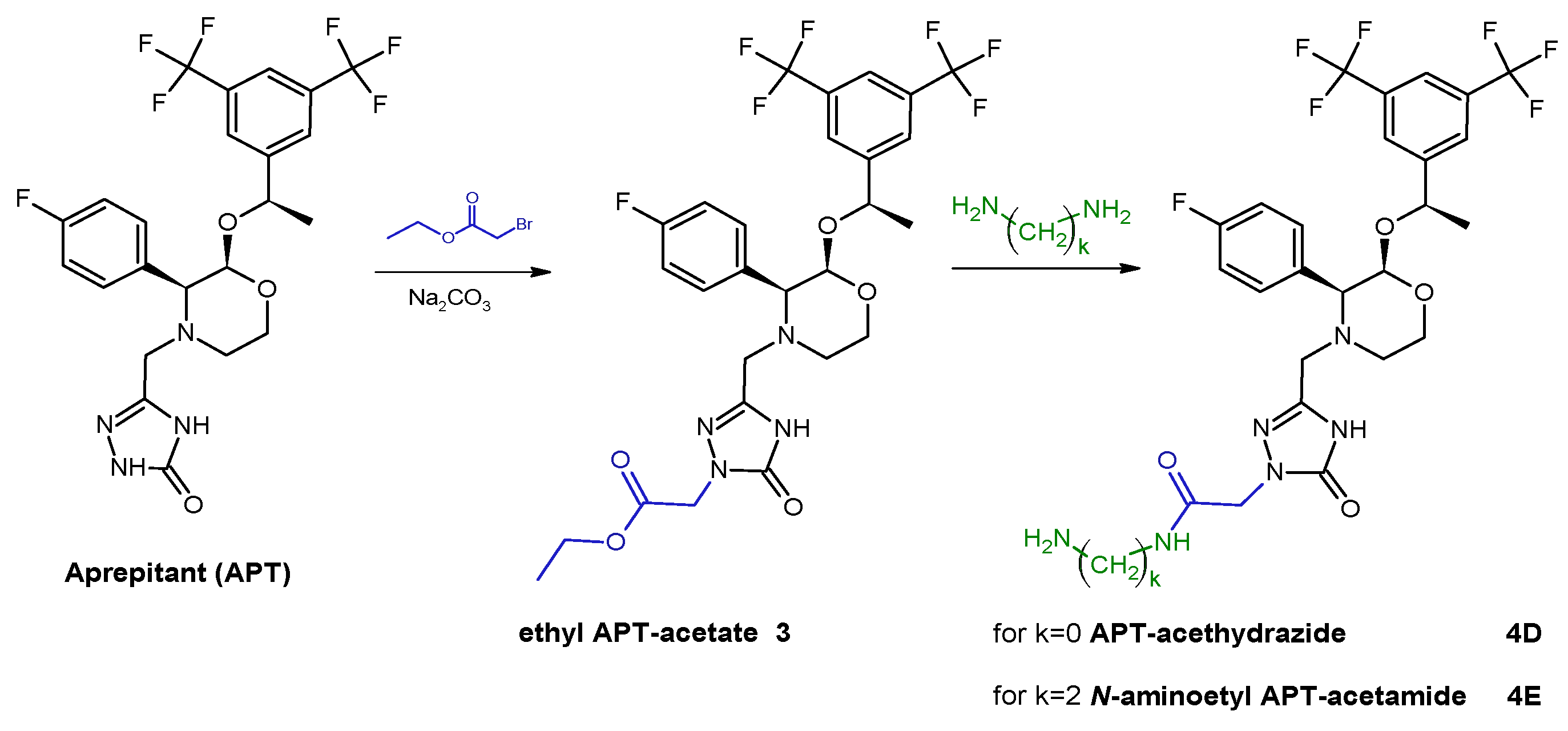
The first stage of synthesis concerned the modification of the APT structure in order to introduce a primary amine group. This was realized according to synthetic pathways presented below by using one of selected alkyl linkers (
Scheme 1) or acetamide linkers (
Scheme 2) so as to receive APT-alkylamine (
2A–
C) or APT-acetamide derivatives (
4D,
E).
2.1.2. Syntheses of Aprepitant Conjugates
The coupling reactions of APT-ethylamine,
2A, with different bifunctional chelating agents, were as follows: DOTA-NHS ester,
p-SCN-Bn-DOTA,
p-SCN-Bn-DOTAGA, or DTPA dianhydride, as presented in
Scheme 3. The use of different chelators allowed for the evaluation of the effect of the chelating moiety on the physicochemical properties of later radioconjugates. Based on the stability results obtained for these radioconjugates (presented in a section below), all other obtained APT derivatives (
2B,
2C,
4D, and
4E) were only conjugated with selected macrocyclic chelator DOTA. The application of different linkers allowed for the evaluation of their influence on the physicochemical properties of later radioconjugates.
2.1.3. Preparation of Radioconjugates
All APT conjugates with DOTA, Bn-DOTA, and Bn-DOTAGA were radiolabeled with
68Ga and
177Lu, while APT conjugates with DTPA were only radiolabeled with
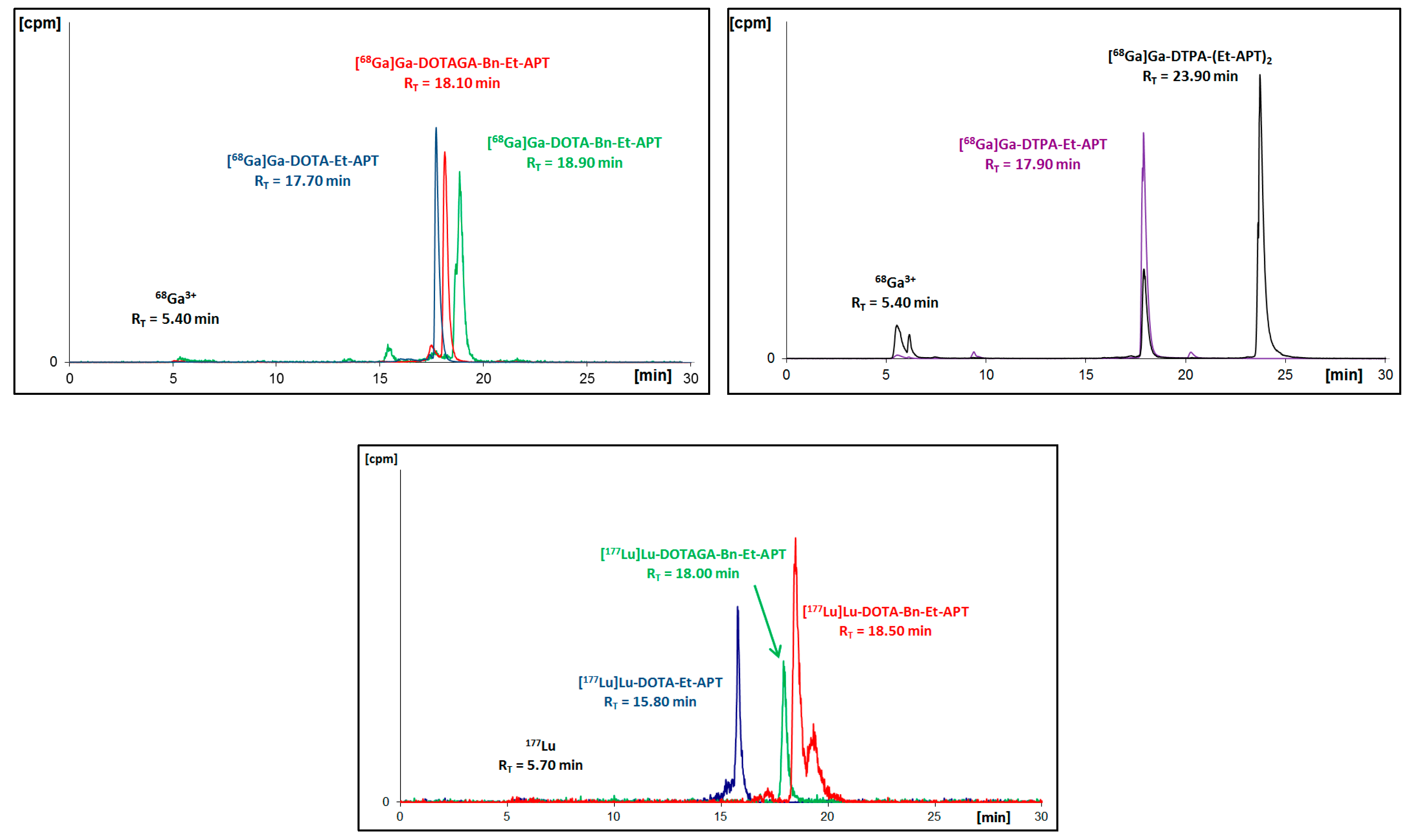
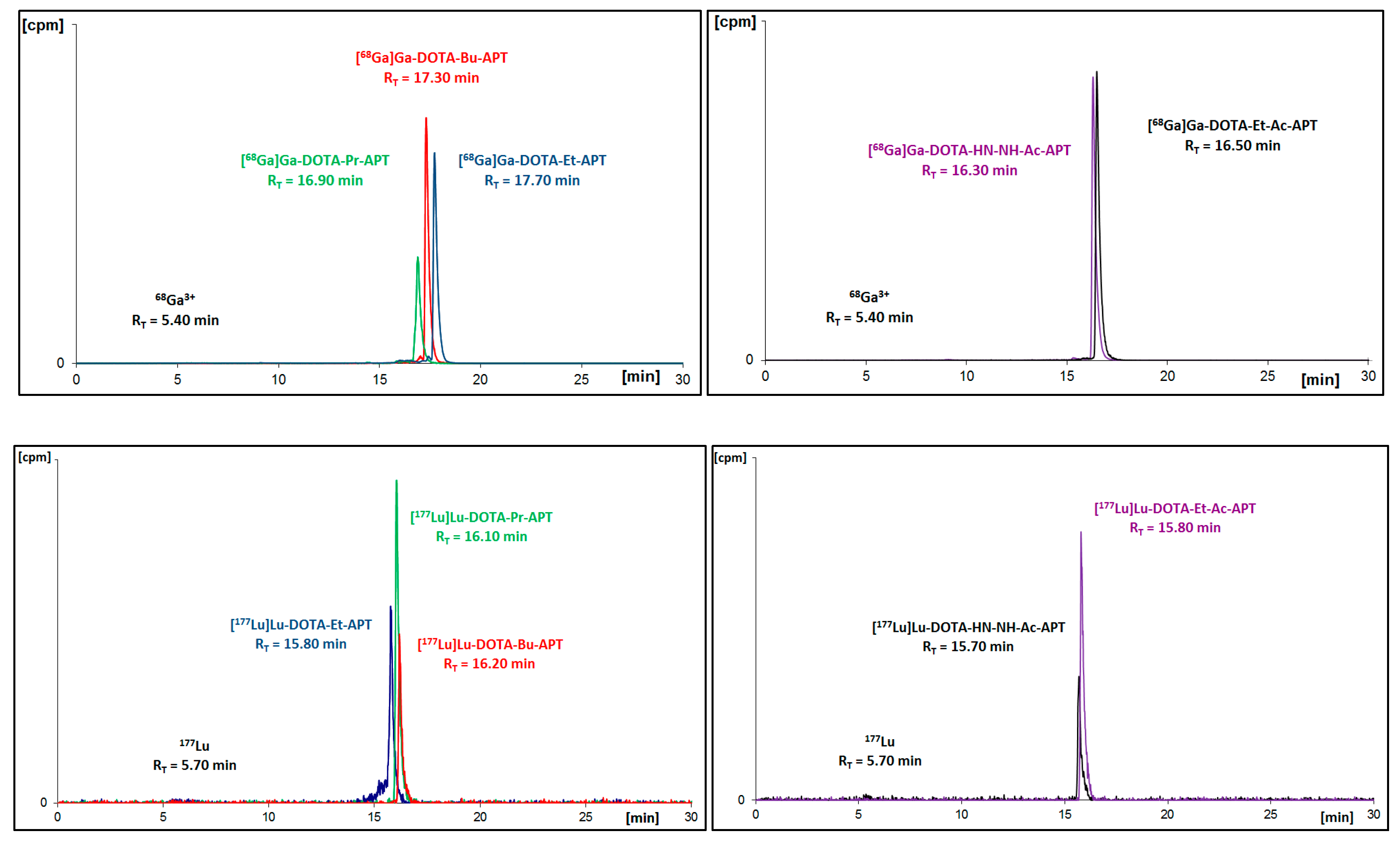
68Ga. Synthesized radioconjugates were purified using the solid phase extraction (SPE) method before HPLC identification (
Figure 2 and
Figure 3) and further analyses.
As a result of the performed radiosyntheses, all radioconjugates were successfully obtained, except for [
68Ga]Ga-DTPA-(Et-APT)
2 (
[68Ga]Ga-9A), which proved to be immediately unstable. Moreover, in the radiochromatogram of [
177Lu]Lu-DOTA-Bn-Et-APT (
[177Lu]Lu-6A) one can see a small additional signal (about 19.3 min) that is recognized as an early by-product of an interaction with solvent (EtOH) from the purification process. To verify the identity of all synthesized [
68Ga]Ga-radioconjugates in a non-carrier added scale, the non-radioactive stable gallium reference compounds (
Ga-5A–
Ga-9A and
Ga-5A–
Ga-5E) were synthesized and characterized by mass spectrometry. The retention time values of the [
68Ga]Ga-radioconjugates and stable references presented below (
Table 1 and
Table 2) overlapped, and the differences between them resulted from the serial connection of UV-Vis and gamma detectors only.
2.2. Physiochemical Evaluation of Radioconjugates
2.2.1. Stability Study
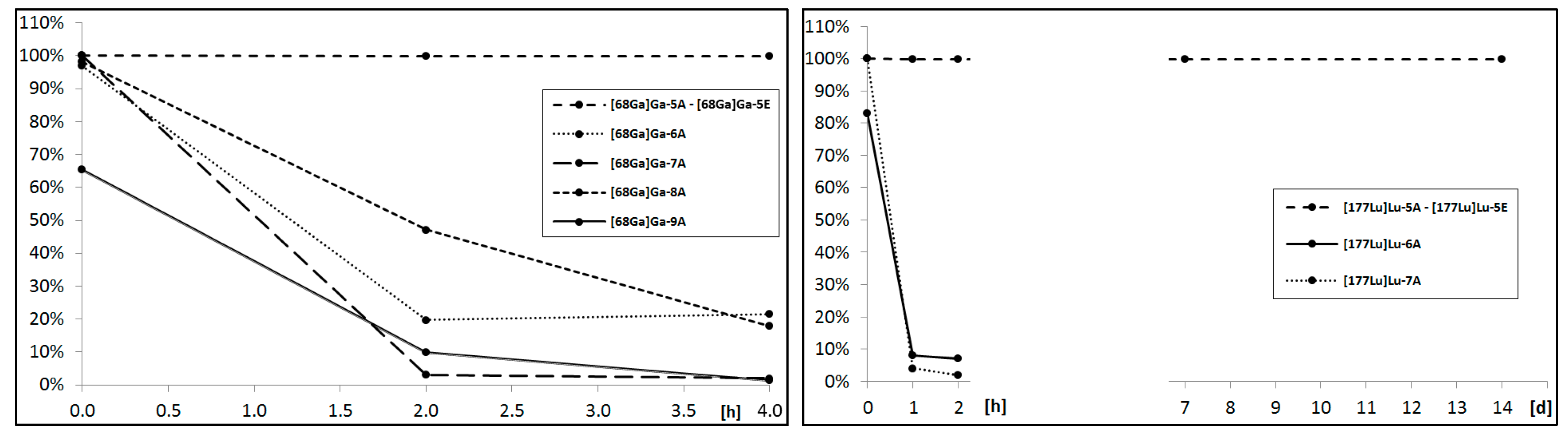
The sine qua non condition of a radionuclide’s application in vivo is its radiopharmaceutical stability in biological fluids like serum or cerebrospinal fluid. For this purpose, each isolated and solvent-free radioconjugate was incubated at 37 °C in human serum (HS). At specific time points, small samples of radioconjugate mixture were analyzed by the HPLC method for the assessment of the radioconjugate condition. The collected data presented on the charts below (
Figure 4) point out that only the DOTA radioconjugates remained stable in the biological fluid; thus, these radioconjugates were selected for further analyses.
We concluded that for the demand of designed aprepitant radioconjugates, the acyclic chelator DTPA showed a poor radionuclide chelating ability during incubation in human serum. DOTA and its analogues presented a satisfactory radionuclide complex stability, however, for the overall stability of the radioconjugate results from the type of the formed chemical bond with the amine terminated aprepitant derivative and the presence of a negative charge on the chelator-metal complex moiety. The amide bond created by the DOTA-NHS ester and uncharged complex in the conjugates remained stable throughout the whole stability study, while the thiourea bonds and negatively charged complexes created by both
p-SCN-Bn-DOTA and
p-SCN-Bn-DOTAGA were found to gradually decompose in time. This phenomenon of instability in HS has been observed previously in various radiopharmaceuticals [
20].
2.2.2. Lipophilicity Study
Drug distribution in vivo is highly related to both the lipophilicity and charge of a drug. The optimal radiotracer lipophilicity value for blood-brain barrier crossing lies within the range from 2.0 to 3.5 [
21]. Non-peptide NK1R antagonists, like aprepitant, are characterized by a high lipophilicity (logD 4.8) [
22], while the DOTA chelator is a highly hydrophilic moiety. In seeking to keep in lipophilicity of radioconjugates in a desired range, the choice of a proper linker (primary aprepitant modification) seems essential for distribution and pharmacokinetic aspects.
In the course of the lipophilicity study, each isolated DOTA radioconjugate (determined as fully stable in HS) was examined for distribution in the system of
n-octanol and a phosphate-buffered saline (PBS) buffer (pH = 7.4) to estimate the lipophilicity of the radiocomplex. The lipophilicity of each radioconjugate (logD), defined as the logarithm of the distribution coefficient (D) is based on the ratio of the radioactivity of the organic phase to the radioactivity of the aqueous phase. The stability of the studied radioconjugate was verified simultaneously during the experiment through the HPLC analysis of the aqueous phase. LogD values of
[68Ga]Ga-5A–[68Ga]Ga-5E and
[177Lu]Lu-5A–[177Lu]Lu-5E are listed below in
Table 3.
The APT-alkylamine derivative-based radioconjugates showed similar lipophilicity values that were higher than those of the APT-acetamide derivative-based radioconjugates. The complexes with lutetium were more lipophilic by (on average) 0.6 logD units. However, the logD values for all radioconjugates significantly decreased in comparison to aprepitant, indicating possible divergences in the pharmacokinetic fate of the radioconjugates and the parent drug.
2.3. Binding Affinity
An important consideration in the search of conjugate vectors for radionuclides is whether the functionalization of a high affinity ligand would not reduce the binding strength for a desired receptor. For the preliminary addressing of this issue in the case of our conjugates, we measured the affinity of compounds
5A–
E (uncomplexed precursors) for the rat neurokinin-1 receptor. The human (hNK1R) and the rat (rNK1R) neurokinin 1 receptors differ in their sequences and pharmacology. It has been established that many (but not all) high affinity NK1R antagonists have a significantly lower affinity for the rat receptor than for that of human origin [
23,
24]. Still, the results presented below give some tentative insight into the affinity changes caused by the functionalization of the aprepitant structure at the triazolinone ring.
The results of the binding affinity determinations are given in
Table 4. The parent compound, aprepitant, was found to exhibit IC
50 = 128.4 nM. This value was roughly consistent with the reported potency of aprepitant in a functional assay. The compound was found to inhibit Substance P-evoked increases in intracellular Ca
2+ mobilization in the cells expressing rNK1R with a pK
B reading 7.3 [
25]. Note that in the assays with cells expressing hNK1R, aprepitant was significantly more potent (pK
B = 8.7), and the reported binding affinities for the human receptor were of the subnanomolar order (e.g., IC
50 = 0.09 nM [
17]).
The aprepitant-based conjugates exhibited a diversified range of affinities. The strongest ligand in the set was the compound bearing a propylamine linker, 5B. It was found to have an IC50 of 0.69 µM. This value was about five times worse than that of the parent compound. Interestingly, decreasing (5A) or increasing (5C) the linker length by one methylene unit was associated with much lower affinity of the micromolar order. The shorter 5A exhibited the lowest binding in the set, with an IC50 of 6.2 µM. The analogue with the butylamine linker (5C) had an IC50 of 1.8 µM. Similar affinities (IC50~2.5 µM) were found for the conjugates with the acylhydrazine (5D) or N-aminoethylacetamide (5E) linkers.
2.4. Molecular Modelling Study
In order to get insight into possible interactions between the aprepitant-DOTA conjugates reported herein and the NK1R, the complexes thereof were modelled by molecular docking. The applied procedure consisted in building the appropriate linker-DOTA fragments into the aprepitant structure crystallized with the receptor (Protein Data Bank (PDB) accession code: 6HLO [
19]), followed by local search docking executed in AutoDock 4.2.6 [
26].
According to this procedure, the presence of a linker-DOTA moiety in the aprepitant-based conjugates did not have a major impact on the interactions between the core of the molecule and the receptor. Only a slight repositioning of the morpholine core, 3,5-bis-trifluoromethylphenyl, or
p-fluorophenyl moieties was observed compared to the 6HLO crystal structure (
Figure 5A). Thus, the conjugates were predicted to bind with the 3,5-bis-trifluoromethylphenyl fragment located at the bottom of the ligand-binding pocket and the DOTA moiety closer to the extracellular side of the receptor (
Figure 5A).
In the part that was common to all studied derivatives (and the parent aprepitant), the complexes were stabilized by (
Figure 5B):
- (i)
hydrogen bonding between Q165 and the ether oxygen,
- (ii)
hydrophobic contacts of the morpholine ring and F268 and I182,
- (iii)
hydrophobic contacts with side chains of N109, P112, and I113,
- (iv)
hydrophobic contacts of the 3,5-bis-trifluoromethylphenyl with W261 and F264,
- (v)
hydrophobic contacts of the p-fluorophenyl ring and H197, V200, T201, I204, and H265.
These interactions were identical to those found for the parent aprepitant in 6HLO structure.
On the other hand, the presence of the linker-DOTA fragment was predicted to weaken the contacts that the triazolinone ring of the parent aprepitant had with the receptor in the crystal structure 6HLO [
19]. In the optimized complexes for all the conjugates, this ring was displaced compared to the parent structure (
Figure 6A,B), so hydrogen bonding to W184 was not possible. On the other hand, a better positioning of this ring for π–π stacking with H197 was predicted for the conjugates.
Regarding the positioning of the linker-DOTA part, in the case of
5A, this fragment docked closely (
Figure 6C) to the extracellular loop 2 (ECL2) and the extracellular terminus of the transmembrane helix 5 (TM5). One of the DOTA’s carboxylate oxygens interacted with the side-chain of K190. For the analogues
5B–
D, the DOTA moiety was predicted to be located between the extracellular tips of TM5 and TM6 (
Figure 6D). Its contacts included residues K194, K190, and P271. In the case of the longest derivative,
5E, the docking placed the DOTA moiety close to TM5 and ECL2 (
Figure 6E). Here, it could interact with K190 and M181.
Since in the crystal structures 6HLO, 6HLL, and 6HLP [
19], several residues by the extracellular end of the receptor were found to adopt different rotamers upon the binding of different ligands, we wanted to see if the flexibility of these residues could affect the docking results. Therefore, the local docking procedure with the enabled flexibility of E193 and H197 was performed. It yielded similar results with only minor adjustments of the side chain rotamers. Its results (in terms of interactions and binding poses) are not discussed herein since they are almost perfectly accounted for by the description of the docking procedure with the rigid receptor.
Regarding the quantitative evaluation (
Table 5), AutoDock scoring function predicted that aprepitant would bind with the free energy of −10.43 kcal/mol. For the conjugates, the estimated energy varied between −9.64 kcal/mol (
5D) and −13.74 kcal/mol (
5E). The predicted energies did not correlate with the experimental data. This was perhaps due to the problems with estimating the entropic contribution because the conjugates differed with respect to the number of the rotatable bonds.
Other sources of significant error may have been the way the DOTA moiety was modelled (aimed at mimicking the presence of the cation in a simplified manner) and the fact that the experimentally evaluated conjugates were uncomplexed.
3. Materials and Methods
Aprepitant (Santa Cruz Biotechnology Inc., Dallas, TX, USA), the DOTA-NHS ester (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid mono-N-hydroxysuccinimide ester), p-SCN-Bn-DOTAGA (2,2′,2″-(10-(1-carboxy-4-((4-isothiocyanatobenzyl)amino)-4-oxobutyl)-1,4,7,10-tetraaza-cyclododecane-1,4,7-triyl)triacetic acid) (CheMatech, Dijon, France), p-SCN-Bn-DOTA (S-2-(4-isothiocyanatobenzyl)-1,4,7,10-tetraazacyclododecane tetraacetic acid) (Macrocyclics, Plano, TX, USA), DTPA dianhydride (diethylenetriaminepentaacetic dianhydride), and other substances and solvents (Sigma Aldrich/Merck, Darmstadt, Germany) were commercially available, defined as reagent grade, and applied without further purification. 68GaCl3 was eluted from the commercially available 68Ge/68Ga generator (Eckert & Ziegler, Berlin, Germany). The 177LuCl3 solution in 0.04 M HCl was purchased at Radioisotope Centre POLATOM, National Centre for Nuclear Research, Otwock-Świerk, Poland. Sep-Pack® Classic Short C18 Cartridges were purchased from WATERS, Milford, MA, USA. Human serum was isolated and purified at the Centre of Radiobiology and Biological Dosimetry, INCT Warsaw, Poland.
The HPLC conditions and gradient were as follows: a semi-preparative Phenomenex Jupiter Proteo column, 4 μm, 90 Å, 250 × 10 mm, with UV/Vis (220 nm) or/and radio γ-detection at gradient elution: 0–20 min 20 to 80% solvent B; 20–30 min 80% solvent B; 2 mL/min; solvent A: 0.1% (v/v) trifluoroacetic acid (TFA) in water; and solvent B: 0.1% (v/v) TFA in acetonitrile.
Mass spectra were measured on a Bruker 3000 Esquire mass spectrometer equipped with electrospray ionization (ESI) (Bruker, Billerica, MA, USA).
3.1. Syntheses of Aprepitant Derivatives and Aprepitant-Based Conjugates
3.1.1. General Procedure of Syntheses of Aprepitant Derivatives with Alkyl Linker, 2A–C
The slight molar excess of the selected n-(terminal-bromoalkyl) phthalimide was added into an equimolar mixture of APT and sodium carbonate in dimethylformamide (DMF). The reaction mixture was vigorously stirred in about 50 °C for 12–18 h. Then, the triple molar excess of hydrazine was added into the reaction mixture for an additional 3 h. The progress of the reaction was monitored by HPLC. The crude reaction mixture was evaporated, dissolved in the HPLC mobile phase, purified by the HPLC method, and lyophilized. The isolated main product was identified as a mono-substituted APT-alkylamine derivative (2A–C, ~75% reaction yield) by MS analysis confirmation.
MS: Calculated monoisotopic mass for APT-Et-NH2, 2A, C25H26F7N5O3: 577.19; found: 578.27 m/z [M + H+]
MS: Calculated monoisotopic mass for APT-Pr-NH2, 2B, C26H28F7N5O3: 591.21; found: 592.12 m/z [M + H+]
MS: Calculated monoisotopic mass for APT-Bu-NH2, 2C, C27H30F7N5O3: 605.22; found: 606.38 m/z [M + H+]
3.1.2. General Procedure of Syntheses of Aprepitant Derivatives with Acetamide Linker, 4D and 4E
The slight molar excess of ethyl 2-bromoacetate was added into an equimolar mixture of APT and sodium carbonate in DMF. The reaction mixture was vigorously stirred in about 50 °C for 24 h. Then, the triple molar excess of hydrazine or ethylenediamine was added into the reaction mixture for an additional 3 h. The progress of the reaction was monitored by HPLC. The crude reaction mixture was evaporated, dissolved in the HPLC mobile phase, purified by the HPLC method, and lyophilized. The isolated main product was identified as a mono-substituted amino-terminated APT-acetamide derivative (4D and 4E, 65–70% reaction yield) by MS analysis confirmation.
MS: Calculated monoisotopic mass for APT-Ac-HN-NH2, 4D, C25H25F7N6O4: 606.19; found: 608.07 m/z [M + H+]
MS: Calculated monoisotopic mass for APT-Ac-Et-NH2, 4E, C27H29F7N6O4: 634.21; found: 635.31 m/z [M + H+]
3.1.3. General Procedure of Syntheses of Aprepitant Conjugates with DOTA, 5A–E
The obtained APT derivative (2A–C, 4D, and 4E) and the DOTA-NHS ester in similar molar ratios were dissolved in DMF purged from oxygen with technical nitrogen and supplemented with a triple molar excess of triethylamine. The reaction mixture was vigorously stirred in about 50 °C for 24 h. The progress of the reaction was monitored by HPLC. The crude reaction mixture was evaporated, dissolved in the HPLC mobile phase, purified by the HPLC method, and lyophilized. The isolated main product was identified as a DOTA conjugate with an APT derivative (5A–E, >90% reaction yield) by MS analysis confirmation.
MS: Calculated monoisotopic mass for APT-Et-DOTA, 5A, C41H52F7N9O10: 963.37; found: 964.27 m/z [M + H+]
MS: Calculated monoisotopic mass for APT-Pr-DOTA, 5B, C42H54F7N9O10: 977.39; found: 978.42 m/z [M + H+]
MS: Calculated monoisotopic mass for APT-Bu-DOTA, 5C, C43H56F7N9O10: 991.40; found: 992.41 m/z [M + H+]
MS: Calculated monoisotopic mass for APT-Ac-HN-NH-DOTA, 5D, C41H51F7N10O11: 992.36; found: 993.17 m/z [M + H+]
MS: Calculated monoisotopic mass for APT-Ac-Et-DOTA, 5E, C43H55F7N10O11: 1020.39; found: 1021.43 m/z [M + H+]
3.1.4. Procedure of Syntheses of Aprepitant-Ethylamine Conjugates with p-SCN-Bn-DOTA and p-SCN-Bn-DOTAGA, 6A and 7A
The APT-ethylamine (2A) and bifunctional chelating agent in similar molar ratios were dissolved in DMF and supplemented with a 5-fold molar excess of triethylamine. The reaction mixture was vigorously stirred in about 50 °C for 24 h. The progress of the reaction was monitored by HPLC. The crude reaction mixture was evaporated, dissolved in the HPLC mobile phase, purified by the HPLC method, and lyophilized. The isolated main product was identified as a DOTA-Bn or DOTAGA-Bn conjugate with an APT derivative (6A and 7A, > 90% reaction yield) by MS analysis confirmation.
MS: Calculated monoisotopic mass for APT-Et-Bn-DOTA, 6A, C49H59F7N10O11S: 1128.40; found: 1129.55 m/z [M + H+]
MS: Calculated monoisotopic mass for APT-Et-Bn-DOTAGA, 7A, C52H64F7N11O12S: 1199.43; found: 1200.66 m/z [M + H+]
3.1.5. Procedure of Syntheses of Aprepitant-Ethylamine Conjugates with DTPA Anhydride, 8A, 9A
The APT-ethylamine (2A) and DTPA anhydride in a 3:2 molar ratio were dissolved in DMF purged from oxygen with technical nitrogen. The reaction mixture was vigorously stirred in room temperature for 2 h. The progress of the reaction was monitored by HPLC. The crude reaction mixture was evaporated, dissolved in the HPLC mobile phase, purified by the HPLC method, and lyophilized. Two isolated main products were identified as DTPA conjugated with one or two molecules of the APT derivative (8A and 9A with ~45% and ~40% reaction yields, respectively) by MS analysis confirmation.
MS: Calculated for monoisotopic mass APT-Et-DTPA, 8A, C39H47F7N8O12: 952.32; found: 953.40 m/z [M + H+]
MS: Calculated for monoisotopic mass APT-Et-DTPA-Et-APT, 9A, C64H71F14N13O14: 1511.50; found: 1512.64 m/z [M + H+]
3.2. Preparation of Radioconjugates
3.2.1. 68Ga Radiolabeling
The 68Ga radiolabeling of the DOTA, Bn-DOTA, and Bn-DOTAGA conjugates of APT was performed according to the following procedure: 145 µL of a concentrated solution of [68Ga]GaCl3 in 0.1 M HCl from the 68Ge/68Ga generator (4.9 ÷ 7.2 MBq) was added into the solution of 25 nmol of the selected conjugate in 200 µL of a 0.2 M acetate buffer (pH = 4.5) and heated for 5–10 min at 95 °C. After this time, each radioconjugate was purified using Sep-Pack® Classic Short C18 Cartridges according to producer recommendations, thereby obtaining an easily vaporized ethanolic solution of each radioconjugate. The effectiveness of the purification was monitored by HPLC. DTPA radioconjugates were obtained via an analogical procedure in room temperature.
3.2.2. 177Lu Radiolabeling
The 177Lu radiolabeling of the DOTA, Bn-DOTA, and Bn-DOTAGA conjugates of APT was performed according to the following procedure: 2.7 ÷ 5.3 µL of a [177Lu]LuCl3 n.c.a. solution in 0.04 M HCl (4.6 ÷ 5.2 MBq) was added into the solution of 2.5 nmol of the selected conjugate in 200 µL of a 0.02 M acetate buffer (pH 4.5) and heated for 10 min at 95 °C. After this time, each radioconjugate was purified using Sep-Pack® C18 Cartridges according to the producer recommendations, thereby obtaining an easily vaporized ethanolic solution of each radioconjugate. The effectiveness of the purification was monitored by HPLC.
3.2.3. Preparation of Non-Radioactive References
The non-radioactive Ga labelling of the DOTA, Bn-DOTA, and Bn-DOTAGA conjugates of APT was performed according to the following procedure: 145 µL of a concentrated solution of 20 mM GaCl3 in 0.1 M HCl was added into the solution of 50 nmol of the selected conjugate in 200 µL of a 0.2 M acetate buffer (pH = 4.5) and heated for 5–10 min at 95 °C. After this time, each reaction mixture was purified by the HPLC method, lyophilized, and characterized by mass spectrometry. DTPA conjugates were obtained via an analogical procedure in room temperature.
MS: Calculated for monoisotopic mass APT-Et-DOTA-Ga, 5A-Ga, C41H50F7N9O10Ga: 1030.80 and 1032.28; found: 1030.40 and 1032.40 m/z [M+]
MS: Calculated for monoisotopic mass APT-Pr-DOTA-Ga, 5B-Ga, C42H52F7N9O10Ga: 1044.30 and 1046.30; found: 1044.38 and 1046.39 m/z [M+]
MS: Calculated for monoisotopic mass APT-Bu-DOTA-Ga, 5C-Ga, C43H54F7N9O10Ga: 1058.31 and 1060.31; found: 1058.37 and 1060.40 m/z [M+]
MS: Calculated for monoisotopic mass APT-Ac-HN-NH-DOTA-Ga, 5D-Ga, C41H49F7N10O11Ga: 1059.27 and 1061.27; found: 1059.31 and 1061.40 m/z [M+]
MS: Calculated for monoisotopic mass APT-Ac-Et-DOTA-Ga, 5E-Ga, C43H53F7N10O11Ga: 1087.30 and 1089.30; found: 1087.37 and 1089.44 m/z [M+]
MS: Calculated for monoisotopic mass APT-Et-Bn-DOTA-Ga, 6A-Ga, C49H57F7N10O11SGa: 1095.31 and 1097.31; found: 1195.54 and 1197.51 m/z [M+]
MS: Calculated for monoisotopic mass APT-Et-Bn-DOTAGA-Ga, 7A-Ga, C52H62F7N11O12Sga: 1266.34 and 1268.34; found: 1266.47 and 1268.47 m/z [M+]
MS: Calculated for monoisotopic mass APT-Et-DTPA-Ga, 8A-Ga, C39H44F7N8O12Ga: 1018.22 and 1020.22; found: 1019.35 and 1021.37 m/z [M + H+]
MS: Calculated for monoisotopic mass APT-Et-DTPA-(Ga)-Et-APT, 9A-Ga, C64H68F14N13O14Ga: 1577.40 and 1579.40; found: 1578.64 and 1580.66 m/z [M + H+]
3.3. Physiochemical Evaluation of Radioconjugates
3.3.1. Stability Study
All obtained radioconjugates (isolated from the reaction mixtures using the SPE method and being solvent-free) were examined in terms of stability in human serum using HPLC analyses. A solution of each isolated selected radioconjugate in 100 µL of a 0.1M PBS buffer pH 7.40 was added to 900 µL of human serum and incubated at 37 °C for 4 h (68Ga radioconjugates) or 14 days (177Lu radioconjugates). At specific time points, 400 µL of the incubated mixture was added into 500 µL of ethanol, vigorously stirred to precipitate serum proteins, and centrifuged (13,500 rpm for 5 min) to separate the supernatant for HPLC analysis.
3.3.2. Lipophilicity Study
The lipophilicity values of the radioconjugates (logD), expressed as the logarithm of its D in the n-octanol/PBS (pH 7.40) system, mimicking the physiological conditions (Product Properties Test Guidelines of the Office of Prevention, Pesticides and Toxic Substances 830.7550, 1996), were determined right after the SPE method purification and ethanol evaporation processes. A solution of isolated selected radioconjugate in 500 µL of a 0.1 M PBS buffer at pH 7.40 and 500 µL of n-octanol was vigorously stirred and centrifuged (13,500 rpm for 5 min) to separate the immiscible phases. The radioactivities of the aqueous and organic layers were determined using a well-type NaI(Tl) detector. The distribution coefficient was calculated as the ratio of the radioactivity of the radioconjugate in the organic phase to that in the aqueous phase. Each measurement was performed in triplicate and averaged. Simultaneously, the aqueous phases were analyzed by HPLC to check whether the studied radioconjugate remained intact during the experiment.
3.4. Binding Affinity Determination
The binding affinity of aprepitant and compounds
5A–
E for rNK1R was determined in a competitive radioligand binding assay using rat brain homogenates, following a previously described method [
27]. In brief, the membrane preparations obtained from rat brains were incubated at 25 °C for 60 min in the presence of a selective radioligand [
3H]-[Sar
9,Met(O
2)
11]-Substance P obtained from PerkinElmer, (Waltham, MA, USA) and the increasing concentrations of the tested compounds (each concentration in duplicate). Non-specific binding was measured in the presence of 10 μM cold Substance P. The assay buffer was composed of 50 mM Tris-HCl (pH 7.4), 5 mM MnCl
2, bovine serum albumin (BSA) (0.1 mg/mL), bacitracin (100 μg/mL), bestatin (30 μM), phenylmethylsulfonyl fluoride (30 μg/mL), and captopril (10 μM). The reaction total volume was 1 mL. With the incubation having been terminated, a rapid filtration through GF/B Whatman glass fiber strips was done with a M-24 Cell Harvester (Brandel, Gaithersburg, MD, USA). The filters were pre-soaked overnight with 0.5% polyethyleneimine so that the extent of non-specific binding could be minimized. After the filtration, the strips were dried, the filter discs were placed separately in 24-well plates, and a Betaplate Scint scintillation solution (PerkinElmer, Waltham, MA, USA) was added to each well. Radioactivity was measured with a MicroBeta LS scintillation counter, Trilux (PerkinElmer, Waltham, MA, USA). The data came from three independent experiments done in duplicate. The results are presented as IC
50 with SEM.
3.5. Docking
In order to obtain the probable structures of the complexes of the neurokinin 1 receptor with the conjugates
5A–
E, the following modelling procedure was performed. The aprepitant structure (with neutral charge) in the complex with the receptor (PDB accession code: 6HLO [
19]) was expanded by attaching to the triazolinone ring the appropriate linkers and the DOTA moiety. Such initial complexes were subjected to local search docking in AutoDock 4.2.6 [
26].
The DOTA geometry was set based on the NOJYIU entry [
28] of The Cambridge Structural Database [
29]. This structure is a DOTA complex with Lu
3+ (diaqua-lutetium(III)-sodium trihydrate). For the purposes of our modelling, DOTA carboxylate arms were protonated and frozen in the conformation found in the crystal structure of lutetium (III) chelate of DOTA (after removing the Lu
3+ cation, Na
+ cations and waters). The rationale behind this gambit was the fact that the carboxylates would be primarily engaged in the interactions with a cation; therefore, they might have been expected to retain the conformation they had in the solid state structure. This approach could also give a rough approximation of the DOTA’s steric influence on the binding of the conjugates despite a lack of properly scaled and validated parameters for modelling and scoring the complexes with the cations of interest.
The used receptor structure was a refined one (as provided by the GPCRdb service [
30]) in order to have the mutated residues replaced with the native ones and to supply the side chains missing in the original PDB structure. The structure was pre-processed in AutoDock Tools [
26]. The box was set around the experimental position of aprepitant in 6HLO and extended towards the extracellular part of the receptor so as to cover the expected length of the expanded conjugate. The grids were calculated with AutoGrid 4 [
26]. We considered two variants of docking with respect to the flexibility of the receptor structure. In the first variant, all receptor residues were rigid. In the second variant, E193 and H197 side-chains were set to be flexible.
The docking procedure was the local search with the following parameters: 500 individuals in population, 500 iterations of the Solis-Wets local search, the
sw_
rho parameter of the local search space set to 20.0, and 1000 local search runs. The structures resulting from the local search were clustered, and the representative models of the lowest scored (on average) cluster were taken for further analysis. For the qualitative assessment of the binding energy, both the lowest and the mean energy of the clusters were collected. The molecular graphics were prepared in PyMol [
31].
For comparative and validation purposes, the very same procedure of local docking (with and without the flexibility of the mentioned two residues) was performed for the parent aprepitant.
4. Conclusions
The presented paper describes the evaluation of aprepitant functionalization in order to provide an application of this NK1R antagonist in nuclear medicine.
Out of the corresponding
68Ga/
177Lu radioconjugates of APT-ethylamine
2A with DOTA, Bn-DOTA, Bn-DOTAGA, and DTPA, only the DOTA amide conjugates showed satisfactory stability in human serum throughout the whole incubation time. The evaluation of the linker effect on radioconjugate lipophilicity indicated APT-alkylamine derivatives as more promising biovectors with features closer to parent aprepitant. The physicochemical properties of obtained APT-alkylamine-DOTA derivatives labelled with
68Ga (
[68Ga]Ga-5A–[68Ga]Ga-5C) can be compared with those of [
67Ga]Ga-NOTA-NK1R radioligands based on another NK1R antagonist—L-733,060 [
32]. The
67/68Ga-radioligands based on these two high affinity NK1R antagonists turned out to be very similar, as evidenced by the following parameters:
- (i)
they were labelled using macrocyclic chelators (DOTA and NOTA) incorporated in the same ‘arm’ of the antagonist molecule core,
- (ii)
the radioconjugates had similar molecular weights (about 1000),
- (iii)
they had comparable logD values (about 0.15 and 0.6 for the APT-radioligands and the L-733,060-radioligands, respectively),
- (iv)
all were fully stable in human serum examinations.
Regarding the affinity studies of the 5A–E conjugates, on the assumption that the human NK1R affinities for aprepitant derivatives were generally much higher than the rat NK1R affinities and that structure-affinity trends were parallel in both species, all the synthesized compounds might be considered to retain reasonable NK1R affinity compared to their parent. In particular, the analogue 5B (which only suffered a few-times decrease in affinity compared to APT) seems to be especially interesting for further development. Obtained results suggest that the functionalizing of the aprepitant structure via the triazolinone ring is the right strategy.
It is also worth mentioning that, in general, radiopharmaceuticals based on small non-peptide molecules (e.g., aprepitant and L-733,060) have many advantages over peptide-based radiopharmaceuticals [
2]. They usually have lower molecular weights, higher lipophilicity values, and, hence, different pharmacokinetics; they are stable in vivo, but, more importantly, their radiosyntheses can be carried out at higher temperatures and in a wider pH range. Moreover, according to the literature, radiopharmaceuticals based on non-peptide antagonists interact with a receptor through more binding sites and accumulate better and for a longer time period in cancer cells [
33,
34]. Even though the further evaluation of aprepitant-based radiopharmaceuticals is still needed, the findings reported herein provide insight on the perspectives of their application in the theranostics paradigm.
5. Patent
In course of this study, the following national patent application was submitted: No. P430136 “The modified drug substance molecule, method of its production, diagnostic or therapeutic receptor radiopharmaceutical based on this molecule, method of its production and its application”.
Author Contributions
Conceptualization, P.K.H. and P.F.J.L.; methodology, P.K.H., P.F.J.L., J.M., and P.K.; investigation, P.K.H. and J.M.; writing—original draft preparation, P.K.H. and P.F.J.L.; writing—review and editing, P.K. and E.G.; visualization, P.F.J.L.; supervision, A.M. and E.G.; project administration, A.M. and E.G.; funding acquisition, A.M. and E.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Science Centre (Poland), grant number 2017/25/B/NZ7/01896.
Acknowledgments
The contribution of Paweł Krzysztof Halik has been done in the frame of the National Centre for Research and Development Project No POWR.03.02.00-00-I009/17 (Radiopharmaceuticals for molecularly targeted diagnosis and therapy, RadFarm. Operational Project Knowledge Education Development 2014–2020 co-financed by European Social Fund).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 6HLO, 6HLL, 6HLP | accession codes of co-ordinates and structure factors in PDB |
| APT | aprepitant |
| BBB | Blood-brain barrier |
| Bn | benzyl moiety |
| BSA | bovine serum albumin |
| DMF | dimethylformamide |
| DOTA | 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid |
| DOTAGA | 1,4,7,10-tetraazacyclododecane, 1 glutaric acid - 4,7,10-acetic acid |
| DTPA | diethylenetriaminepentaacetic acid |
| ECL | extracellular loop |
| ESI | electrospray ionization |
| GAMG | human glioblastoma cell line |
| HEK 293 | human embryonic kidney 293 cell line |
| hNK1R | human neurokinin 1 receptor |
| HS | human serum |
| IC100 | maximal inhibitory concentration |
| IC50 | half-maximal inhibitory concentration |
| logD | logarithm of distribution coefficient |
| MS | mass spectrometry |
| n.c.a. | non-carrier added |
| NHS | N-hydroxysuccinimide moiety |
| NK1R | neurokinin 1 receptor, tachykinin 1 receptor |
| PBS | phosphate-buffered saline |
| PDB | Protein Data Bank |
| pKB | negative logarithm of Boltzmann constant |
| p-SCN | para-isocyanate group |
| rNK1R | rat neurokinin 1 receptor |
| RT | retention time |
| SEM | standard error of the means |
| SP | Substance P |
| SPE | solid phase extraction |
| TFA | trifluoroacetic acid |
| TM | transmembrane helix |
References
- Muñoz, M.; Rosso, M.; Coveñas, R. The NK-1 receptor: A new target in cancer therapy. Curr. Drug Targets 2011, 12, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Majkowska-Pilip, A.; Halik, P.K.; Gniazdowska, E. The Significance of NK1 receptor ligands and their application in targeted radionuclide tumour therapy. Pharmaceutics 2019, 11, 443. [Google Scholar] [CrossRef] [PubMed]
- Coveñas, R.; Muñoz, M. Cancer progression and substance P. Histol. Histopathol. 2014, 29, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, T.; Kwatra, S.G.; Goldsmith, L.E.; Richardson, M.D.; Cox, E.A.; Sampson, J.H.; Kwatra, M.M. A constitutively active form of neurokinin 1 receptor and neurokinin 1 receptor-mediated apoptosis in glioblastomas. J. Neurochem. 2009, 109, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Neth, O.; Ilmer, M.; Garnier, A.; Salinas-Martín, M.V.; de Agustín Asencio, J.C.; von Schweinitz, D.; Kappler, R. Hepatoblastoma cells express truncated neurokinin-1 receptor and can be inhibited by aprepitant in vitro and in vivo. J. Hepatol. 2014, 60, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Quartara, L.; Altamura, M.; Evangelista, S.; Maggi, C.A. Tachykinin receptor antagonists in clinical trials. Expert Opin. Investig. Drugs 2009, 18, 1843–1864. [Google Scholar] [CrossRef]
- Muñoz, M.; Rosso, M.; Aguilar, F.J.; González-Moles, M.A.; Redondo, M.; Esteban, F. NK-1 receptor antagonists induce apoptosis and counteract substance P-related mitogenesis in human laryngeal cancer cell line HEp-2. Investig. New Drugs 2008, 26, 111–118. [Google Scholar] [CrossRef]
- Muñoz, M.; Rosso, M.; Robles-Frías, M.J.; Salinas-Martín, M.V.; Coveñas, R. The NK-1 receptor is expressed in human melanoma and is involved in the antitumor action of the NK-1 receptor antagonist aprepitant on melanoma cell lines. Lab Investig. 2010, 90, 1259–1269. [Google Scholar] [CrossRef]
- Muñoz, M.; González-Ortega, A.; Coveñas, R. The NK-1 receptor is expressed in human leukemia and is involved in the antitumor action of aprepitant and other NK-1 receptor antagonists on acute lymphoblastic leukemia cell lines. Investig. New Drugs 2012, 30, 529–540. [Google Scholar] [CrossRef]
- Muñoz, M.; González-Ortega, A.; Salinas-Martín, M.V.; Carranza, A.; Garcia-Recio, S.; Almendro, V.; Coveñas, R. The neurokinin-1 receptor antagonist aprepitant is a promising candidate for the treatment of breast cancer. Int. J. Oncol. 2014, 45, 1658–1672. [Google Scholar] [CrossRef]
- Muñoz, M.; Berger, M.; Rosso, M.; Gonzalez-Ortega, A.; Carranza, A.; Coveñas, R. Antitumor activity of neurokinin-1 receptor antagonists in MG-63 human osteosarcoma xenografts. Int. J. Oncol. 2014, 44, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Dikmen, M. Antiproliferative and apoptotic effects of aprepitant on human glioblastoma U87MG cells. Marmara Pharm. J. 2017, 21, 156–164. [Google Scholar] [CrossRef]
- Kitchens, C.A.; McDonald, P.R.; Pollack, I.F.; Wipf, P.; Lazo, J.S. Synergy between microtubule destabilizing agents and neurokinin 1 receptor antagonists identified by an siRNA synthetic lethal screen. FASEB J. 2009, 23, 756–813. [Google Scholar]
- Kast, R.E.; Ramiro, S.; Llado, S.; Toro, S.; Coveñas, R.; Muñoz, M. Antitumour action of temozolomide, ritonavir and aprepitant against human glioma cells. J. Neurooncol. 2016, 126, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Rosso, M. The NK-1 receptor antagonist aprepitant as a broad-spectrum antitumor drug. Investig. New Drugs 2010, 28, 187–193. [Google Scholar] [CrossRef]
- Hale, J.J.; Mills, S.G.; MacCoss, M.; Shah, S.K.; Qi, H.; Mathre, D.J.; Cascieri, M.A.; Sadowski, S.; Strader, C.D.; MacIntyre, D.E.; et al. 2(S)-((3,5-Bis(trifluoromethyl)benzyl)oxy)-3(S)-phenyl-4-((3-oxo-1,2,4-triazol-5-yl)methyl)morpholine (1): A potent, orally active, morpholine-based human neurokinin-1 receptor antagonist. J. Med. Chem. 1996, 39, 1760–1762. [Google Scholar] [CrossRef]
- Hale, J.J.; Mills, S.G.; MacCoss, M.; Finke, P.E.; Cascieri, M.A.; Sadowski, S.; Ber, E.; Chicchi, G.G.; Kurtz, M.; Metzger, J.; et al. Structural optimization affording 2-(R)-(1-(R)-3,5-Bis(trifluoromethyl) phenylethoxy)-3-(S)-(4-fluoro)phenyl-4-(3-oxo-1,2,4-triazol-5-yl)methylmorpholine, a potent, orally active, long-acting Morpholine Acetal human NK-1 receptor antagonist. J. Med. Chem. 1998, 41, 4607–4614. [Google Scholar] [CrossRef]
- Harrison, T.; Owens, A.P.; Williams, B.J.; Swain, C.J.; Williams, A.; Carlson, A.; Rycroft, W.; Tattersall, F.D.; Cascieri, M.A.; Chicchi, G.G.; et al. An orally active, water-soluble Neurokinin-1 receptor antagonist suitable for both intravenous and oral clinical administration. J. Med. Chem. 2001, 44, 4296–4299. [Google Scholar] [CrossRef]
- Schöppe, J.; Ehrenmann, J.; Klenk, C.; Rucktooa, P.; Schütz, M.; Doré, A.S.; Plückthun, A. Crystal structures of the human neurokinin 1 receptor in complex with clinically used antagonists. Nat. Commun. 2019, 10, 17–27. [Google Scholar] [CrossRef]
- Giannini, G.; Milazzo, F.M.; Battistuzzi, G.; Rosi, A.; Anastasi, A.M.; Petronzelli, F.; Albertoni, C.; Tei, L.; Leone, L.; Salvini, L.; et al. Synthesis and preliminary in vitro evaluation of DOTA-Tenatumomabconjugates for theranostic applications in tenascin expressing tumors. Bioorg. Med. Chem. 2019, 27, 3248–3253. [Google Scholar] [CrossRef]
- Waterhouse, R.N. Determination of Lipophilicity and its use as a predictor of blood–brain barrier penetration of molecular imaging agents. Mol. Imaging Biol. 2003, 5, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Loper, A.; Landis, E.; Hettrick, L.; Novak, L.; Lynn, K.; Chen, C.; Thompson, K.; Higgins, R.; Batra, U.; et al. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: A Beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int. J. Pharm. 2004, 285, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Appell, K.C.; Fragale, B.J.; Loscig, J.; Singh, S.; Tomczuk, B.E. Antagonists that demonstrate species differences in neurokinin-1 receptors. Mol. Pharmacol. 1992, 41, 772–778. [Google Scholar] [PubMed]
- Pradier, L.; Habert-Ortoli, E.; Emile, L.; Le Guern, J.; Loquet, I.; Bock, M.D.; Clot, J.; Mercken, L.; Fardin, V.; Garret, C. Molecular determinants of the species selectivity of neurokinin type 1 receptor antagonists. Mol. Pharmacol. 1995, 47, 314–321. [Google Scholar]
- Leffer, A.; Ahlstedt, I.; Engberg, S.; Svensson, A.; Billger, M.; Oberg, L.; Bjursell, M.K.; Lindström, E.; von Mentzer, B. Characterization of species-related differences in the pharmacology of tachykinin NK receptors 1, 2 and 3. Biochem. Pharmacol. 2009, 77, 1522–1530. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Matalińska, J.; Lipiński, P.F.J.; Kotlarz, A.; Kosson, P.; Muchowska, A.; Dyniewicz, J. Evaluation of receptor affnity, analgesic activity and cytotoxicity of a hybrid peptide, AWL3020. Int. J. Pept. Res. Ther. 2020. [Google Scholar] [CrossRef]
- Aime, S.; Barge, A.; Botta, M.; Fasano, M.; Ayala, J.D.; Bombieri, G. Crystal structure and solution dynamics of the lutetium(III) chelate of DOTA. Inorg. Chmica Acta 1996, 246, 423–429. [Google Scholar] [CrossRef]
- Allen, F.H. The cambridge structural database: A quarter of a million crystal structures and rising. Acta Crystallogr. B 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 4, D440–D446. [Google Scholar] [CrossRef]
- Schrödinger LLC. The PyMOL Molecular Graphics System 2018. Available online: https://github.com/schrodinger/pymol-open-source (accessed on 31 July 2020).
- Zhang, H.; Kanduluru, A.K.; Desai, P.; Ahad, A.; Carlin, S.; Tandon, N.; Weber, W.A.; Low, P.S. Synthesis and evaluation of a Novel 64Cu- and 67Ga-Labeled Neurokinin 1 receptor antagonist for in Vivo targeting of NK1R-Positive tumor Xenografts. Bioconjug. Chem. 2018, 29, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Ginj, M.; Zhang, H.; Waser, B.; Cescato, R.; Wild, D.; Wang, X.; Erchegyi, J.; Rivier, J.; Mäcke, H.R.; Reubi, J.C. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc. Natl. Acad. Sci. USA 2006, 103, 16436–16441. [Google Scholar] [CrossRef] [PubMed]
- Cescato, R.; Maina, T.; Nock, B.; Nikolopoulou, A.; Charalambidis, D.; Piccand, V.; Reubi, J.C. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J. Nucl. Med. 2008, 49, 318–326. [Google Scholar] [CrossRef] [PubMed]
| Sample Availability: Samples of the compounds 2A–C, 4D, E are available from the authors. |
Figure 1.
Structure of aprepitant with its key elements marked.
Figure 1.
Structure of aprepitant with its key elements marked.
Scheme 1.
Synthetic route of aprepitant derivatives with aminoalkyl linkers; where n = {2; 3; 4}.
Scheme 1.
Synthetic route of aprepitant derivatives with aminoalkyl linkers; where n = {2; 3; 4}.
Scheme 2.
Synthetic route of aprepitant derivatives with acetamide linkers; where k = {0; 2}.
Scheme 2.
Synthetic route of aprepitant derivatives with acetamide linkers; where k = {0; 2}.
Scheme 3.
Synthetic routes of conjugations of selected chelators to aprepitant-ethylamine 2A.
Scheme 3.
Synthetic routes of conjugations of selected chelators to aprepitant-ethylamine 2A.
Figure 2.
Radiochromatograms of aprepitant (APT)-ethylamine 2A conjugates with DOTA, Bn-DOTA, Bn-DOTAGA or DTPA radiolabeled with gallium-68 (upper two) or with lutetium-177 (bottom).
Figure 2.
Radiochromatograms of aprepitant (APT)-ethylamine 2A conjugates with DOTA, Bn-DOTA, Bn-DOTAGA or DTPA radiolabeled with gallium-68 (upper two) or with lutetium-177 (bottom).
Figure 3.
Radiochromatograms of DOTA conjugates with all APT derivatives radiolabeled with gallium-68 (upper two) or with lutetium-177 (bottom two).
Figure 3.
Radiochromatograms of DOTA conjugates with all APT derivatives radiolabeled with gallium-68 (upper two) or with lutetium-177 (bottom two).
Figure 4.
Percentage of intact [68Ga]Ga-radioconjugates (left) and [177Lu]Lu-radioconjugates (right) determined at specific time points during incubation in 37 °C human serum.
Figure 4.
Percentage of intact [68Ga]Ga-radioconjugates (left) and [177Lu]Lu-radioconjugates (right) determined at specific time points during incubation in 37 °C human serum.
Figure 5.
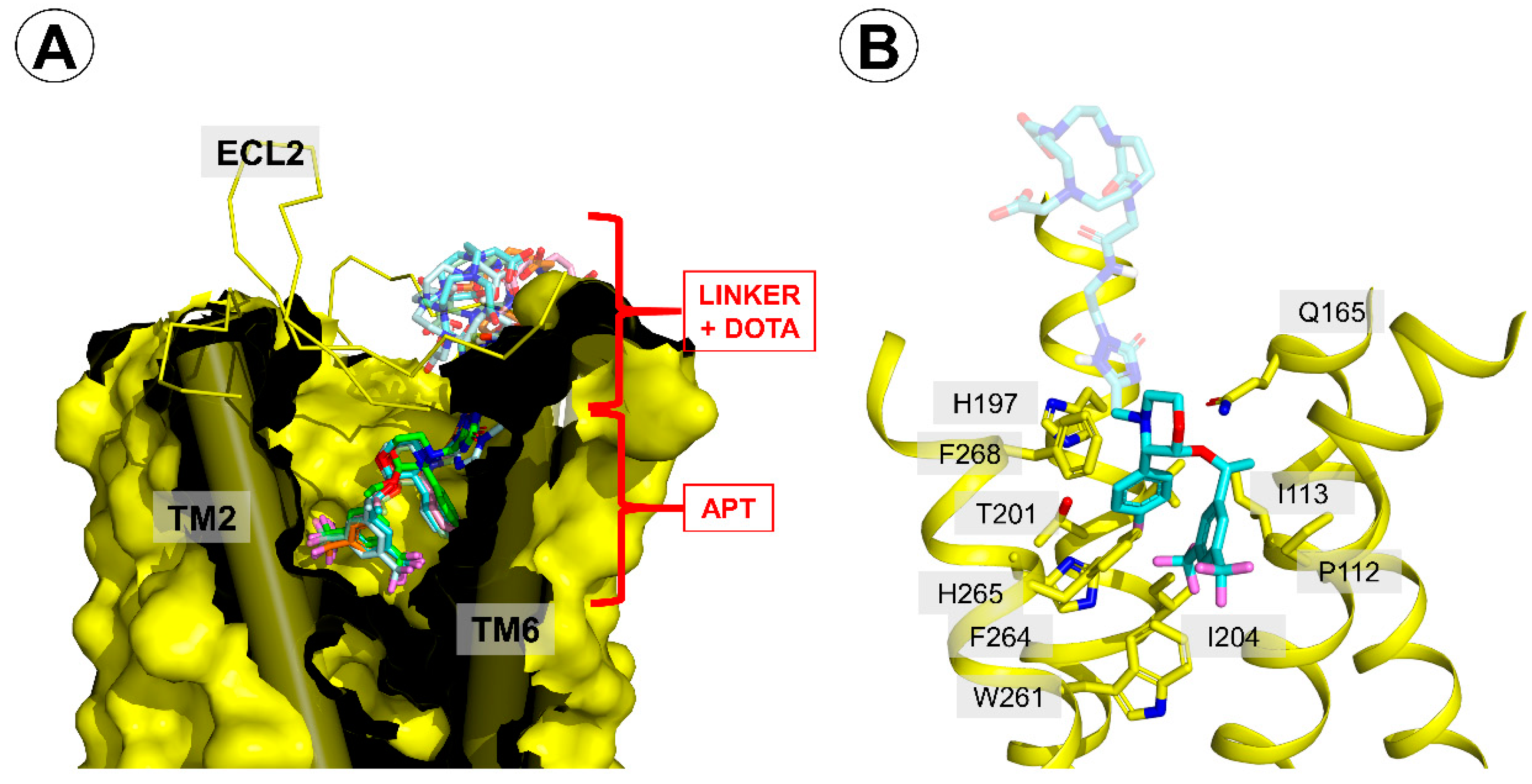
Binding mode of the reported conjugates in the neurokinin 1 receptor (NK1R) binding site. (A) A generalized view on the binding mode. The receptor is displayed as a yellow surface, with transmembrane helices (TMs) 2 and 6 shown as cylinders. The extracellular loop 2 (ECL2) is shown as a yellow ribbon. The conjugates are represented as colored sticks. (B) A view focused on the interactions of the part common to aprepitant and the conjugates. The conjugate shown is compound 5A (pale blue sticks). Only several residues of the receptor are shown (yellow sticks).
Figure 5.
Binding mode of the reported conjugates in the neurokinin 1 receptor (NK1R) binding site. (A) A generalized view on the binding mode. The receptor is displayed as a yellow surface, with transmembrane helices (TMs) 2 and 6 shown as cylinders. The extracellular loop 2 (ECL2) is shown as a yellow ribbon. The conjugates are represented as colored sticks. (B) A view focused on the interactions of the part common to aprepitant and the conjugates. The conjugate shown is compound 5A (pale blue sticks). Only several residues of the receptor are shown (yellow sticks).
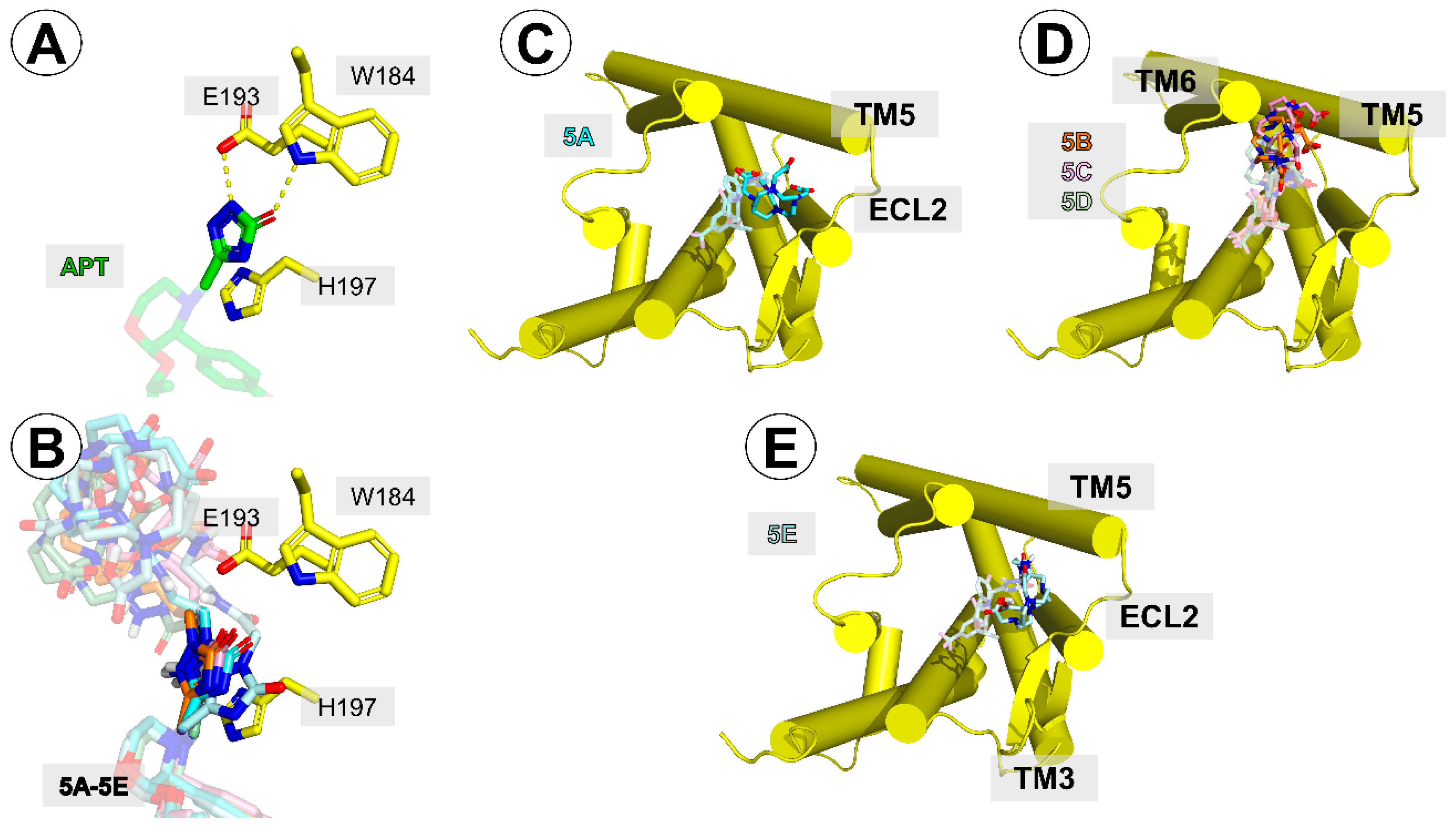
Figure 6.
(A) Interactions of aprepitant’s triazolinone ring with W184, E193, and H197 side-chains. (B) positioning of the triazolinone ring of the conjugates in the same projection as in (A). (C–E) Relative position of the DOTA moiety in conjugates 5A (C), 5B–D (D), and 5E (E). The receptor helices are shown as yellow cylinders.
Figure 6.
(A) Interactions of aprepitant’s triazolinone ring with W184, E193, and H197 side-chains. (B) positioning of the triazolinone ring of the conjugates in the same projection as in (A). (C–E) Relative position of the DOTA moiety in conjugates 5A (C), 5B–D (D), and 5E (E). The receptor helices are shown as yellow cylinders.
Table 1.
Retention times (RT) of stable gallium conjugates and [68Ga]Ga-radioconjugates of aprepitant-ethylamine 2A.
Table 1.
Retention times (RT) of stable gallium conjugates and [68Ga]Ga-radioconjugates of aprepitant-ethylamine 2A.
| Stable Ga-Conjugate | RT | [68Ga]Ga-Radioconjugate | RT |
|---|
| Ga-DOTA-Et-APT, Ga-5A | 17.4 min | [68Ga]Ga-DOTA-Et-APT, [68Ga]Ga-5A | 17.7 min |
| Ga-DOTA-Bn-Et-APT, Ga-6A | 18.5 min | [68Ga]Ga-DOTA-Bn-Et-APT, [68Ga]Ga-6A | 18.9 min |
| Ga-DOTAGA-Bn-Et-APT, Ga-7A | 17.7 min | [68Ga]Ga-DOTAGA-Bn-Et-APT, [68Ga]Ga-7A | 18.1 min |
| Ga-DTPA-Et-APT, Ga-8A | 17.6 min | [68Ga]Ga-DTPA-Et-APT, [68Ga]Ga-8A | 17.9 min |
| Ga-DTPA-(Et-APT)2, Ga-9A | 23.6 min | [68Ga]Ga-DTPA-(Et-APT)2, [68Ga]Ga-9A | 23.9 min |
Table 2.
RT of stable gallium conjugates and [68Ga]Ga-radioconjugates of all aprepitant derivatives.
Table 2.
RT of stable gallium conjugates and [68Ga]Ga-radioconjugates of all aprepitant derivatives.
| Stable Ga-Conjugate | RT | [68Ga]Ga-Radioconjugate | RT |
|---|
| Ga-DOTA-Et-APT, Ga-5A | 17.4 min | [68Ga]Ga-DOTA-Et-APT, [68Ga]Ga-5A | 17.7 min |
| Ga-DOTA-Pr-APT, Ga-5B | 16.8 min | [68Ga]Ga-DOTA-Pr-APT, [68Ga]Ga-5B | 17.0 min |
| Ga-DOTA-Bu-APT, Ga-5C | 16.9 min | [68Ga]Ga-DOTA-Bu-APT, [68Ga]Ga-5C | 17.3 min |
| Ga-DOTA-HN-NH-Ac-APT, Ga-5D | 15.8 min | [68Ga]Ga-DOTA-HN-NH-Ac-APT, [68Ga]Ga-5D | 16.3 min |
| Ga-DOTA-Et-Ac-APT, Ga-5E | 16.1 min | [68Ga]Ga-DOTA-Et-Ac-APT, [68Ga]Ga-5E | 16.5 min |
Table 3.
LogD values of human serum stable radioconjugates determined in n-octanol/PBS buffer system.
Table 3.
LogD values of human serum stable radioconjugates determined in n-octanol/PBS buffer system.
| Radioconjugate | logD |
|---|
| 68Ga- | 177Lu- |
|---|
| APT-Et-DOTA, 5A | 0.141 ± 0.019 | 0.708 ± 0.021 |
| APT-Pr-DOTA, 5B | 0.058 ± 0.016 | 0.654 ± 0.022 |
| APT-Bu-DOTA, 5C | 0.290 ± 0.018 | 0.777 ± 0.021 |
| APT-Ac-HN-NH-DOTA, 5D | −1.012 ± 0.017 | −0.401 ± 0.015 |
| APT-Ac-Et-DOTA, 5E | −0.231 ± 0.015 | 0.500 ± 0.017 |
Table 4.
Binding affinity of aprepitant and compounds 5A–E for the rat neurokinin 1 receptor.
Table 4.
Binding affinity of aprepitant and compounds 5A–E for the rat neurokinin 1 receptor.
| Compound | IC50 ± SEM a [µM] | Ratio to APT |
|---|
| Aprepitant | 0.13 ± 0.06 | 1.0 |
| APT-Et-DOTA, 5A | 6.2 ± 2.6 | 48.2 |
| APT-Pr-DOTA, 5B | 0.69 ± 0.07 | 5.3 |
| APT-Bu-DOTA, 5C | 1.8 ± 0.7 | 14.3 |
| APT-Ac-HN-NH-DOTA, 5D | 2.5 ± 0.7 | 19.1 |
| APT-Ac-Et-DOTA, 5E | 2.5 ± 0.5 | 19.7 |
Table 5.
Scoring results from molecular docking. The values are the estimated free energy of binding (kcal/mol).
Table 5.
Scoring results from molecular docking. The values are the estimated free energy of binding (kcal/mol).
| Rigid Receptor | Enabled Flexibility of E193 and H197 |
|---|
| Compound | Lowest a | Mean b | Lowest a | Mean b |
|---|
| APT-Et-DOTA, 5A | −11.26 | −11.26 | −12.37 | −10.24 |
| APT-Pr-DOTA, 5B | −10.84 | −9.86 | −10.74 | −9.27 |
| APT-Bu-DOTA, 5C | −12.26 | −11.90 | −12.21 | −11.83 |
| APT-Ac-HN-NH-DOTA, 5D | −9.64 | −9.07 | −9.19 | −8.68 |
| APT-Ac-Et-DOTA, 5E | −13.74 | −13.16 | −14.31 | −13.60 |
| Aprepitant (APT) | −10.43 | −10.31 | −10.57 | −10.40 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}