IDH1 Targeting as a New Potential Option for Intrahepatic Cholangiocarcinoma Treatment—Current State and Future Perspectives

 , , and
, , and
Abstract
1. Cholangiocarcinoma: From Classification to Treatment Strategies
2. Molecular Profile of iCCA
IDH1 Mutations in Intrahepatic Cholangiocarcinoma
3. Biological Impact of IDH1 Mutation in Cellular Processes and Its Contribution to Carcinogenesis
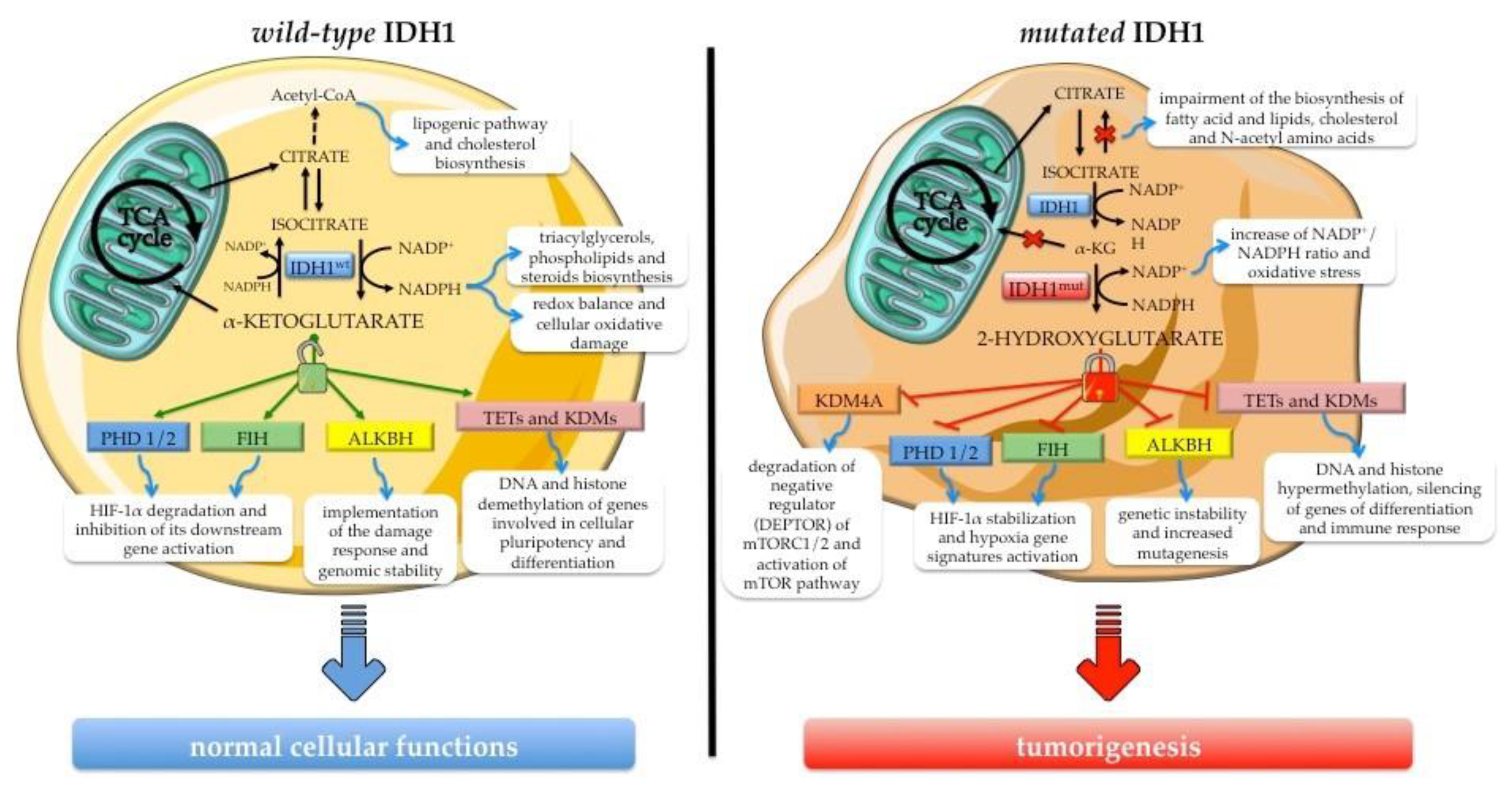
3.1. The Role of Wild-Type IDH1 in Cellular Processes
3.2. Biological Effects of Neomorphic Activity of Mutated IDH1
3.3. Biological Impact of IDH1 Mutation in Intrahepatic Cholangiocarcinoma
4. IDH1 Inhibitors for Intrahepatic Cholangiocarcinoma Treatment
5. Future Perspectives of IDH1 Inhibitors Use in Intrahepatic Cholangiocarcinoma
Author Contributions
Funding
Conflicts of Interest
References
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Blechacz, B. Cholangiocarcinoma: Current knowledge and new developments. Gut Liver 2017, 11, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma-evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef]
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39, 19–31. [Google Scholar] [CrossRef]
- Kendall, T.; Verheij, J.; Gaudio, E.; Evert, M.; Guido, M.; Goeppert, B.; Carpino, G. Anatomical, histomorphological and molecular classification of cholangiocarcinoma. Liver Int. 2019, 39, 7–18. [Google Scholar] [CrossRef]
- Cillo, U.; Fondevila, C.; Donadon, M.; Gringeri, E.; Mocchegiani, F.; Schlitt, H.J.; Ijzermans, J.N.M.; Vivarelli, M.; Zieniewicz, K.; Olde, S.W.M.; et al. Surgery for cholangiocarcinoma. Liver Int. 2019, 39, 143–155. [Google Scholar] [CrossRef]
- Kelley, R.K.; Bridgewater, J.; Gores, G.J.; Zhu, A.X. Systemic therapies for intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 72, 353–363. [Google Scholar] [CrossRef]
- Cotton, J.; Lamarca, A.; McNamara, M.G.; Valle, J.W. Novel treatments for advanced cholangiocarcinoma. In Liver Cancers; Springer: New York, NY, USA, 2019; pp. 227–243. [Google Scholar]
- Stobiecka, M.; Hepel, M. Double-shell gold nanoparticle-based DNA-carriers with poly-l-lysine binding surface. Biomaterials 2011, 32, 3312–3321. [Google Scholar] [CrossRef]
- Stobiecka, M.; Dworakowska, B.; Jakiela, S.; Lukasiak, A.; Chalupa, A.; Zembrzycki, K. Sensing of survivin mRNA in malignant astrocytes using graphene oxide nanocarrier-supported oligonucleotide molecular beacons. Sens. Actuators B Chem. 2016, 235, 136–145. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Hubner, R.A.; David Ryder, W.; Valle, J.W. Second-line chemotherapy in advanced biliary cancer: A systematic review. Ann. Oncol. 2014, 25, 2328–2338. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.E.; Law, L.; Nguyen, V.D.; Qiao, W.; Javle, M.M.; Kaseb, A.; Shroff, R.T. Second-line systemic treatment for advanced cholangiocarcinoma. J. Gastrointest. Oncol. 2014, 5, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Fornaro, L.; Vivaldi, C.; Cereda, S.; Leone, F.; Aprile, G.; Lonardi, S.; Silvestris, N.; Santini, D.; Milella, M.; Caparello, C.; et al. Second-line chemotherapy in advanced biliary cancer progressed to first-line platinum-gemcitabine combination: A multicenter survey and pooled analysis with published data. J. Exp. Clin. Cancer Res. 2015, 34, 156. [Google Scholar] [CrossRef]
- Guion-Dusserre, J.F.; Lorgis, V.; Vincent, J.; Bengrine, L.; Ghiringhelli, F. FOLFIRI plus bevacizumab as a second-line therapy for metastatic intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2015, 21, 2096–2101. [Google Scholar] [CrossRef]
- Lowery, M.A.; Goff, L.W.; Keenan, B.P.; Jordan, E.; Wang, R.; Bocobo, A.G.; Chou, J.F.; O’Reilly, E.M.; Harding, J.J.; Kemeny, N.; et al. Second-line chemotherapy in advanced biliary cancers: A retrospective, multicenter analysis of outcomes. Cancer 2019, 125, 4426–4434. [Google Scholar] [CrossRef]
- Condelli, V.; Maddalena, F.; Sisinni, L.; Lettini, G.; Matassa, D.S.; Piscazzi, A.; Palladino, G.; Amoroso, M.R.; Esposito, F.; Landriscina, M. Targeting TRAP1 as a downstream effector of BRAF cytoprotective pathway: A novel strategy for human BRAF-driven colorectal carcinoma. Oncotarget 2015, 6, 22298–22309. [Google Scholar] [CrossRef]
- Spagnoletti, G.; Li Bergolis, V.; Piscazzi, A.; Giannelli, F.; Condelli, V.; Sisinni, L.; Bove, G.; Storto, G.; Landriscina, M. Cyclin-dependent kinase 1 targeting improves sensitivity to radiation in BRAF V600E colorectal carcinoma cells. Tumor Biol. 2018, 40. [Google Scholar] [CrossRef]
- Stobiecka, M.; Ratajczak, K.; Jakiela, S. Toward early cancer detection: Focus on biosensing systems and biosensors for an anti-apoptotic protein survivin and survivin mRNA. Biosens. Bioelectron. 2019, 137, 58–71. [Google Scholar] [CrossRef]
- Criscuolo, D.; Avolio, R.; Calice, G.; Laezza, C.; Paladino, S.; Navarra, G.; Maddalena, F.; Crispo, F.; Pagano, C.; Bifulco, M.; et al. Cholesterol homeostasis modulates platinum sensitivity in human ovarian cancer. Cells 2020, 9, 828. [Google Scholar] [CrossRef]
- Marin, J.J.G.; Lozano, E.; Herraez, E.; Asensio, M.; Di Giacomo, S.; Romero, M.R.; Briz, O.; Serrano, M.A.; Efferth, T.; Macias, R.I.R. Chemoresistance and chemosensitization in cholangiocarcinoma. Biochim. Biophys. Acta—Mol. Basis Dis. 2018, 1864, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant isocitrate dehydrogenase inhibitors as targeted cancer therapeutics. Front. Oncol. 2019, 9, 417. [Google Scholar] [CrossRef] [PubMed]
- Moeini, A.; Sia, D.; Bardeesy, N.; Mazzaferro, V.; Llovet, J.M. Molecular pathogenesis and targeted therapies for intrahepatic cholangiocarcinoma. Clin. Cancer Res. 2016, 22, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.B.; Thorgeirsson, S.S. Genetic profiling of intrahepatic cholangiocarcinoma. Curr. Opin. Gastroenterol. 2012, 28, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Borad, M.J.; Gores, G.J.; Roberts, L.R. Fibroblast growth factor receptor 2 fusions as a target for treating cholangiocarcinoma. Curr. Opin. Gastroenterol. 2015, 31, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef]
- Yang, B.; House, M.G.; Guo, M.; Herman, J.G.; Clark, D.P. Promoter methylation profiles of tumor suppressor genes in intrahepatic and extrahepatic cholangiocarcinoma. Mod. Pathol. 2005, 18, 412–420. [Google Scholar] [CrossRef][Green Version]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef]
- Zender, S.; Nickeleit, I.; Wuestefeld, T.; Sörensen, I.; Dauch, D.; Bozko, P.; El-Khatib, M.; Geffers, R.; Bektas, H.; Manns, M.P.; et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 2013, 23, 784–795. [Google Scholar] [CrossRef]
- Boulter, L.; Guest, R.V.; Kendall, T.J.; Wilson, D.H.; Wojtacha, D.; Robson, A.J.; Ridgway, R.A.; Samuel, K.; Van Rooijen, N.; Barry, S.T.; et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J. Clin. Investig. 2015, 125, 1269–1285. [Google Scholar] [CrossRef]
- Boscoe, A.N.; Rolland, C.; Kelley, R.K. Frequency and prognostic significance of isocitrate dehydrogenase 1 mutations in cholangiocarcinoma: A systematic literature review. J. Gastrointest. Oncol. 2019, 10, 751–765. [Google Scholar] [CrossRef] [PubMed]
- De Botton, S.; Mondesir, J.; Willekens, C.; Touat, M. IDH1 and IDH2 mutations as novel therapeutic targets: Current perspectives. J. Blood Med. 2016, 7, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Govindan, A.; Sheth, R.A.; Nardi, V.; Blaszkowsky, L.S.; Faris, J.E.; Clark, J.W.; Ryan, D.P.; Kwak, E.L.; Allen, J.N.; et al. Prognosis and clinicopathologic features of patients with advanced stage isocitrate dehydrogenase (IDH) mutant and IDH wild-type intrahepatic cholangiocarcinoma. Oncologist 2015, 20, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Kipp, B.R.; Voss, J.S.; Kerr, S.E.; Barr Fritcher, E.G.; Graham, R.P.; Zhang, L.; Edward Highsmith, W.; Zhang, J.; Roberts, L.R.; Gores, G.J.; et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum. Pathol. 2012, 43, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
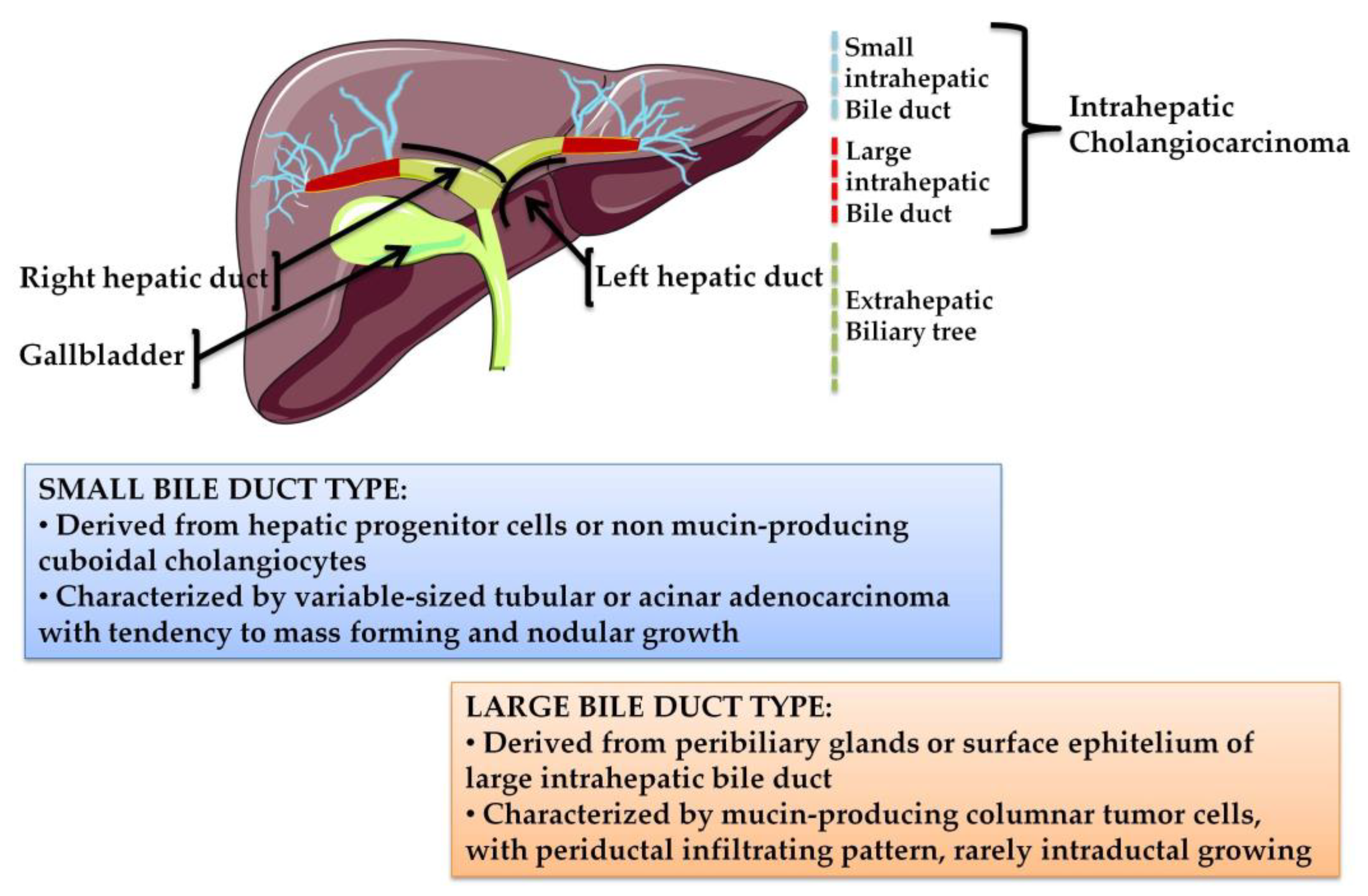
- Aishima, S.; Oda, Y. Pathogenesis and classification of intrahepatic cholangiocarcinoma: Different characters of perihilar large duct type versus peripheral small duct type. J. Hepato Biliary Pancreat. Sci. 2015, 22, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Misumi, K.; Shibahara, J.; Arita, J.; Sakamoto, Y.; Hasegawa, K.; Kokudo, N.; Fukayama, M. Distinct clinicopathologic and genetic features of 2 histologic subtypes of intrahepatic cholangiocarcinoma. Am. J. Surg. Pathol. 2016, 40, 1021–1030. [Google Scholar] [CrossRef]
- Liau, J.Y.; Tsai, J.H.; Yuan, R.H.; Chang, C.N.; Lee, H.J.; Jeng, Y.M. Morphological subclassification of intrahepatic cholangiocarcinoma: Etiological, clinicopathological, and molecular features. Mod. Pathol. 2014, 27, 1163–1173. [Google Scholar] [CrossRef]
- Akita, M.; Fujikura, K.; Ajiki, T.; Fukumoto, T.; Otani, K.; Azuma, T.; Itoh, T.; Ku, Y.; Zen, Y. Dichotomy in intrahepatic cholangiocarcinomas based on histologic similarities to hilar cholangiocarcinomas. Mod. Pathol. 2017, 30, 986–997. [Google Scholar] [CrossRef]
- Zou, S.; Li, J.; Zhou, H.; Frech, C.; Jiang, X.; Chu, J.S.C.; Zhao, X.; Li, Y.; Li, Q.; Wang, H.; et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 2014, 5, 5696. [Google Scholar] [CrossRef]
- Akita, M.; Sofue, K.; Fujikura, K.; Otani, K.; Itoh, T.; Ajiki, T.; Fukumoto, T.; Zen, Y. Histological and molecular characterization of intrahepatic bile duct cancers suggests an expanded definition of perihilar cholangiocarcinoma. HPB 2019, 21, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Wang, K.; Gay, L.; Al-Rohil, R.; Rand, J.V.; Jones, D.M.; Lee, H.J.; Sheehan, C.E.; Otto, G.A.; Palmer, G.; et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 2014, 19, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Kirstein, M.M.; Vogel, A. Epidemiology and risk factors of cholangiocarcinoma. Visc. Med. 2016, 32, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef]
- Chan-On, W.; Nairismägi, M.L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef]
- Fujimoto, A.; Furuta, M.; Shiraishi, Y.; Gotoh, K.; Kawakami, Y.; Arihiro, K.; Nakamura, T.; Ueno, M.; Ariizumi, S.I.; Nguyen, H.H.; et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat. Commun. 2015, 6, 6120. [Google Scholar] [CrossRef]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: Potential targets for intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef]
- Simbolo, M.; Fassan, M.; Ruzzenente, A.; Mafficini, A.; Wood, L.D.; Corbo, V.; Melisi, D.; Malleo, G.; Vicentini, C.; Malpeli, G.; et al. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget 2014, 5, 2839–2852. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, J.; Xu, Z.; Peng, B.; Huang, Q.; Arnold, E.; Ding, J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 2004, 279, 33946–33957. [Google Scholar] [CrossRef]
- Zhao, S.; Guan, K.L. IDH1 mutant structures reveal a mechanism of dominant inhibition. Cell Res. 2010, 20, 1279–1281. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Cross, J.R.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.A.; Thompson, C.B. Identification of additional IDH mutations associated with oncometabolite R(−)-2-hydroxyglutarate production. Oncogene 2012, 31, 2491–2498. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Reitman, Z.J.; Duncan, C.G.; Spasojevic, I.; Gooden, D.M.; Rasheed, B.A.; Yang, R.; Lopez, G.Y.; He, Y.; McLendon, R.E.; et al. Disruption of wild-type IDH1 suppresses d-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2013, 73, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Lu, C.; Cross, J.R.; Abdel-Wahab, O.; Levine, R.L.; Schwartz, G.K.; Thompson, C.B. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J. Biol. Chem. 2013, 288, 3804–3815. [Google Scholar] [CrossRef]
- Matteo, D.A.; Grunseth, A.J.; Gonzalez, E.R.; Anselmo, S.L.; Kennedy, M.A.; Moman, P.; Scott, D.A.; Hoang, A.; Sohl, C.D. Molecular mechanisms of isocitrate dehydrogenase 1 (IDH1) mutations identified in tumors: The role of size and hydrophobicity at residue 132 on catalytic efficiency. J. Biol. Chem. 2017, 292, 7971–7983. [Google Scholar] [CrossRef]
- Pusch, S.; Schweizer, L.; Beck, A.C.; Lehmler, J.M.; Weissert, S.; Balss, J.; Miller, A.K.; von Deimling, A. d-2-Hydroxyglutarate producing neo-enzymatic activity inversely correlates with frequency of the type of isocitrate dehydrogenase 1 mutations found in glioma. Acta Neuropathol. Commun. 2014, 2, 19. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef]
- Rzem, R.; Veiga-Da-Cunha, M.; Noël, G.; Goffette, S.; Nassogne, M.C.; Tabarki, B.; Schöller, C.; Marquardt, T.; Vikkula, M.; Van Schaftingen, E. A gene encoding a putative FAD-dependent l-2-hydroxyglutarate dehydrogenase is mutated in l-2-hydroxyglutaric aciduria. Proc. Natl. Acad. Sci. USA 2004, 101, 16849–16854. [Google Scholar] [CrossRef]
- Achouri, Y.; Noël, G.; Vertommen, D.; Rider, M.H.; Veiga-Da-Cunha, M.; Van Schaftingen, E. Identification of a dehydrogenase acting on d-2-hydroxyglutarate. Biochem. J. 2004, 381, 35–42. [Google Scholar] [CrossRef]
- Tommasini-Ghelfi, S.; Murnan, K.; Kouri, F.M.; Mahajan, A.S.; May, J.L.; Stegh, A.H. Cancer-associated mutation and beyond: The emerging biology of isocitrate dehydrogenases in human disease. Sci. Adv. 2019, 5, eaaw4543. [Google Scholar] [CrossRef] [PubMed]
- Abla, H.; Sollazzo, M.; Gasparre, G.; Iommarini, L.; Porcelli, A.M. The multifaceted contribution of α-ketoglutarate to tumor progression: An opportunity to exploit? Semin. Cell Dev. Biol. 2020, 98, 26–33. [Google Scholar] [CrossRef] [PubMed]
- McDonough, M.A.; Loenarz, C.; Chowdhury, R.; Clifton, I.J.; Schofield, C.J. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr. Opin. Struct. Biol. 2010, 20, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Finley, L.W.S.; Cross, J.R.; Allis, C.D.; Craig, B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Reid, M.A.; Lowman, X.H.; Kulkarni, R.P.; Tran, T.Q.; Liu, X.; Yang, Y.; Hernandez-davies, J.E.; Rosales, K.K.; Li, H.; et al. Regional glutamine deficiency in tumors promotes de-differentiation through inhibition of histone demethylation. Nat. Cell Biol. 2016, 18, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Kwak, S.; Cho, E.; Youn, H.; Hwang, I.; Kwak, S.; Lee, S.; Kim, H.; Lee, S.E.; Kim, J.; et al. Psat1-dependent fluctuations in α-ketoglutarate affect the timing of ESC differentiation. Cell Metab. 2016, 24, 494–501. [Google Scholar] [CrossRef]
- Tran, T.Q.; Hanse, E.A.; Habowski, A.N.; Li, H.; Ishak Gabra, M.B.; Yang, Y.; Lowman, X.H.; Ooi, A.M.; Liao, S.Y.; Edwards, R.A.; et al. α-ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nat. Cancer 2020, 1, 345–358. [Google Scholar] [CrossRef]
- Lu, H.; Dalgard, C.L.; Mohyeldin, A.; McFate, T.; Tait, A.S.; Verma, A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J. Biol. Chem. 2005, 280, 41928–41939. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain: A hypoxic switch. Science. 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef]
- Tennant, D.A.; Frezza, C.; MacKenzie, E.D.; Nguyen, Q.D.; Zheng, L.; Selak, M.A.; Roberts, D.L.; Dive, C.; Watson, D.G.; Aboagye, E.O.; et al. Reactivating HIF prolyl hydroxylases under hypoxia results in metabolic catastrophe and cell death. Oncogene 2009, 28, 4009–4021. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, J.; Shenghong, M.; Zhang, L.; Yao, J.; Hoadley, K.A.; Wilkerson, M.D.; Perou, C.M.; Guan, K.-L.; Ye, D.; et al. Oncometabolite d-2-hydroxyglutarate inhibits ALKBH DNA repair enzymes and sensitizes IDH-mutant cells to alkylating agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Bian, K.; Tang, Q.; Fedeles, B.I.; Singh, V.; Humulock, Z.T.; Essigmann, J.M.; Li, D. Oncometabolites d- and l-2-hydroxyglutarate inhibit the Alkb family DNA repair enzymes under physiological conditions. Chem. Res. Toxicol. 2017, 30, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; Van Noorden, C.J.F. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.Q.; Ishak Gabra, M.B.; Lowman, X.H.; Yang, Y.; Reid, M.A.; Pan, M.; O’Connor, T.R.; Kong, M. Glutamine deficiency induces DNA alkylation damage and sensitizes cancer cells to alkylating agents through inhibition of ALKBH enzymes. PLoS Biol. 2017, 15, e2002810. [Google Scholar] [CrossRef] [PubMed]
- Herr, C.Q.; Hausinger, R.P. Amazing diversity in biochemical roles of Fe(II)/2-oxoglutarate oxygenases. Trends Biochem. Sci. 2018, 43, 517–532. [Google Scholar] [CrossRef]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; Deberardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef]
- Chen, Q.; Kirk, K.; Shurubor, Y.I.; Zhao, D.; Arreguin, A.J.; Shahi, I.; Valsecchi, F.; Primiano, G.; Calder, E.L.; Carelli, V.; et al. Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab. 2018, 27, 1007–1025.e5. [Google Scholar] [CrossRef]
- Agledal, L.; Niere, M.; Ziegler, M. The phosphate makes a difference: Cellular functions of NADP. Redox Rep. 2010, 15, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Wang, B.; Liu, H.; Shah, H.; Carmona-fontaine, C.; Rustenburg, A.S.; Salah, S.; Gunner, M.R.; Chodera, J.D.; Cross, J.R.; et al. l-2-hydroxyglutarate production arises from non-canonical enzyme function at acidic pH. Nat. Chem. Biol. 2017, 13, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Turcan, S.; Fabius, A.W.M.; Borodovsky, A.; Pedraza, A.; Brennan, C.; Huse, J.; Viale, A.; Riggins, G.J.; Chan, T.A. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT inhibitor decitabine. Oncotarget 2013, 4, 1729–1736. [Google Scholar] [CrossRef]
- Zhang, X.; Rao, A.; Sette, P.; Deibert, C.; Pomerantz, A.; Kim, W.J.; Kohanbash, G.; Chang, Y.; Park, Y.; Engh, J.; et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro Oncol. 2016, 18, 1402–1412. [Google Scholar] [CrossRef]
- Mu, L.; Long, Y.; Yang, C.; Jin, L.; Tao, H.; Ge, H.; Chang, Y.E.; Karachi, A.; Kubilis, P.S.; De Leon, G.; et al. The IDH1 mutation-induced oncometabolite, 2-hydroxyglutarate, may affect DNA methylation and expression of PD-L1 in gliomas. Front. Mol. Neurosci. 2018, 11, 82. [Google Scholar] [CrossRef]
- Colvin, H.; Nishida, N.; Konno, M.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; Hata, T.; Kawamoto, K.; Asai, A.; Tsunekuni, K.; et al. Oncometabolite d-2-hydroxyglurate directly induces epithelial-mesenchymal transition and is associated with distant metastasis in colorectal cancer. Sci. Rep. 2016, 6, 36289. [Google Scholar] [CrossRef]
- Toh, T.B.; Lim, J.J.; Chow, E.K.H. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29. [Google Scholar] [CrossRef]
- Turcan, S.; Makarov, V.; Taranda, J.; Wang, Y.; Fabius, A.W.M.; Wu, W.; Zheng, Y.; El-Amine, N.; Haddock, S.; Nanjangud, G.; et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 2018, 50, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Raineri, S.; Mellor, J. IDH1: Linking metabolism and epigenetics. Front. Genet. 2018, 9, 493. [Google Scholar] [CrossRef] [PubMed]
- Gagné, L.M.; Boulay, K.; Topisirovic, I.; Huot, M.É.; Mallette, F.A. Oncogenic activities of IDH1/2 mutations: From epigenetics to cellular signaling. Trends Cell Biol. 2017, 27, 738–752. [Google Scholar]
- Ye, D.; Guan, K.L.; Xiong, Y. Metabolism, activity, and targeting of d- and l-2-hydroxyglutarates. Trends Cancer 2018, 4, 151–165. [Google Scholar] [CrossRef]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 2009, 324, 261–265. [Google Scholar] [CrossRef]
- Jiang, B.; Zhao, W.; Shi, M.; Zhang, J.; Chen, A.; Ma, H.; Suleman, M.; Lin, F.; Zhou, L.; Wang, J.; et al. IDH1 Arg-132 mutant promotes tumor formation through down-regulating p53. J. Biol. Chem. 2018, 293, 9747–9758. [Google Scholar] [CrossRef]
- Tarhonskaya, H.; Rydzik, A.M.; Leung, I.K.H.; Loik, N.D.; Chan, M.C.; Kawamura, A.; McCullagh, J.S.O.; Claridge, T.D.W.; Flashman, E.; Schofield, C.J. Non-enzymatic chemistry enables 2-hydroxyglutarate-mediated activation of 2-oxoglutarate oxygenases. Nat. Commun. 2014, 5, 3423. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Itsumi, M.; Elia, A.J.; Harris, I.S.; Chio, I.I.C.; Cairns, R.A.; Mccracken, S.; Wakeham, A.; Haight, J.; et al. d-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012, 26, 2038–2049. [Google Scholar] [CrossRef]
- Abdollahi, A.; Hahnfeldt, P.; Maercker, C.; Gröne, H.J.; Debus, J.; Ansorge, W.; Folkman, J.; Hlatky, L.; Huber, P.E. Endostatin’s antiangiogenic signaling network. Mol. Cell 2004, 13, 649–663. [Google Scholar] [CrossRef]
- Kalluri, R. Basement membranes: Structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 2003, 3, 422–433. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Carbonneau, M.; Gagne, L.M.; Lalonde, M.E.; Germain, M.A.; Motorina, A.; Guiot, M.C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7, 12700. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef] [PubMed]
- Grassian, A.R.; Parker, S.J.; Davidson, S.M.; Divakaruni, A.S.; Green, C.R.; Zhang, X.; Slocum, K.L.; Pu, M.; Lin, F.; Vickers, C.; et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014, 74, 3317–3331. [Google Scholar] [CrossRef]
- Parker, S.J.; Metallo, C.M. Metabolic consequences of oncogenic IDH mutations. Pharmacol. Ther. 2015, 152, 54–62. [Google Scholar] [CrossRef]
- Li, F.; He, X.; Ye, D.; Lin, Y.; Yu, H.; Yao, C.; Huang, L.; Zhang, J.; Wang, F.; Xu, S.; et al. NADP+-IDH mutations promote hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Mol. Cell 2015, 60, 661–675. [Google Scholar] [CrossRef]
- Izquierdo-Garcia, J.L.; Viswanath, P.; Eriksson, P.; Cai, L.; Radoul, M.; Chaumeil, M.M.; Blough, M.; Luchman, H.A.; Weiss, S.; Cairncross, J.G.; et al. IDH1 mutation induces reprogramming of pyruvate metabolism. Cancer Res. 2015, 75, 2999–3009. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell 2018, 175, 101–116.e25. [Google Scholar] [CrossRef]
- Chaumeil, M.M.; Radoul, M.; Najac, C.; Eriksson, P.; Viswanath, P.; Blough, M.D.; Chesnelong, C.; Luchman, H.A.; Cairncross, J.G.; Ronen, S.M. Hyperpolarized 13C MR imaging detects no lactate production in mutant IDH1 gliomas: Implications for diagnosis and response monitoring. Neuroimage Clin. 2016, 12, 180–189. [Google Scholar] [CrossRef]
- Viswanath, P.; Chaumeil, M.M.; Ronen, S.M. Molecular imaging of metabolic reprograming in mutant IDH cells. Front. Oncol. 2016, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Chesnelong, C.; Chaumeil, M.M.; Blough, M.D.; Al-Najjar, M.; Stechishin, O.D.; Chan, J.A.; Pieper, R.O.; Ronen, S.M.; Weiss, S.; Luchman, H.A.; et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro. Oncol. 2014, 16, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Goyal, L.; Yau, T.; Poon, R.T.; Ancukiewicz, M.; Deshpande, V.; Christiani, D.C.; Liebman, H.M.; Yang, H.; Kim, H.; et al. Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin. Cancer Res. 2014, 20, 1884–1890. [Google Scholar] [CrossRef]
- Delahousse, J.; Verlingue, L.; Broutin, S.; Legoupil, C.; Touat, M.; Doucet, L.; Ammari, S.; Lacroix, L.; Ducreux, M.; Scoazec, J.Y.; et al. Circulating oncometabolite d-2-hydroxyglutarate enantiomer is a surrogate marker of isocitrate dehydrogenase–mutated intrahepatic cholangiocarcinomas. Eur. J. Cancer 2018, 90, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Dong, Q.; Zhang, C.; Kuan, P.F.; Liu, Y.; Jeck, W.R.; Andersen, J.B.; Jiang, W.; Savich, G.L.; Tan, T.X.; et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013, 32, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Parachoniak, C.A.; Ghanta, K.S.; Fitamant, J.; Ross, K.N.; Najem, M.S.; Gurumurthy, S.; Akbay, E.A.; Sia, D.; Cornella, H.; et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014, 513, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Miao, R.; Liu, T.; Xiang, X.; Gu, J.; Jia, Y.; Li, Z.; Fu, Y.; He, Y.; Zhang, Y.; et al. IDH1 as a frequently mutated gene has potential effect on exosomes releasement by epigenetically regulating P2RX7 in intrahepatic cholangiocarcinoma. Biomed. Pharmacother. 2019, 113, 108774. [Google Scholar] [CrossRef]
- Nepal, C.; O’Rourke, C.J.; Oliveira, D.V.N.P.; Taranta, A.; Shema, S.; Gautam, P.; Calderaro, J.; Barbour, A.; Raggi, C.; Wennerberg, K.; et al. Genomic perturbations reveal distinct regulatory networks in intrahepatic cholangiocarcinoma. Hepatology 2018, 68, 949–963. [Google Scholar] [CrossRef]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 2017, 18, 2780–2794. [Google Scholar] [CrossRef]
- Fujiwara, H.; Tateishi, K.; Misumi, K.; Hayashi, A.; Igarashi, K.; Kato, H.; Nakatsuka, T.; Suzuki, N.; Yamamoto, K.; Kudo, Y.; et al. Mutant IDH1 confers resistance to energy stress in normal biliary cells through PFKP-induced aerobic glycolysis and AMPK activation. Sci. Rep. 2019, 9, 18859. [Google Scholar] [CrossRef]
- Ding, N.; Che, L.; Li, X.L.; Liu, Y.; Jiang, L.J.; Fan, B.; Tao, J.Y.; Chen, X.; Ji, J.F. Oncogenic potential of IDH1 R132C mutant in cholangiocarcinoma development in mice. World J. Gastroenterol. 2016, 22, 2071–2080. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, K.J.; Luo, L.; Hsu, V.; Gudi, R.; Dorff, S.E.; Przepiorka, D.; Deisseroth, A.; Shen, Y.L.; Sheth, C.M.; Charlab, R.; et al. FDA approval summary: Ivosidenib for relapsed or refractory acute myeloid leukemia with an isocitrate dehydrogenase-1 mutation. Clin. Cancer Res. 2019, 25, 3205–3209. [Google Scholar] [CrossRef] [PubMed]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Cho, Y.S.; Levell, J.R.; Liu, G.; Caferro, T.; Sutton, J.; Shafer, C.M.; Costales, A.; Manning, J.R.; Zhao, Q.; Sendzik, M.; et al. Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor. ACS Med. Chem. Lett. 2017, 8, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Caravella, J.A.; Lin, J.; Diebold, R.B.; Campbell, A.M.; Ericsson, A.; Gustafson, G.; Wang, Z.; Castro, J.; Clarke, A.; Gotur, D.; et al. Structure-based design and identification of FT-2102 (Olutasidenib), a potent mutant-selective IDH1 inhibitor. J. Med. Chem. 2020, 63, 1612–1623. [Google Scholar]
- Pusch, S.; Krausert, S.; Fischer, V.; Balss, J.; Ott, M.; Schrimpf, D.; Capper, D.; Sahm, F.; Eisel, J.; Beck, A.C.; et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 2017, 133, 629–644. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Herbst, L.; Pusch, S.; Klett, L.; Goparaju, R.; Stichel, D.; Kaulfuss, S.; Panknin, O.; Zimmermann, K.; Toschi, L.; et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia 2017, 31, 2020–2028. [Google Scholar] [CrossRef]
- Deng, G.; Shen, J.; Yin, M.; McManus, J.; Mathieu, M.; Gee, P.; He, T.; Shi, C.; Bedel, O.; McLean, L.R.; et al. Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. J. Biol. Chem. 2015, 290, 762–774. [Google Scholar] [CrossRef]
- Fan, B.; Mellinghoff, I.K.; Wen, P.Y.; Lowery, M.A.; Goyal, L.; Tap, W.D.; Pandya, S.S.; Manyak, E.; Jiang, L.; Liu, G.; et al. Clinical pharmacokinetics and pharmacodynamics of ivosidenib, an oral, targeted inhibitor of mutant IDH1, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 433–444. [Google Scholar] [CrossRef]
- Lowery, M.A.; Burris, H.A.; Janku, F.; Shroff, R.T.; Cleary, J.M.; Azad, N.S.; Goyal, L.; Maher, E.A.; Gore, L.; Hollebecque, A.; et al. Safety and activity of ivosidenib in patients with IDH1-mutant advanced cholangiocarcinoma: A phase 1 study. Lancet Gastroenterol. Hepatol. 2019, 4, 711–720. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Crispo, F.; Condelli, V.; Lepore, S.; Notarangelo, T.; Sgambato, A.; Esposito, F.; Maddalena, F.; Landriscina, M. Metabolic dysregulations and epigenetics: A bidirectional interplay that drives tumor progression. Cells 2019, 8, 798. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Gordan, J.D.; Kleinstiver, B.P.; Vu, P.; Najem, M.S.; Yeo, J.C.; Shi, L.; Kato, Y.; Levin, R.S.; Webber, J.T.; et al. Isocitrate dehydrogenase mutations confer dasatinib hypersensitivity and SRC dependence in intrahepatic cholangiocarcinoma. Cancer Discov. 2016, 6, 727–739. [Google Scholar] [CrossRef]
- Fujiwara, H.; Tateishi, K.; Kato, H.; Nakatsuka, T.; Yamamoto, K.; Tanaka, Y.; Ijichi, H.; Takahara, N.; Mizuno, S.; Kogure, H.; et al. Isocitrate dehydrogenase 1 mutation sensitizes intrahepatic cholangiocarcinoma to the BET inhibitor JQ1. Cancer Sci. 2018, 109, 3602–3610. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wild, A.T.; Turcan, S.; Wu, W.H.; Sigel, C.; Klimstra, D.S.; Ma, X.; Gong, Y.; Holland, E.C.; Huse, J.T.; et al. Targeting therapeutic vulnerabilities with PARP inhibition and radiation in IDH-mutant gliomas and cholangiocarcinomas. Sci. Adv. 2020, 6, eaaz3221. [Google Scholar] [CrossRef]
- Johannessen, T.C.A.; Mukherjee, J.; Viswanath, P.; Ohba, S.; Ronen, S.M.; Bjerkvig, R.; Pieper, R.O. Rapid conversion of mutant IDH1 from driver to passenger in a model of human gliomagenesis. Mol. Cancer Res. 2016, 14, 976–983. [Google Scholar] [CrossRef]
- Su, L.; Zhang, X.; Zheng, L.; Wang, M.; Zhu, Z.; Li, P. Mutation of isocitrate dehydrogenase 1 in cholangiocarcinoma impairs tumor progression by inhibiting isocitrate metabolism. Front. Endocrinol. (Lausanne) 2020, 11, 189. [Google Scholar] [CrossRef]
- Tan, F.; Jiang, Y.; Sun, N.; Chen, Z.; Lv, Y.; Shao, K.; Li, N.; Qiu, B.; Gao, Y.; Li, B.; et al. Identification of isocitrate dehydrogenase 1 as a potential diagnostic and prognostic biomarker for non-small cell lung cancer by proteomic analysis. Mol. Cell. Proteom. 2012, 11, M111.008821. [Google Scholar] [CrossRef]
- Sun, N.; Chen, Z.; Tan, F.; Zhang, B.; Yao, R.; Zhou, C.; Li, J.; Gao, Y.; Liu, Z.; Tan, X.; et al. Isocitrate dehydrogenase 1 is a novel plasma biomarker for the diagnosis of non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 5136–5145. [Google Scholar] [CrossRef]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-associated IDH1 promotes growth and resistance to targeted therapies in the absence of mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef]
- Zarei, M.; Lal, S.; Parker, S.J.; Nevler, A.; Vaziri-Gohar, A.; Dukleska, K.; Mambelli-Lisboa, N.C.; Moffat, C.; Blanco, F.F.; Chand, S.N.; et al. Posttranscriptional upregulation of IDH1 by HuR establishes a powerful survival phenotype in pancreatic cancer cells. Cancer Res. 2017, 77, 4460–4471. [Google Scholar] [CrossRef] [PubMed]
- Al-Amodi, H.S.A.B.; Nabih, E.S.; Kamel, H.F.M.; El Sayed, M.A.; Dwedar, I.A.M. Wild-type isocitrate dehydrogenase 1 over-expression is related to cancer stem cells survival in lung adenocarcinoma. Cancer Investig. 2018, 36, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, F.; Liu, X.; Zhang, T.; Liu, F.; Ge, X.; Mao, Y.; Hua, D. Correlation of IDH1 and B7–H3 expression with prognosis of CRC patients. Eur. J. Surg. Oncol. 2018, 44, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Xiao, L.; Zhao, Y.; Shi, J.; Yuan, Y.; Gu, Y.; Zhang, F.; Gao, X.; Yang, Y.; Yang, R.; et al. Wild-type IDH1 and mutant IDH1 opposingly regulate podoplanin expression in glioma. Transl. Oncol. 2020, 13, 100758. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| IC50 (µM) | ||||||
|---|---|---|---|---|---|---|
| Drug | IDH1 Mutation | Mutant | Wild-Type | Ref | Clinical Trial Number | Phase |
| AG-120 | R132H R132C R132G R132L R132S | 0.012 0.013 0.008 0.013 0.012 | 0.072 | [124] | NCT02073994 NCT02989857 NCT04088188 2015-005117-72 | I III I III |
| IDH305 | R132H R132C | 0.027 0.028 | 6.14 | [125] | NCT02381886 | I |
| FT-2102 | R132H R132C | 0.0212 0.0094 | >20.0 | [126] | NCT03684811 2018-001796-21 | I/II Ib/II |
| BAY1436032 | R132H R132C R132G R132L R132S | 0.015 0.015 0.004 * 0.003 * 0.016 * | 20.0 | [127,128] | NCT02746081 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crispo, F.; Pietrafesa, M.; Condelli, V.; Maddalena, F.; Bruno, G.; Piscazzi, A.; Sgambato, A.; Esposito, F.; Landriscina, M. IDH1 Targeting as a New Potential Option for Intrahepatic Cholangiocarcinoma Treatment—Current State and Future Perspectives. Molecules 2020, 25, 3754. https://doi.org/10.3390/molecules25163754
Crispo F, Pietrafesa M, Condelli V, Maddalena F, Bruno G, Piscazzi A, Sgambato A, Esposito F, Landriscina M. IDH1 Targeting as a New Potential Option for Intrahepatic Cholangiocarcinoma Treatment—Current State and Future Perspectives. Molecules. 2020; 25(16):3754. https://doi.org/10.3390/molecules25163754
Chicago/Turabian StyleCrispo, Fabiana, Michele Pietrafesa, Valentina Condelli, Francesca Maddalena, Giuseppina Bruno, Annamaria Piscazzi, Alessandro Sgambato, Franca Esposito, and Matteo Landriscina. 2020. "IDH1 Targeting as a New Potential Option for Intrahepatic Cholangiocarcinoma Treatment—Current State and Future Perspectives" Molecules 25, no. 16: 3754. https://doi.org/10.3390/molecules25163754
APA StyleCrispo, F., Pietrafesa, M., Condelli, V., Maddalena, F., Bruno, G., Piscazzi, A., Sgambato, A., Esposito, F., & Landriscina, M. (2020). IDH1 Targeting as a New Potential Option for Intrahepatic Cholangiocarcinoma Treatment—Current State and Future Perspectives. Molecules, 25(16), 3754. https://doi.org/10.3390/molecules25163754