Abstract
Cholangiocarcinoma is a primary malignancy of the biliary tract characterized by late and unspecific symptoms, unfavorable prognosis, and few treatment options. The advent of next-generation sequencing has revealed potential targetable or actionable molecular alterations in biliary tumors. Among several identified genetic alterations, the IDH1 mutation is arousing interest due to its role in epigenetic and metabolic remodeling. Indeed, some IDH1 point mutations induce widespread epigenetic alterations by means of a gain-of-function of the enzyme, which becomes able to produce the oncometabolite 2-hydroxyglutarate, with inhibitory activity on α-ketoglutarate-dependent enzymes, such as DNA and histone demethylases. Thus, its accumulation produces changes in the expression of several key genes involved in cell differentiation and survival. At present, small-molecule inhibitors of IDH1 mutated enzyme are under investigation in preclinical and clinical phases as promising innovative treatments for IDH1-mutated intrahepatic cholangiocarcinomas. This review examines the molecular rationale and the results of preclinical and early-phase studies on novel pharmacological agents targeting mutant IDH1 in cholangiocarcinoma patients. Contextually, it will offer a starting point for discussion on combined therapies with metabolic and epigenetic drugs, to provide molecular support to target the interplay between metabolism and epigenetics, two hallmarks of cancer onset and progression.
1. Cholangiocarcinoma: From Classification to Treatment Strategies
Cholangiocarcinoma (CCA) is a heterogeneous group of hepatobiliary malignancy that originates from biliary epithelium, at any portion of the tree, and shows features of cholangiocyte differentiation [1]. Cholangiocarcinoma represents almost 3% of all gastrointestinal tumors and the global CCA incidence rate shows geographic variation, probably as a result of differences in genetic characteristics and/or exposure to risk factors of the world’s populations [2]. Intriguingly, Eastern countries, particularly the northeast of Thailand, exhibit higher age-standardized incidence rates (ASIRs) than in the West (Europe, United States and Australia), where the incidence of this disease is <6 per 100,000 cases [2], so much so that CCA is considered a rare cancer. Generally, a slightly smaller incidence and mortality is observed in women compared to men (the male-to-female ratio is 1:1.2–1.5) [3,4,5].
On the basis of their anatomical location, CCAs can be classified into three clinically distinct types of cancers: intrahepatic (iCCA), perihilar (pCCA), and distal (dCCA) cholangiocarcinoma (Figure 1).
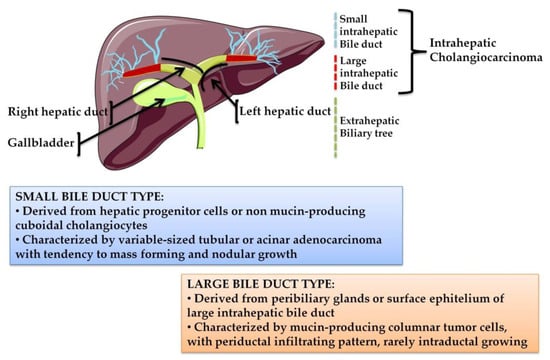
Figure 1.
The biliary tree, based on anatomical classification, is subdivided into intrahepatic and extrahepatic portions. The extrahepatic tract comprises the right and left hepatic ducts, from which arises a perihilar CCA (pCCA), the common bile duct that accounts for distal CCA (dCCA), and the gallbladder. The intrahepatic counterpart derives from the second-order bile ducts. The sub-classification of intrahepatic CCA (iCCA) is based on the size of the duct from which the tumor originates: The small bile duct type arises from the interlobular and septal ducts, while the large duct type arises from the segmental ducts.
iCCA arises in the second-degree bile ducts, specifically from segmental bile ducts to smaller branches of the intrahepatic part of biliary tree. In contrast, pCCA and dCCA are cancers of the extrahepatic biliary tree because the first originates from the right and/or left hepatic duct and/or the common hepatic duct proximally to the cystic duct origin, while the second occurs below the insertion of the cystic duct into the common bile duct, but not including the ampulla Vater [1,6]. pCCA accounts approximately 50–60% of CCAs cases and together with dCCA (20–30%) represents 80–90% of all CCAs diagnosed in the United States; the remaining 10–20% is represented by iCCA, which is the less frequent subtype of CCA, but the second most common primary intrahepatic malignancy after hepatocellular carcinoma (HCC) [5]. Although pCCA and dCCA represent the majority of CCA cases, over the last two decades, iCCA has shown a progressive increase of incidence in the world, whereas the incidence rates of other CCA subtypes have decreased in the same period [2,5].
This change in epidemiological trends is transforming the iCCA subtype into a global health problem that warrants attention and investigations for several reasons: (i) Incidence and mortality rates have risen significantly since the end of the past century; (ii) the knowledge about molecular mechanisms underlying iCCA onset is lacking and incomplete, thus a series of questions remain unanswered; (iii) this CCA subtype displays the highest inter-tumor heterogeneity, making its diagnosis complex, subsequently affecting the prognosis and management of patients; (iv) no effective therapies are available—thus far, iCCA is recognized as an orphan-drug disease.
Unlike pCCAs and dCCAs, which are mucin-producing adenocarcinomas (conventional type) or papillary tumors, iCCAs are characterized by highly variable morphological aspects, distinguishing a mucin-producing adenocarcinoma (bile duct (mucinous) type iCCAs or large bile duct type iCCAs), which originates from cholangiocytes and peribiliary glands, and a mixed subtype (bile ductular (mixed)-type iCCAs or small bile duct type iCCAs), in which areas of adenocarcinoma coexist with areas of hepatocytic differentiation, suggesting it originated from hepatic progenitor cells [2,6]. iCCA is characterized by clinical aggressiveness, like all CCA subtypes, but unlike pCCA and dCCA, it is adversely affected by several difficulties in patient management, probably due to its histological heterogeneity which results in divergent clinicopathological features. Less than 10% of patients survive over five year after diagnosis, mostly due to late diagnosis and limited treatment options [4]. Minimal and unspecific symptoms appear in early and/or pre-invasive stages, when surgical resection could represent the best therapeutic option combined with chemotherapy or radiation [3]. As a consequence, the diagnosis frequently occurs in the advanced stage of the disease, due to the “silent” clinical character of the tumor. Curative surgery with complete resection and/or liver transplantation is the preferred treatment option for iCCA, but only approximately 35% of patients are eligible for these approaches [2,4] and, after surgery, over half of resected patients have recurrence of disease in few years (generally 1–2 years depending on CCA subtype) [7]. Capecitabine as adjuvant chemotherapy showed efficacy in iCCA patients after surgical resection, with overall survival (OS) increased to 51 months in the treatment arm compared with 36 months in the observation arm (BilCap study). The PRODIGE12 study demonstrated that the adjuvant chemotherapy with gemcitabine and oxaliplatin (GEMOX), initiated three months after resection of biliary tract cancer, is ineffective in improving recurrence-free survival of CCA patients compared to placebo [8]. Other valid options for patients with localized unresectable CCAs are locoregional therapies, based on the focalized delivery of chemotherapy and radiotherapy. Techniques such as transarterial chemoembolization (TACE) and transarterial radioembolization (TARE), hepatic arterial-based therapies (HAT), radiofrequency ablation and photodynamic therapy (PDT) can increase survival and improve local control in locally advanced-metastatic iCCAs [9], although recurrence rates remain high [3]. In the field of innovative potential cancer treatment strategies, the use of non-toxic functionalized nanostructures, such as gold nanoparticles (AuNPs) [10] or graphene oxide nanosheets (GOxNs) [11], represent promising alternative systems to deliver chemotherapeutic agents selectively to tumor cells, reducing the drug exposure of normal cells. The potential application of these nanosized materials in targeted drug delivery and controlled release applications could improve the outcome of aggressive and poorly responsive cancers like iCCA.
For inoperable patients with metastasis disease at diagnosis, the median OS ranges from 5 to 12 months [3] and systemic therapies are the only curative opportunity, even though they have limited effectiveness [8]. The standard chemotherapeutic regimen for advanced iCCA is based on a gemcitabine-cisplatin combination (CisGem), which increases survival by about three months compared to gemcitabine alone, achieving an OS of 11.7 months [12]. However, this cancer is highly chemoresistant and, at present, no second-line therapy with established benefits is available, even though several studies were carried out for this purpose [13,14,15,16,17]. Chemoresistance is the major cause of treatment failure in this setting and is often caused by the activation of several anti-apoptotic pathways [18,19,20,21], which may act alone or synergistically to sustain cancer cell escape from cytotoxic or cytostatic effects of anticancer agents. Certainly, a better knowledge and/or comprehension of complex mechanisms responsible for drug resistance may improve the management of iCCA patients, suggesting targeted therapy strategies to enhance the response to chemotherapy [22].
iCCA is a lethal disease with an extremely poor response to conventional anticancer therapies and this consideration highlights the urgency of precision medicine approaches to go beyond current protocols. The advent of molecular profiling of human cancers and the discovery of driver mutations offers the opportunity to develop novel anticancer agents for clinical use, designed to hit specific molecular targets. In the iCCA subtype, few but distinctive genetic aberrations have been characterized; in particular, the isocitrate dehydrogenase 1 (IDH1) mutation, which is the most frequent, may represent the starting point for innovative individualized therapies. Indeed, several inhibitors of IDH1 enzyme are available and they have already been tested in other malignancies with promising results [23]. In the scenario of personalized treatment, this review discusses the opportunity of using IDH1 inhibitors as novel therapeutic options for IDH1-mutated iCCA patients and speculates on the advantage of their use in treatment as well as the potential of combined treatments for more successful approaches.
2. Molecular Profile of iCCA
The characterization of iCCA’s biological traits represents a milestone to better understand the complex framework of molecular alterations and improves the limits of the clinic-pathological classification. While some other human cancers benefit from targeted therapies that are widely accessible in clinical practice, at present no oncogene addiction has been defined for iCCA [24]. A plethora of papers has been published with the aim of identifying a genetic map, which could explain the mechanisms underlying iCCA’s pathogenesis, including rare and common genetic variants, chromosomal aberrations, and alterations of epigenomic and transcriptomic profiling [25]. Regarding genetic variants, the most prevalent in iCCA are KRAS, BRAF, IDH1/2, EGFR, PTPN3, PIK3CA, and loss of function TP53 mutations. While KRAS, TP53, EGFR, and PIK3CA mutations are common in all CCAs, BRAF, IDH1/2, and PTPN3 mutations prevail in iCCA [2]. Evidence of chromosomal aberrations, which results in both the loss and gain of gene copy numbers at different chromosomal loci, has been widely reviewed [24], but the best characterized is the FGFR2 translocation and its related fusion proteins because of its potential amenability to targeted therapy [26]. The portion of the FGF receptor involved in translocation is the same in all CCA variants, determining constitutive activation of the downstream signaling pathway [27], but the fusion protein is almost exclusive of iCCA because these rearrangements are approximately absent in pCCA/dCCA and rare in HCC [27].
Investigating iCCA epigenomic profiling, several gene promoters have been found hypermethylated. Although it is not just a peculiarity of this cancer type, genes interested by epigenetic silencing are tumor suppressors involved in cell cycle control (e.g., P16INK4A, P14ARF and RASSF1A), regulation of cell adhesion/attachment and signaling (e.g., CDH1, APC and SOCS3), redox homeostasis (e.g., GSTP), and regulation of DNA damage (e.g., MLH-1) [28]. In addition, several chromatin remodeling factors, such as ARID1A, BAP1 and PBRM1, have been found with inactivating mutations [24], contributing to iCCA epigenetic rewiring.
Transcriptomic profiling allowed the sub-classification of iCCAs into two distinct molecular subclasses. The inflammatory subtype is enriched in IL-10/-6 and STAT3 pathway, while the “proliferation” subtype is characterized by the constitutive activation of RAS and MET signaling cascade [29]. Finally, emerging pathways are NOTCH signaling, which has a key role in the development of cholangiocyte [30], and WNT signaling, which achieves a central role in the development and progression of iCCA, as observed in other cancers [31]. Altogether, these alterations represent the current knowledge on both the genetic defects and pathways involved in the pathogenesis of iCCA and may provide putative candidates for targeted approaches.
IDH1 Mutations in Intrahepatic Cholangiocarcinoma
A limited number of cancer types (e.g., glioma, acute myeloid leukemia and chondrosarcoma) harbor frequent mutations in IDH1 and IDH2 genes, among which iCCA is one. The expression of mutated forms of these genes is mostly absent in p/dCCA and HCC [32], thus IDH1/2 mutations could potentially be considered not only a therapeutic target for iCCA, but also a putative biomarker useful from a diagnostic perspective. IDH1/2 mutations have been detected in 25% of iCCA cases. Even if some studies considered their combined frequencies [33], IDH1 is more frequently mutated respect to IDH2 and IDH1 gene mutations, which range from 4.5 to 55.6% in iCCA [32], while the IDH2 contribution is only partial, with a mutation rate that ranges from 2 to 6% [33]. Intriguingly, IDH1 and IDH2 mutations are mutually exclusive [32].
IDH1 harbors missense mutations confined predominantly to a single residue (e.g., R132) in the active site of the enzyme. Five mutations have been described (i.e., p.R132H, p.R132C, p.R132G, p.R132S, and p.R132L) in IDH1-mutated cancers, but R132C is the most frequent in iCCA [32]. Among IDH1/2-mutated iCCA, the majority of cancers are poorly differentiated [34,35,36].
Considering the subset of IDH1-mutated iCCAs, a discrepancy was observed in the IDH1 hotspot mutation frequency in two iCCA subtypes, small bile duct type and large bile duct type [6,37]. Hayashi et al. reported that only the small bile duct subtype harbors IDH1/2 mutations and FGFR2 translocation with an incidence of 21/53 and 6/53, respectively [38]. One small bile duct subtype case displayed co-occurring mutations in KRAS and IDH1 genes [38]. Liau et al. obtained similar results observing a higher IDH1/2 mutation rate (13/77) in cholangiolar-type, corresponding to the small bile duct iCCA, compared to the bile duct type, corresponding to the large bile duct iCCA [39]. In contrast, Akita et al. observed that the frequency of the KRAS mutation between the two groups of iCCA was not significantly different, but IDH1 mutations remained specific for peripheral iCCA [40], and no mutation in IDH2 was found in that cohort [40,41]. A similar stratification of iCCA subtypes based on mutational profiling was reported by Akita et al., who observed that SMAD4 and MDM2 mutations were restricted to pCCA and large duct type iCCA, whereas BAP1 and IDH1 were more specific to the small duct type iCCA [42], associated with lower CA19-9 levels [34,42]. Nevertheless, the clinical significance of lower level of CA19-9 in patients with IDH mutations needs to be clarified. No significant association of IDH1/2 mutation with TNM stage, age or gender was reported in iCCA [34,35,36,43].
The mutation prevalence of IDH1/2 in iCCA is mostly variable depending on the size and geographical location of the object of the studies [32]. The prognostic value of IDH1 is still debated due to discordant results observed in different cohorts [32]. These discrepancies could depend on the distribution of risk factors in heterogeneous cohorts investigated in these studies. Indeed, the risk factors associated with iCCA, such as liver fluke infection and chronic viral hepatitis (e.g., HBV), are more prevalent in Asian countries than the West [44]. Jusakul et al. reported different genomic profiles among fluke-negative and positive iCCA, with the negatives characterized by BAP1, IDH1/2 mutations, FGFR alterations and up-regulated PI3K signatures [45]. Chan-on et al. reported that IDH1/2 genes were more commonly mutated in non-Opisthorchis viverrini flukes iCCA (Singapore cohort with 22.2% of cases) compared to O. viverrini iCCA (Thailand cohort with 3.2% of cases) [46]. Another study demonstrated a higher IDH1/2 mutation prevalence in patients without hepatitis compared to those with hepatitis (20% vs. 2%, respectively) [47].
The advent of new technologies and high-throughput sequencing enabled genome mapping of iCCA patients, leading to the identification of a clear molecular iCCA subtype harboring IDH1 mutations, with distinctive biological features. It also revealed a complex and highly variable genomic scenario in iCCA, which makes it difficult to define an unambiguous signature for this cancer type. Indeed, Lowery et al. described a tendency towards mutual exclusivity between genetic alterations such as TP53:IDH1, IDH1:KRAS, and IDH1:FGFR2 [48], while the recent systematic literature review of Boscoe et al. showed that IDH1 mutations were associated with other mutations of characteristic genes frequently found in iCCA [32]. Beyond ARID1A (22%), BAP1 mutation or loss (13.3%); and PBRM1 (13.3%), which were the most common, PINK3A, CDKN2A, TP53, MAP2K1, SMAD4, KRAS, BRAF, and PTEN showed a frequency ranging from 7% to 1% [32]. However, two independent studies found that IDH1 and BAP1 mutations were mutually exclusive [45,49].
Any conclusion about the differences observed in the frequency of mutations, association with prognosis and co-occurring/mutually exclusive mutation profiling must be taken with caution, due to cohort heterogeneity, a lack of rigorous inclusion criteria, and sampling difficulties due to the intrinsic and extrinsic heterogeneity of iCCA. Future studies are need for a better characterization of morpho-molecular subtypes of iCCA, which can be realized only if the identification of anatomical subtypes and clinicopathological features of the cohorts are scrupulously conducted. However, the discovery of IDH1 mutations, even if restricted to an iCCA subtype, opens the way to use a precision therapeutic approach, potentially more effective than existent therapies, for the subclass of IDH1-harboring iCCAs.
3. Biological Impact of IDH1 Mutation in Cellular Processes and Its Contribution to Carcinogenesis
IDH1 is a multifaceted enzyme with a crucial role in cellular metabolism, epigenetic regulation, cellular redox homeostasis, and DNA repair. Specific hot-spot mutations influence its catalytic activity, promoting a gain-of-function of the mutant form with aberrant and uncontrollable consequences on normal cell activities. Branching out into different physiological functions in the cells, IDH1 mutations play a role in carcinogenesis processes, gaining value as novel targets for anticancer therapies (Figure 2).
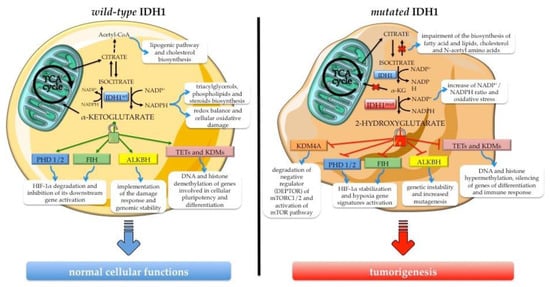
Figure 2.
Overview of IDH1’s biological role in normal cellular functions and the consequence of its neomorphic activity acquired upon hot-spot mutation. The wild-type enzyme (IDH1wt) ensures normal cellular processes, sustaining metabolic pathways and the activity of α-ketoglutarate-dependent dioxygenases such as epigenetic enzymes (e.g., ten eleven translocation hydroxylases (TETs) and Jumonji C domain-containing lysine demethylases—JmjC-KDMs or JHDMs), HIF-1α regulators (e.g., factor inhibiting HIF (FIH) and prolyl hydroxylase domain-containing protein (PHD1/2)), and the DNA repair enzyme alkB homolog (ALKBH). The gain-of-function mutant enzyme (IDH1mut) produces the oncometabolite 2-hydroxyglutarate (2HG), a potent inhibitor of α-ketoglutarate-dependent dioxygenases, with concomitant depletion of the NADPH pool. The consequences of 2HG accumulation are the metabolic and epigenetic reprogramming and the aberrant activation of signaling pathways sustaining cancer onset and progression.
IDH1 is located in both the cytoplasm and peroxisomes, and catalyzes the reaction that leads to α-ketoglutarate (α-KG) production starting from oxidative decarboxylation of isocitrate (ICT). The reaction is reversible and dependent on nicotinamide adenine dinucleotide phosphate (NADP+), Mg2+ or Mn2+. IDH1 acts as a homodimer and each monomer is characterized by three domains: The large domain (residues 1–103 and 286–414) characterized by a Rossmann fold; the small domain (residues 104–136 and 186–285) composed of an α/β sandwich structure; and the claps domain (residues 137–185) with two two-stranded and anti-parallel β-sheets [50]. IDH1 has three biological conformational states, substantially differing in the structure of the active site. The regulation, from the transition active/closed conformation to the inactive/opened structure, is independent of post-transduction events; instead, a substrate-dependent self-regulatory mechanism controls this switch [50].
Regarding IDH1 somatic mutations, no truncation or frameshift mutants have been reported and all mutations are heterozygous [51]. The mutations are confined to the catalytic site, especially at residue Arg132 (R132) level, leading to a gain of function of the enzyme [52]. This residue is responsible for forming hydrogen bonds with the α-carboxyl and β-carboxyl groups of isocitrate and mediates IDH-isocitrate binding [50]. The acquired mutation decreases the affinity for ICT with the concomitant increase of the binding affinity for NADPH. Thus, the IDH1 mutant (IDH1mut) active site is shifted to reduce α-KG to d-2-hydroxyglutarate (d-2HG), acquiring a new catalytic function [52].
Although IDH1 mutations with loss-of-function have been reported, all mutations harboring neomorphic activity produce the (D) isomer of 2HG [53,54]. Dang et al. proposed a model in which the wild-type/mutated IDH1 (IDH1wt/IDH1mut) heterodimer catalyzes two reactions simultaneously: Conversion of ICT to α-KG reducing NADP+ by the wild-type monomer and conversion of α-KG to d-2HG with NADPH oxidation by the mutant monomer catalysis [52]. It has been demonstrated that wild-type IDH1 (IDH1wt) gliomas are characterized by lower levels of d-2HG compared to heterozygous IDH1mut tumors depending on the loss of the wild-type allele [55]. However, another study showed that the production of d-2HG is increased in IDH1mut tumors if IDH1wt is co-expressed [56]. Additionally, there is evidence that d-2HG concentration may vary among different IDH1 mutations [57,58]. R132G exhibits the highest levels followed by R132C and R132H [57,58], while R132S/L catalyze the conversion of α-KG to d-2HG at a rate similar to R132C/H [57].
2HG is an intermediary metabolite referred to as an oncometabolite for its oncogenic intracellular signaling functions [59] and because of its involvement in reprogramming of several cellular processes, such as gene expression and cellular metabolism. In nature, it exists as two enantiomers—S-2-HG (or l-2HG) and R-2-HG (or d-2HG)—generally produced at low concentrations in healthy mammalian cells through the activity of two FAD-dependent mitochondrial enzymes—d- and l-2-hydroxyglutarate dehydrogenase (D2HGDH and L2HGDH)—that convert the corresponding enantiomer into α-KG [60,61]. Several studies demonstrated that the aberrant accumulation of 2-HG within cells caused by neomorphic IDH1mut activity or IDH1wt transcriptional deregulation contributes to the onset of cancer [62].
3.1. The Role of Wild-Type IDH1 in Cellular Processes
The direction of IDH1-mediated reaction depends on physiological and/or environmental conditions and changes both the intracellular level of αKG and the NADP+/NADPH ratio, with remarkable consequences on a plethora of cellular pathways.
αKG is a metabolite with pleiotropic activity at the crossroad of a wide range of physiological activities beyond cellular metabolism [63]. It is an obligatory cofactor of numerous 2-oxoglutarate-dependent dioxygenases (2-OGDOs), a superfamily of phylogenetically conserved enzymes containing mononuclear non-heme iron sites that catalyzes hydroxylation and demethylation of proteins, which provide different biological functions [64]. In particular, some epigenetic effectors, such as the ten eleven translocation hydroxylases (TETs), enzymes promoting DNA demethylation, and the Jumonji C domain-containing lysine demethylases (JmjC-KDMs or JHDMs), acting on lysine residues of histone proteins, are members of 2-OGDO family and their activity is αKG-dependent. Thus, αKG availability influences and regulates epigenetic processes with consequences for the expression of some regulatory genes involved in cellular pluripotency and differentiation [65,66,67], as well as in oncogenic signaling correlated with stemness [68].
Other interesting members of 2-OGDOs are the prolyl hydroxylases (PHDs) and the factor inhibiting HIF (FIH, also known as HIF1AN), an asparaginyl hydroxylase. PHDs are involved in hypoxia inducible factors α-subunit paralog 1 (HIF-1α) destabilization by hydroxylation of conserved proline residues (Pro402 and Pro564), which triggers pVHL-mediated ubiquitination and the consequent proteasomal degradation of HIF-1α subunit [69]. Instead, hydroxylation of HIF-1α by FIH blocks its association with the transcriptional co-activators CREB-binding protein (CBP) and p300, preventing the activation of downstream genes [70,71]. Even modest variations of intracellular αKG abundance influence PHDs and FIH activity, leading to altered HIF-1α stabilization associated with profound changes in cells metabolism [72].
αKG is interwoven with DNA repair mechanisms due to the involvement of αKG-dependent dioxygenases in this cellular process. Indeed, the DNA repair enzyme alkB homolog (ALKBH) [73,74] and the DNA damage response proteins lysine-specific demethylase 4A/B (KDM4A/B) [75] require this metabolite to carry out their activity and implement damage response. Variability in cellular αKG pool, derived by glutamine catabolism, alters the steady-state DNA repair system with consequent genomic instability [76]. Other cellular processes for which αKG supply is important are fatty acid homeostasis [62] and the translational machinery [77], due to the involvement of some 2-OGDO enzymes [62,77].
Under hypoxia or mitochondrial metabolic impairment, IDH1 activity becomes fundamental since cytosolic αKG is reductively carboxylate to generate citric acid and acetyl-CoA by the reversible IDH1-mediated reaction, to refill the lipogenesis pathway and the cholesterol biosynthesis of their precursors [78,79,80,81]. Beyond αKG, IDH1 catalysis is the main source of non-mitochondrial NADPH, a crucial element of cell redox balance. This co-factor is a key reducing agent required for detoxification processes through the reduction of glutathione and thioredoxins, and the activation of catalase tetramer and cytochrome P450, which are together involved in cellular protection against reactive oxygen species [75]. In addition, as an electrons donor, NADPH is essential for the biosynthesis of triacylglycerols, phospholipids, steroids (e.g., cholesterol and steroid hormones), amino acids (glutamate and proline) and deoxyribonucleotides [82], in this way supporting the DNA damage repair system.
In this scenario, it is evident that IDH1 acts at the intersection of different cellular processes and its dysregulation, in terms of both expression and activity, may be the starting point for the carcinogenetic process.
3.2. Biological Effects of Neomorphic Activity of Mutated IDH1
In the last decade, the attention on hot-spot mutations of IDH1 has escalated because IDH1mut produces d-2HG isomer that, despite being less potent than l-2HG in inhibiting some α-KG-dependent dioxygenases [83,84], has a notable impact on epigenetic reprogramming of IDH1 mutated cells, due to the competitive block of TETs and JmjC-KDMs activity [83]. IDH1mut cancers exhibit an hypermethylated phenotype characterized by CpG island hypermethylation and higher global DNA and histone methylation, in particular an increase in marks such as H3K4me3, H3K9me3 and H3K27me3 [85]. The biological effect of high DNA methylation and histone modifications of H3K9me3 and H3K27me3, associated with repression of transcription, is the silencing of genes involved in differentiation [86,87] and immune response [88,89]. Concerning the increase of H3K4me3, an activation mark, it could support the aberrant expression of the gene involved in oncogenesis and stem-like features [90,91,92], triggering carcinogenesis. Moreover, although genome methylation is generally considered a reversible and dynamic process, Turcan et al. demonstrated that the reprogramming of epigenome and transcriptome, driven by IDH1mut, shows partial persistence in specific genome regions even when the expression of mutated IDH1 is suppressed [92]. This observation adds a new piece to the puzzle of IDH1mut commitment to gene expression deregulation, which spreads to the control of chromatin domain formation. Indeed, beyond gene silencing, the increase of DNA methylation induced by 2-HG accumulation causes a rearrangement of chromatin domains and structures, such as insulator binding sites and/or enhancer-associated chromatin marks, leading to aberrant expression of oncogenes [93]. d-2HG may also predispose IDH1mut cells to oncogenic transformation by direct inhibition of ALKBH 1 and 2, other α-KG-dependent dioxygenases involved in DNA repair pathways, and/or aberrant expression of DNA repair genes [94,95]. Although 2-HG produces genetic instability, which may contribute to cancer initiation by favoring mutagenesis, the accumulation of DNA damage could be an advantage for patients harboring IDH1mut because it increases their vulnerability to chemotherapeutics and radiotherapy [75,85].
The mechanism underlying IDH1 mutation and d-2HG tumorigenesis goes beyond DNA repair failure and epigenetic alterations. As a competitive inhibitor of 2-OGDO enzymes, d-2HG affects cellular signaling and pathways. Indeed, the oncometabolite inhibits two regulator enzymes of HIF-1 transcription factors, as mentioned above, even though conflicting observations have been reported. Some studies demonstrated a significant accumulation of HIF-1α [96] or β [97] in IDH1mut cell lines, while others obtained contrasting results when examining the effect of 2-HG accumulation on HIF1 induction/stabilization and/or hypoxia gene signatures activation in different tumor types and in vivo models [94]. Additionally, Tarhonskaya et al. demonstrated a d-2HG-enabled activation of PHD2, assaying its activity in vitro. In a reducing environment and at physiologically relevant concentrations of Fe (II/III), the apparent activation of PHD2 by d-2HG is caused by a non-enzymatic conversion of 2-HG to α-KG [98]. It is likely that these HIF-1 results reflect different mechanisms for its stabilization depending on the cell type or contest/environment, and this may explain discrepancies in the literature. Moreover, it was demonstrated that d-2HG may contribute to enhanced HIF1 signaling directly by the inhibition of FIH [70] and/or indirectly by fault in collagen protein maturation [99]. Inhibition of collagen-4-prolyl hydroxylase by d-2HG reduces endostatin production [83], potentially favoring the induction of the hypoxia pathway. Indeed, endostatin, a secreted anti-angiogenic peptide generated by proteolytic processing of collagen XVIII within the extracellular matrix, downregulates HIF1 signaling reducing HIF1A expression and simultaneously increases FIH mRNA levels [100]. Additionally, in IDH1mut cancers, collagen maturation defects were observed due to the 2HG-mediated hindrance to α-KG-dependent prolyl 4-hydroxylases 1, 2, and 3 (P4HA1/2/3), and procollagen-lysine 2-oxoglutarate 5-dioxygenases 1, 2, and 3 (PLOD1/2/3) activity [99]. This group of collagen hydroxylases mediates the hydroxylation of proline and lysine residues of collagen proteins driving their correct folding. An impairment of hydroxylysine-mediated glycosylation results in instability and major solubility of different type IV collagen proteins, with consequent fragility of vessels’ basal membranes, which could favor tumor progression by allowing epithelial cell invasion and angiogenesis [101].
Besides HIF1 signaling, another important pathway regulated by IDH1mut is the mammalian/mechanistic target of rapamycin (mTOR), which is responsive to mitogenic signals and/or the availability of nutrients/cellular energy, and regulates cell growth, proliferation, autophagy, survival, and metabolism on the basis of signal inputs [102]. In various cell types with IDH1 mutations, an unscheduled activation of mTOR pathway was found due to d-2-HG inhibitor effect on α-KG-dependent dioxygenases. Indeed, d-2HG-mediated inhibition of the lysine demethylase KDM4A causes destabilization of DEPTOR protein, the endogenous negative regulator of mTORC1/2, promoting its degradation by the proteasome and leading to mTOR being turned on in a PTEN-independent manner [103]. The physiological consequences of this unconventional activation of mTOR cascade are numerous and include cell growth and metabolism [104].
The acquisition of IDH1 mutations results in substantial reprogramming of cellular metabolism. Reitman et al. demonstrated alterations in cellular concentration of several metabolites (e.g., some amino acids, glutathione metabolites, choline derivatives, and TCA cycle intermediates) for which the 2-HG production by IDH1R132H neomorphic activity was not the unique factor responsible [105], but α-KG and NADPH availability became crucial in the rewiring of the metabolic landscape of cells carrying the mutation.
One of the most important pieces of evidence is that IDH1mut is unable to catalyze the reductive carboxylation reaction and the glutaminolysis pathway becomes the pre-eminent source from which to generate α-KG for 2-HG in mutated cells [106]. Indeed, to compensate cytosolic α-KG depletion and increase d-2HG production, these cells redirect oxidative flux through the TCA cycle, increase respiration, and modulate the conversion of glutamine to citrate, acetyl-CoA, and fatty acids under various conditions, impairing the biosynthesis of fatty acid and lipids, cholesterol and N-acetyl amino acids [107]. In addition, the conversion reaction of α-KG to d-2HG consumes NADPH resulting in a change in the cellular NADP+/NADPH ratio that leads to an increase in oxidative stress by decreasing GSH pools [107]. The decrease of reducing equivalents may be compensated by transfers between the cytosolic and mitochondrial pyridine pools, so any alteration in redox homeostasis induced by low NADPH availability may also have implications for mitochondrial metabolism [106]. In addition, IDH1 mutations may indirectly interfere with mitochondrial respiration, because d-2HG is able to inhibit the succinate dehydrogenase (SDH) enzyme. SDH inactivation causes an increase in cellular succinate levels and respiration inhibition with the concomitant onset of cancerous metabolism and mitochondrial dysfunction, leading to apoptosis resistance [108].
Ronen et al. reported the reprogramming of pyruvate metabolism in genetically engineered IDH1mut cells via modulation of pyruvate dehydrogenase (PHD) [109], the enzyme that catalyzes the decarboxylation of pyruvate to acetyl CoA prior to entry into the TCA cycle. Via HIF1 signaling activation, IDH1mut cells are able to reduce PHD activity, upregulating the expression of PDK1 and PDK3, HIF1-regulated genes, which mediate the inhibitory phosphorylation of PHD. In this way, the flux of pyruvate into the TCA cycle is dropped; thus, the contribution of glucose to glutamate synthesis is reduced, as confirmed by the low levels of glutamate in mutant cells [105]. A decrease in glutamate is also obtained by 2HG-mediated inhibition of transaminases BCAT1 and BCAT2 in cells with IDH1 mutations [110]. Cells could utilize excess glucose as an alternative source for 2-HG production [109]. As a matter of fact, glioma tumors and cells harboring IDH1mut are characterized by lower intracellular lactate levels compared to IDH1wt [111,112], probably due to hypermethylation of the lactate dehydrogenase A (LDHA) gene promoter [113] driven by 2-HG increase.
The tangled mechanisms by which IDH1mut becomes able to interfere with normal cell functions remain unclear, but it is evident that the balance of intracellular levels of many factors, particularly d-2HG, αKG and NADPH, is crucial in contributing to cancer development and progression.
3.3. Biological Impact of IDH1 Mutation in Intrahepatic Cholangiocarcinoma
The biological defects of IDH1 mutations have been investigated predominantly in gliomas and glioblastomas. Nonetheless, some downstream effects of both neomorphic enzyme activity and d-2HG accumulation may be seen in other cancer types with similar consequences on cellular physiopathology.
As previously reported, iCCA is one of the cancer types that frequently harbors mutations in the IDH1 gene. The relatively small number of iCCA patients enrolled in studies and the difficulty for a correct discrimination between iCCA and other CCA subtypes has hindered the investigation of the biological role of IDH1mut in iCCA carcinogenesis, and results are often contradictory in terms of prognostic significance. However, one indisputable observation is that iCCA patients with IDH1mut are characterized by significantly higher levels of d-2HG, compared to wild-type tumors [114,115], and this may lead to alterations of normal cellular functions.
In 2013, Wang et al. demonstrated that IDH1mut/IDH2mut iCCAs show a hypermethylated phenotype, at both the DNA and histone level, and half the hypermethylated genes found in iCCA overlap with epigenetically silenced genes identified in glioblastomas [116]. They speculated that some methylated genes could be involved in hepatocyte and cholangiocyte differentiation due to the higher levels of hepatic stem cell lineage markers in IDH1mut/IDH2mut patients’ samples [116]. One year later, Saha et al. reported that an increase of 2-HG in hepatoblasts, expressing IDH1R123C form, epigenetically controls the expression of HNF-4α, a master regulator of hepatocyte lineage progression [117]. They demonstrated the involvement of d-2HG in the inhibition of hepatocellular differentiation and uncontrolled proliferation of liver progenitor cells, with cooperative function of activated KRas, establishing that IDH1mut may represent an early event in iCCA carcinogenesis, as observed in glioblastoma and acute myeloid leukemia (AML). During iCCA transcriptome reprogramming driven by IDH1/2 mutations, an increase in levels of p53 protein was observed as a consequence of cellular stress induced by the elevated mutation rate; HIF-1α levels also increased, and this was caused by the 2-HG inhibitory effect on hypoxia regulator enzymes [116].
Recently, an additional role of IDH1 mutations in CCA progression was established. Starting with a meta-analysis, Zhang et al. observed that the P2RX7 gene is epigenetically regulated by IDH1R132C mutation in CCA samples, demonstrating its involvement in cancer progression by affecting exosomes release from tumor cells [118].
Beyond DNA/histone methylome dysregulation, IDH1mut is strictly associated with metabolic reprogramming in iCCA patients. Analyzing genomic (whole-exome sequencing, targeted exome sequencing) and epigenomic data from 496 iCCA patients, perturbation of purine, glutathione and citric acid pathways was revealed, suggesting an oncogenic function of IDH1mut, which implies cellular metabolism [119]. By an integrated analysis of CCA samples extracted by The Cancer Genome Atlas (TCGA), another research group identified a class of IDH mutant-enriched clusters with high expression of mitochondrial genes, including components of the electron transport chain and citric acid cycle, low expression of chromatin modifiers, in particular epigenetic silencing of ARID1A, and relatively high mitochondrial DNA copy numbers [120].
Additionally, an unexpected control of IDH1mut on glycolytic flux has been observed in intrahepatic biliary organoids. The epigenetic remodeling prompted by oncogenic activity of the mutated enzyme causes an increase in H3K4me3 levels on the promoter region of PFKP gene with its consequent active transcription. The up-regulation of the rate-limiting glycolytic enzyme PFKP enhances glycolysis, promoting the formation of biliary organoids and conferring resistance to stress [121]. These results were confirmed by analyzing the expression of PFKP in surgically resected iCCA specimens: Elevated levels of enzyme were found in 68% of cases and the majority carried out IDH1mut [121].
It is unclear whether hot-spot IDH1 mutations are sufficient to support the initiation of the carcinogenetic process in iCCA or whether it needs to occur in combination with other genetic events, such as the activation of the Notch pathway and loss of p53 tumor suppressor [122]. Due to the rarity of these tumors, until now knowledge regarding the role of IDH1mut in iCCA biology is limited and almost confined to epigenetic rewiring driven by 2-HG accumulation.
4. IDH1 Inhibitors for Intrahepatic Cholangiocarcinoma Treatment
The discovery of gain-of-function mutations in the IDH1 enzyme has revolutionized pharmaceutical approaches, making targeted therapies against mutated enzymatic forms a productive research field. Since mutations in IDH1 occur in a broad spectrum of cancer types, both solid and hematologic, over recent years research has focused on the development of small synthetic molecules capable of inhibiting the aberrant activity of IDH1mut.
Many inhibitors are active against common IDH1 mutations (e.g., R132H, R132C, R132G, R132S, and R132L), but few are specific for the IDH1mut isoform (e.g., R140Q or R132C), and only one compound is a pan-inhibitor capable of blocking both IDH1 and IDH2 mutants. The majority of these small molecules acts as allosteric inhibitors, occupying the catalytic pocket of the enzyme in the open conformation, thus promoting structural changes that result in enzyme inactivity. However, other compounds bind specific amino acid residues, blocking the NADPH binding or chelating Mg2+/Mn2+ essentials for catalysis and for adopting the catalytically competent conformation [23].
Numerous pre-clinical and clinical investigations were conducted, particularly in AML and glioblastoma, and all of them demonstrated the positive effect of IDH1mut inhibition on reversing epigenetic changes as a consequence of significant reductions of d-2HG levels. The biological effect of enzymatic inhibition included enhanced progenitor cell differentiation, decreased stem cell marker expression and a reduction of proliferation in both in vitro and derivative tumor models [62]. The successes obtained in clinical trials, in terms of both safety and efficacy, helped obtain FDA approval in 2018 of the first IDH1 inhibitor—AG-120 (Ivosidenib), used to treat for refractory/relapsed AML adult patients harboring susceptible IDH1 mutations [123].
Due to both the promising results obtained by IDH1 inhibitors in clinical trials in other malignancies and the limitations in current treatment options for metastatic iCCA, IDH1mut and its associated molecular pathways have become attractive therapeutic targets in the management of these patients. Four IDH1mut-inhibitors (e.g., pan-IDH1mut AG-120 and BAY1436032; specific-IDH1mut FT-2102 and IDH305) are under investigation for iCCA patients in six active clinical trials (Table 1). These molecules are chemically and pharmacodynamically different, although all of them are active against the same IDH1 mutant isoform.

Table 1.
IDH1 inhibitors under clinical investigation in metastatic iCCA.
AG-120 (Tibsovo®, Agios Pharmaceuticals (Cambridge, MA, USA) is an oral allosteric reversible inhibitor active against various IDH1R132 mutants with comparable potency [124]. AG-120 is the result of a chemical structure optimization of the first IDH1-inhibitor AG-5198, a phenyl-glycine based compound with a strong inhibitory effect on the IDH1 enzyme but poor metabolic stability. With the substitution of specific functional groups, AG-120 was obtained, a molecule characterized by high polarity and solubility; good stability in human liver microsomes; low human pregnane X receptor (PXR) activation, which regulates genes involved in drug metabolism and efflux; good permeability; and low efflux ratio [124]. At the dimer interface, the inhibitor competes with high specificity for binding with Mg2+ co-factor, a catalytically essential divalent ion, preventing the enzyme from achieving the catalytically competent conformation [129]. AG-120 also demonstrated slow-tight binding inhibition against the IDH1wt homodimer [124] (Table 1), offering a new potential application in IDH1-overexpressing cancers.
At present, three clinical trials (NCT02073994, NCT02989857, NCT04088188) investigating AG-120 in CCA are underway. NCT02073994 is a Phase I multicenter open-label, dose escalation and expansion study, which enrolled patients with advanced solid tumors including CCA, 43.5% of the solid tumor cohort. The study has been conducted in order to assess the safety, tolerability, pharmacokinetics, pharmacodynamics and clinical activity of AG-120 as a single agent for treatment of patients harboring an IDH1 mutation. Ivosidenib demonstrated rapid oral absorption and a long half-life, due to a slow elimination rate. After multiple doses at the optimum regimen of 500 mg administrated once daily (QD), patients with CCA showed plasma 2-HG levels decrease by up to 98%, a concentration similar to that seen in healthy subjects, and the inhibition effect was persistent over the whole treatment period [130]. In the CCA subgroup, AG-120 demonstrated good tolerability and no dose-limiting toxicities. Patients treated with ivosidenib at the regimen of 500 mg QD showed a median progression-free survival (PFS) of 3.8 months and a median OS of 13.8 months. Stable disease was noted in 56% of patients, which is clinically relevant considering that this result is comparable to the proportion of patients with stable disease receiving gemcitabine-cisplatin chemotherapy [131]. One limit of this study is represented by its non-randomized design, although Agios Pharmaceuticals sponsored a global Phase III randomized, double-blind, placebo-controlled study known as ClarIDHy (NCT02989857). This is intended to determine the efficacy and safety of orally administered Tibsovo® (AG-120) in patients with previously treated IDH1-mutant CCA after progression on standard chemotherapy. The first results of this Phase III trial are encouraging because a significant increase of PFS for the ivosidenib group compared with the placebo group was observed. Under a regimen of 500 mg QD, patients treated with IDH1 inhibitor showed a median PFS of 2.7 months compared to 1.4 months for the placebo group. Although this could appear a modest improvement in terms of absolute values, the results are statistically robust and indicate a notable reduction in risk of disease progression, independently of the number of previous lines of therapy. In addition, a difference of 4.8 months in median OS was observed between ivosidenib and the placebo group [132].
An independent non-randomized Phase I clinical trial was initiated in 2019 to assess the side effects and the optimal dose for treatment of unresectable and/or metastatic CCA patients harboring IDH1 mutations, with gemcitabine and cisplatin in combination with AG-120 (NCT04088188). The study is still in its initial phase and is not yet recruiting.
IDH305 is a selective IDH1R132H/C inhibitor (Table 1) developed by Novartis Pharmaceuticals (Basel, Switzerland). This pyrimidin-5-yl-oxazolidine-2-one acts as allosteric non-competitive inhibitor occupying the binding pocket of the enzyme and stabilizing IDH1 mutant in a catalytically inactive open conformation via steric hindrance. In pre-clinical studies, this inhibitor showed an interesting brain penetration compared to other IDH1-inibiting compounds, low liver microsomal clearance values and comparable 2-HG reduction efficacy with respect to other inhibitors [125]. In 2015, a Phase I non-randomized clinical trial (NCT02381886) was started to estimate the safety and tolerability of IDH305, to assess the pharmacodynamics and plasma pharmacokinetic of the compound and evaluate the overall response rate in order to test the anti-tumor activity of this inhibitor. Results are not yet available but 166 patients with advanced malignancies harboring IDH1R132 mutations, including iCCA, have been enrolled.
Very few pre-clinical studies are available regarding FT-2102 (olutasidenib), a potent, orally active and brain penetrant inhibitor of IDH1mut (Table 1), the development of which was supported by Forma Therapeutics (Watertown, MA, United States). FT-2102 is an optimized quinoline with potent and selective inhibitory activity against IDH1 mutants and a good oral bioavailability. This molecule efficiently reduces 2-HG production in xenograft IDH1R132H in vivo models, binding competitively with the isocitrate-binding pocket of each monomer of IDH1mut, near the dimer interface, and blocking the conformational changes fundamental for catalysis. Moreover, FT-2102 showed good cell permeability and microsomal stability as well as low efflux ratio in xenograft models [126]. At present, FT-2102 is undergoing clinical investigation for advanced solid tumors and gliomas (NCT03684811). The study was initiated to assess the safety, efficacy, pharmacokinetic and pharmacodynamic of FT-2102 as a single agent and in combination with other anti-cancer drugs, among them the DNA-demethylating agent azacytidine. As a consequence, the clinical trial is planned to be structured in two parts: iCCA patients will be enrolled for a Phase I/II trial for dose determination and clinical activity will take place in the single-agent arm and/or in the gemcitabine/cisplatin combination arm.
BAY-1436032 is a potent pan-inhibitor equally active against all mutant isoforms of IDH1 (Table 1), the development of which has been sponsored by Bayer AG (Leverkusen, Germany). Development was conducted upon screening of over 3 million of compounds. It performs its inhibitory effect in an allosteric manner, binding the pocket located between the two IDH1mut molecules of the homodimeric IDH1R132 complex, and stabilizing the inactive open conformation [127]. The reduced 2-HG production induced by BAY 1,436,032 has a positive effect on proliferation and differentiation in primary glioma cultures [127] and in patient-derived AML mouse xenografts harboring IDH1 mutations [128]. In 2016, an open-label, non-randomized, multicenter Phase I study (NCT02746081) began to evaluate the tolerability and safety of BAY1436032, and to investigate the pharmacokinetics and preliminary pharmacodynamic of this inhibitor in patients with IDH1R132-mutant advanced solid tumors, including IDH1-mutated iCCAs. The cohort of enrolled patients is still limited (88 patients); thus, preliminary results are lacking.
Two independent clinical trials are also being conducted in the European Union and have been registered in the EU Clinical Trials Register. One is a Phase 1b/2 study (EudraCT Number: 2018-001796-21) designed for the assessment of FT-2102 in a heterogeneous group of patients with advanced solid tumors, including iCCA, characterized by IDH1 mutations. The second is focused on CCA; indeed, it is a Phase 3, multicenter, randomized trial (EudraCT Number: 2015-005117-72) activated to investigate the efficacy of AG-120 in the treatment of non-resectable or metastatic CCAs harboring IDH1mut.
5. Future Perspectives of IDH1 Inhibitors Use in Intrahepatic Cholangiocarcinoma
The status of “rare cancer” and the difficulties for clinicians to obtain a correct molecular diagnosis for iCCA patients have contributed to the lack of adequate treatments for this type of aggressive tumor. In addition, conventional therapies (e.g., chemotherapy and radiotherapy) produced unsatisfying results, particularly in the adjuvant and second-line settings, supporting an immediate demand of effective alternative treatment options.
The advent of NGS technologies suggested that iCCA represents a subgroup of CCAs with discrete driver mutations, some of which are targetable with novel therapies. In particular, IDH1 mutations have aroused increasing interest in the field of targeted therapies because of their involvement in both metabolic and epigenetic rewiring, two hallmarks of cancer interwoven with tumor initiation and progression [133]. Although the prognostic significance of IDH1mut in iCCA is still debated [32,34], the promising results obtained by clinical trials are indisputable [132]. However, this field of investigation is still in its early phase and several issues are still debated. An unsolved open question is the potential contribute of IDH1mut in favoring sensitivity to agents targeting specific pathways involved in cancer growth and progression. Indeed, in vitro studies revealed that iCCA cell lines harboring different hot-spot mutations IDH1R132 are more sensitive to dasatanib [134], a multi-tyrosine kinase inhibitor, JQ1 [135], a selective inhibitor of bromodomain and extraterminal domain (BET) proteins, and olaparib [136], a poly(adenosine 5′-diphosphate) ribose polymerase (PARP) inhibitor, when compared to wild-type lines. However, none of these studies looked deeply into the characterization of molecular mechanisms underlying the anti-tumor effects of these drugs on IDH1mut iCCA cells, limiting their translational application. In fact, the phase II trial (NCT02428855), activated in 2015 to investigate the safety and effectiveness of dasatanib in CCA treatment, was closed because it was not approved by the FDA for this specific purpose.
Despite significant preclinical and clinical successes obtained by IDH1mut inhibitors as single agents for the management of cancers with IDH1 mutations, there is an opportunity to improve their efficacy. It was demonstrated that the global DNA hypermethylated pattern, driven by IDH1 neomorphic activity, may not be rapidly and completely reversed upon single-agent inhibition of the mutated enzyme and/or depletion of 2-HG accumulation [92,137]. Consequently, it may be worthwhile to investigate combination therapies with IDH1 inhibitors and demethylating and/or histone modifying agents in order to improve the beneficial effect of an IDH1mut targeted monotherapy. In such a context, the NCT03684811 trial has already included a cohort of FT-2102 in combination with azacytidine but enrolling only glioma and chondrosarcoma patients. However, the encouraging utility of combining epigenetic drugs with small-molecule inhibitors of mutant IDH1 emerged by several trials active for AML and glioblastoma, and this may provide a robust rationale to extend this strategy in advanced and metastatic iCCA.
A fascinating unexplored research field is the potential oncogenic role that IDH1wt overexpression may play in iCCA pathogenesis. Indeed, a recent study by Su et al. highlighted that IDH1wt (not the mutated form IDH1R132C) promotes cell proliferation, invasion and migration in iCCA cell lines by regulation of αKG and NADPH levels [138]. These results are not surprising because over recent years it has emerged that an aberrant expression of non-mutated IDH1 in numerous cancers (e.g., non-small cell lung carcinoma, squamous cell lung cancer, pancreatic ductal adenocarcinoma, primary glioblastoma) [139,140,141,142] correlates with therapy resistance, an aggressive phenotype and poor prognosis [141,143,144,145]. The biological consequences of IDH1 up-regulation are ascribable to the central role of this enzyme in the regulation of both metabolic and epigenetic processes. Recently, the IDH1wt enzyme has become an interesting actionable target for cancer therapy equal to the mutated isoforms. Some developed IDH1mut inhibitors, such as AG-120 and GSK321, are active against the wild-type enzyme at relatively low concentrations (IDH1wt IC50: 0.046 μM vs. IDH1mut IC50: 0.0046 μM for GSK321) [62], allowing their use for IDH1-overexpressing cancers as single agents or in combination with other anticancer therapies. To our knowledge, at present, investigations focused on IDH1wt expression in iCCA have been not yet started, even though IDH1wt-dependent regulation of the expression of EMT markers was reported in CCA [138].
In conclusion, it is evident that IDH1 enzyme is entangled involved in many cellular functions and is an interesting therapeutic option for orphan-drug cancer types such as iCCA. However, patients should be evaluated not only for the mutational profile of IDH1 enzyme, but also for its expression and activity, in order to improve the applicability and effectiveness of IDH1 inhibitors in advanced iCCA management.
Author Contributions
Conceptualization, M.L., F.E., A.S., M.P., V.C., F.C.; Writing—original draft preparation, F.C., M.P., V.C.; Writing—review and editing, F.C., M.P., V.C., F.M., G.B., A.P., A.S., F.E., M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by current research funds from the Italian Ministry of Health to IRCCS CROB.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Blechacz, B. Cholangiocarcinoma: Current knowledge and new developments. Gut Liver 2017, 11, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma-evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef]
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39, 19–31. [Google Scholar] [CrossRef]
- Kendall, T.; Verheij, J.; Gaudio, E.; Evert, M.; Guido, M.; Goeppert, B.; Carpino, G. Anatomical, histomorphological and molecular classification of cholangiocarcinoma. Liver Int. 2019, 39, 7–18. [Google Scholar] [CrossRef]
- Cillo, U.; Fondevila, C.; Donadon, M.; Gringeri, E.; Mocchegiani, F.; Schlitt, H.J.; Ijzermans, J.N.M.; Vivarelli, M.; Zieniewicz, K.; Olde, S.W.M.; et al. Surgery for cholangiocarcinoma. Liver Int. 2019, 39, 143–155. [Google Scholar] [CrossRef]
- Kelley, R.K.; Bridgewater, J.; Gores, G.J.; Zhu, A.X. Systemic therapies for intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 72, 353–363. [Google Scholar] [CrossRef]
- Cotton, J.; Lamarca, A.; McNamara, M.G.; Valle, J.W. Novel treatments for advanced cholangiocarcinoma. In Liver Cancers; Springer: New York, NY, USA, 2019; pp. 227–243. [Google Scholar]
- Stobiecka, M.; Hepel, M. Double-shell gold nanoparticle-based DNA-carriers with poly-l-lysine binding surface. Biomaterials 2011, 32, 3312–3321. [Google Scholar] [CrossRef]
- Stobiecka, M.; Dworakowska, B.; Jakiela, S.; Lukasiak, A.; Chalupa, A.; Zembrzycki, K. Sensing of survivin mRNA in malignant astrocytes using graphene oxide nanocarrier-supported oligonucleotide molecular beacons. Sens. Actuators B Chem. 2016, 235, 136–145. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Hubner, R.A.; David Ryder, W.; Valle, J.W. Second-line chemotherapy in advanced biliary cancer: A systematic review. Ann. Oncol. 2014, 25, 2328–2338. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.E.; Law, L.; Nguyen, V.D.; Qiao, W.; Javle, M.M.; Kaseb, A.; Shroff, R.T. Second-line systemic treatment for advanced cholangiocarcinoma. J. Gastrointest. Oncol. 2014, 5, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Fornaro, L.; Vivaldi, C.; Cereda, S.; Leone, F.; Aprile, G.; Lonardi, S.; Silvestris, N.; Santini, D.; Milella, M.; Caparello, C.; et al. Second-line chemotherapy in advanced biliary cancer progressed to first-line platinum-gemcitabine combination: A multicenter survey and pooled analysis with published data. J. Exp. Clin. Cancer Res. 2015, 34, 156. [Google Scholar] [CrossRef]
- Guion-Dusserre, J.F.; Lorgis, V.; Vincent, J.; Bengrine, L.; Ghiringhelli, F. FOLFIRI plus bevacizumab as a second-line therapy for metastatic intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2015, 21, 2096–2101. [Google Scholar] [CrossRef]
- Lowery, M.A.; Goff, L.W.; Keenan, B.P.; Jordan, E.; Wang, R.; Bocobo, A.G.; Chou, J.F.; O’Reilly, E.M.; Harding, J.J.; Kemeny, N.; et al. Second-line chemotherapy in advanced biliary cancers: A retrospective, multicenter analysis of outcomes. Cancer 2019, 125, 4426–4434. [Google Scholar] [CrossRef]
- Condelli, V.; Maddalena, F.; Sisinni, L.; Lettini, G.; Matassa, D.S.; Piscazzi, A.; Palladino, G.; Amoroso, M.R.; Esposito, F.; Landriscina, M. Targeting TRAP1 as a downstream effector of BRAF cytoprotective pathway: A novel strategy for human BRAF-driven colorectal carcinoma. Oncotarget 2015, 6, 22298–22309. [Google Scholar] [CrossRef]
- Spagnoletti, G.; Li Bergolis, V.; Piscazzi, A.; Giannelli, F.; Condelli, V.; Sisinni, L.; Bove, G.; Storto, G.; Landriscina, M. Cyclin-dependent kinase 1 targeting improves sensitivity to radiation in BRAF V600E colorectal carcinoma cells. Tumor Biol. 2018, 40. [Google Scholar] [CrossRef]
- Stobiecka, M.; Ratajczak, K.; Jakiela, S. Toward early cancer detection: Focus on biosensing systems and biosensors for an anti-apoptotic protein survivin and survivin mRNA. Biosens. Bioelectron. 2019, 137, 58–71. [Google Scholar] [CrossRef]
- Criscuolo, D.; Avolio, R.; Calice, G.; Laezza, C.; Paladino, S.; Navarra, G.; Maddalena, F.; Crispo, F.; Pagano, C.; Bifulco, M.; et al. Cholesterol homeostasis modulates platinum sensitivity in human ovarian cancer. Cells 2020, 9, 828. [Google Scholar] [CrossRef]
- Marin, J.J.G.; Lozano, E.; Herraez, E.; Asensio, M.; Di Giacomo, S.; Romero, M.R.; Briz, O.; Serrano, M.A.; Efferth, T.; Macias, R.I.R. Chemoresistance and chemosensitization in cholangiocarcinoma. Biochim. Biophys. Acta—Mol. Basis Dis. 2018, 1864, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant isocitrate dehydrogenase inhibitors as targeted cancer therapeutics. Front. Oncol. 2019, 9, 417. [Google Scholar] [CrossRef] [PubMed]
- Moeini, A.; Sia, D.; Bardeesy, N.; Mazzaferro, V.; Llovet, J.M. Molecular pathogenesis and targeted therapies for intrahepatic cholangiocarcinoma. Clin. Cancer Res. 2016, 22, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.B.; Thorgeirsson, S.S. Genetic profiling of intrahepatic cholangiocarcinoma. Curr. Opin. Gastroenterol. 2012, 28, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Borad, M.J.; Gores, G.J.; Roberts, L.R. Fibroblast growth factor receptor 2 fusions as a target for treating cholangiocarcinoma. Curr. Opin. Gastroenterol. 2015, 31, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef]
- Yang, B.; House, M.G.; Guo, M.; Herman, J.G.; Clark, D.P. Promoter methylation profiles of tumor suppressor genes in intrahepatic and extrahepatic cholangiocarcinoma. Mod. Pathol. 2005, 18, 412–420. [Google Scholar] [CrossRef][Green Version]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef]
- Zender, S.; Nickeleit, I.; Wuestefeld, T.; Sörensen, I.; Dauch, D.; Bozko, P.; El-Khatib, M.; Geffers, R.; Bektas, H.; Manns, M.P.; et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 2013, 23, 784–795. [Google Scholar] [CrossRef]
- Boulter, L.; Guest, R.V.; Kendall, T.J.; Wilson, D.H.; Wojtacha, D.; Robson, A.J.; Ridgway, R.A.; Samuel, K.; Van Rooijen, N.; Barry, S.T.; et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J. Clin. Investig. 2015, 125, 1269–1285. [Google Scholar] [CrossRef]
- Boscoe, A.N.; Rolland, C.; Kelley, R.K. Frequency and prognostic significance of isocitrate dehydrogenase 1 mutations in cholangiocarcinoma: A systematic literature review. J. Gastrointest. Oncol. 2019, 10, 751–765. [Google Scholar] [CrossRef] [PubMed]
- De Botton, S.; Mondesir, J.; Willekens, C.; Touat, M. IDH1 and IDH2 mutations as novel therapeutic targets: Current perspectives. J. Blood Med. 2016, 7, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Govindan, A.; Sheth, R.A.; Nardi, V.; Blaszkowsky, L.S.; Faris, J.E.; Clark, J.W.; Ryan, D.P.; Kwak, E.L.; Allen, J.N.; et al. Prognosis and clinicopathologic features of patients with advanced stage isocitrate dehydrogenase (IDH) mutant and IDH wild-type intrahepatic cholangiocarcinoma. Oncologist 2015, 20, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Kipp, B.R.; Voss, J.S.; Kerr, S.E.; Barr Fritcher, E.G.; Graham, R.P.; Zhang, L.; Edward Highsmith, W.; Zhang, J.; Roberts, L.R.; Gores, G.J.; et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum. Pathol. 2012, 43, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
- Aishima, S.; Oda, Y. Pathogenesis and classification of intrahepatic cholangiocarcinoma: Different characters of perihilar large duct type versus peripheral small duct type. J. Hepato Biliary Pancreat. Sci. 2015, 22, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Misumi, K.; Shibahara, J.; Arita, J.; Sakamoto, Y.; Hasegawa, K.; Kokudo, N.; Fukayama, M. Distinct clinicopathologic and genetic features of 2 histologic subtypes of intrahepatic cholangiocarcinoma. Am. J. Surg. Pathol. 2016, 40, 1021–1030. [Google Scholar] [CrossRef]
- Liau, J.Y.; Tsai, J.H.; Yuan, R.H.; Chang, C.N.; Lee, H.J.; Jeng, Y.M. Morphological subclassification of intrahepatic cholangiocarcinoma: Etiological, clinicopathological, and molecular features. Mod. Pathol. 2014, 27, 1163–1173. [Google Scholar] [CrossRef]
- Akita, M.; Fujikura, K.; Ajiki, T.; Fukumoto, T.; Otani, K.; Azuma, T.; Itoh, T.; Ku, Y.; Zen, Y. Dichotomy in intrahepatic cholangiocarcinomas based on histologic similarities to hilar cholangiocarcinomas. Mod. Pathol. 2017, 30, 986–997. [Google Scholar] [CrossRef]
- Zou, S.; Li, J.; Zhou, H.; Frech, C.; Jiang, X.; Chu, J.S.C.; Zhao, X.; Li, Y.; Li, Q.; Wang, H.; et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 2014, 5, 5696. [Google Scholar] [CrossRef]
- Akita, M.; Sofue, K.; Fujikura, K.; Otani, K.; Itoh, T.; Ajiki, T.; Fukumoto, T.; Zen, Y. Histological and molecular characterization of intrahepatic bile duct cancers suggests an expanded definition of perihilar cholangiocarcinoma. HPB 2019, 21, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Wang, K.; Gay, L.; Al-Rohil, R.; Rand, J.V.; Jones, D.M.; Lee, H.J.; Sheehan, C.E.; Otto, G.A.; Palmer, G.; et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 2014, 19, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Kirstein, M.M.; Vogel, A. Epidemiology and risk factors of cholangiocarcinoma. Visc. Med. 2016, 32, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef]
- Chan-On, W.; Nairismägi, M.L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef]
- Fujimoto, A.; Furuta, M.; Shiraishi, Y.; Gotoh, K.; Kawakami, Y.; Arihiro, K.; Nakamura, T.; Ueno, M.; Ariizumi, S.I.; Nguyen, H.H.; et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat. Commun. 2015, 6, 6120. [Google Scholar] [CrossRef]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: Potential targets for intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef]
- Simbolo, M.; Fassan, M.; Ruzzenente, A.; Mafficini, A.; Wood, L.D.; Corbo, V.; Melisi, D.; Malleo, G.; Vicentini, C.; Malpeli, G.; et al. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget 2014, 5, 2839–2852. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, J.; Xu, Z.; Peng, B.; Huang, Q.; Arnold, E.; Ding, J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 2004, 279, 33946–33957. [Google Scholar] [CrossRef]
- Zhao, S.; Guan, K.L. IDH1 mutant structures reveal a mechanism of dominant inhibition. Cell Res. 2010, 20, 1279–1281. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Cross, J.R.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.A.; Thompson, C.B. Identification of additional IDH mutations associated with oncometabolite R(−)-2-hydroxyglutarate production. Oncogene 2012, 31, 2491–2498. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Reitman, Z.J.; Duncan, C.G.; Spasojevic, I.; Gooden, D.M.; Rasheed, B.A.; Yang, R.; Lopez, G.Y.; He, Y.; McLendon, R.E.; et al. Disruption of wild-type IDH1 suppresses d-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2013, 73, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Lu, C.; Cross, J.R.; Abdel-Wahab, O.; Levine, R.L.; Schwartz, G.K.; Thompson, C.B. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J. Biol. Chem. 2013, 288, 3804–3815. [Google Scholar] [CrossRef]
- Matteo, D.A.; Grunseth, A.J.; Gonzalez, E.R.; Anselmo, S.L.; Kennedy, M.A.; Moman, P.; Scott, D.A.; Hoang, A.; Sohl, C.D. Molecular mechanisms of isocitrate dehydrogenase 1 (IDH1) mutations identified in tumors: The role of size and hydrophobicity at residue 132 on catalytic efficiency. J. Biol. Chem. 2017, 292, 7971–7983. [Google Scholar] [CrossRef]
- Pusch, S.; Schweizer, L.; Beck, A.C.; Lehmler, J.M.; Weissert, S.; Balss, J.; Miller, A.K.; von Deimling, A. d-2-Hydroxyglutarate producing neo-enzymatic activity inversely correlates with frequency of the type of isocitrate dehydrogenase 1 mutations found in glioma. Acta Neuropathol. Commun. 2014, 2, 19. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef]
- Rzem, R.; Veiga-Da-Cunha, M.; Noël, G.; Goffette, S.; Nassogne, M.C.; Tabarki, B.; Schöller, C.; Marquardt, T.; Vikkula, M.; Van Schaftingen, E. A gene encoding a putative FAD-dependent l-2-hydroxyglutarate dehydrogenase is mutated in l-2-hydroxyglutaric aciduria. Proc. Natl. Acad. Sci. USA 2004, 101, 16849–16854. [Google Scholar] [CrossRef]
- Achouri, Y.; Noël, G.; Vertommen, D.; Rider, M.H.; Veiga-Da-Cunha, M.; Van Schaftingen, E. Identification of a dehydrogenase acting on d-2-hydroxyglutarate. Biochem. J. 2004, 381, 35–42. [Google Scholar] [CrossRef]
- Tommasini-Ghelfi, S.; Murnan, K.; Kouri, F.M.; Mahajan, A.S.; May, J.L.; Stegh, A.H. Cancer-associated mutation and beyond: The emerging biology of isocitrate dehydrogenases in human disease. Sci. Adv. 2019, 5, eaaw4543. [Google Scholar] [CrossRef] [PubMed]
- Abla, H.; Sollazzo, M.; Gasparre, G.; Iommarini, L.; Porcelli, A.M. The multifaceted contribution of α-ketoglutarate to tumor progression: An opportunity to exploit? Semin. Cell Dev. Biol. 2020, 98, 26–33. [Google Scholar] [CrossRef] [PubMed]
- McDonough, M.A.; Loenarz, C.; Chowdhury, R.; Clifton, I.J.; Schofield, C.J. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr. Opin. Struct. Biol. 2010, 20, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Finley, L.W.S.; Cross, J.R.; Allis, C.D.; Craig, B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Reid, M.A.; Lowman, X.H.; Kulkarni, R.P.; Tran, T.Q.; Liu, X.; Yang, Y.; Hernandez-davies, J.E.; Rosales, K.K.; Li, H.; et al. Regional glutamine deficiency in tumors promotes de-differentiation through inhibition of histone demethylation. Nat. Cell Biol. 2016, 18, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Kwak, S.; Cho, E.; Youn, H.; Hwang, I.; Kwak, S.; Lee, S.; Kim, H.; Lee, S.E.; Kim, J.; et al. Psat1-dependent fluctuations in α-ketoglutarate affect the timing of ESC differentiation. Cell Metab. 2016, 24, 494–501. [Google Scholar] [CrossRef]
- Tran, T.Q.; Hanse, E.A.; Habowski, A.N.; Li, H.; Ishak Gabra, M.B.; Yang, Y.; Lowman, X.H.; Ooi, A.M.; Liao, S.Y.; Edwards, R.A.; et al. α-ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nat. Cancer 2020, 1, 345–358. [Google Scholar] [CrossRef]
- Lu, H.; Dalgard, C.L.; Mohyeldin, A.; McFate, T.; Tait, A.S.; Verma, A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J. Biol. Chem. 2005, 280, 41928–41939. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain: A hypoxic switch. Science. 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef]
- Tennant, D.A.; Frezza, C.; MacKenzie, E.D.; Nguyen, Q.D.; Zheng, L.; Selak, M.A.; Roberts, D.L.; Dive, C.; Watson, D.G.; Aboagye, E.O.; et al. Reactivating HIF prolyl hydroxylases under hypoxia results in metabolic catastrophe and cell death. Oncogene 2009, 28, 4009–4021. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, J.; Shenghong, M.; Zhang, L.; Yao, J.; Hoadley, K.A.; Wilkerson, M.D.; Perou, C.M.; Guan, K.-L.; Ye, D.; et al. Oncometabolite d-2-hydroxyglutarate inhibits ALKBH DNA repair enzymes and sensitizes IDH-mutant cells to alkylating agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Bian, K.; Tang, Q.; Fedeles, B.I.; Singh, V.; Humulock, Z.T.; Essigmann, J.M.; Li, D. Oncometabolites d- and l-2-hydroxyglutarate inhibit the Alkb family DNA repair enzymes under physiological conditions. Chem. Res. Toxicol. 2017, 30, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; Van Noorden, C.J.F. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.Q.; Ishak Gabra, M.B.; Lowman, X.H.; Yang, Y.; Reid, M.A.; Pan, M.; O’Connor, T.R.; Kong, M. Glutamine deficiency induces DNA alkylation damage and sensitizes cancer cells to alkylating agents through inhibition of ALKBH enzymes. PLoS Biol. 2017, 15, e2002810. [Google Scholar] [CrossRef] [PubMed]
- Herr, C.Q.; Hausinger, R.P. Amazing diversity in biochemical roles of Fe(II)/2-oxoglutarate oxygenases. Trends Biochem. Sci. 2018, 43, 517–532. [Google Scholar] [CrossRef]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; Deberardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef]
- Chen, Q.; Kirk, K.; Shurubor, Y.I.; Zhao, D.; Arreguin, A.J.; Shahi, I.; Valsecchi, F.; Primiano, G.; Calder, E.L.; Carelli, V.; et al. Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab. 2018, 27, 1007–1025.e5. [Google Scholar] [CrossRef]
- Agledal, L.; Niere, M.; Ziegler, M. The phosphate makes a difference: Cellular functions of NADP. Redox Rep. 2010, 15, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Wang, B.; Liu, H.; Shah, H.; Carmona-fontaine, C.; Rustenburg, A.S.; Salah, S.; Gunner, M.R.; Chodera, J.D.; Cross, J.R.; et al. l-2-hydroxyglutarate production arises from non-canonical enzyme function at acidic pH. Nat. Chem. Biol. 2017, 13, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Turcan, S.; Fabius, A.W.M.; Borodovsky, A.; Pedraza, A.; Brennan, C.; Huse, J.; Viale, A.; Riggins, G.J.; Chan, T.A. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT inhibitor decitabine. Oncotarget 2013, 4, 1729–1736. [Google Scholar] [CrossRef]
- Zhang, X.; Rao, A.; Sette, P.; Deibert, C.; Pomerantz, A.; Kim, W.J.; Kohanbash, G.; Chang, Y.; Park, Y.; Engh, J.; et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro Oncol. 2016, 18, 1402–1412. [Google Scholar] [CrossRef]
- Mu, L.; Long, Y.; Yang, C.; Jin, L.; Tao, H.; Ge, H.; Chang, Y.E.; Karachi, A.; Kubilis, P.S.; De Leon, G.; et al. The IDH1 mutation-induced oncometabolite, 2-hydroxyglutarate, may affect DNA methylation and expression of PD-L1 in gliomas. Front. Mol. Neurosci. 2018, 11, 82. [Google Scholar] [CrossRef]
- Colvin, H.; Nishida, N.; Konno, M.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; Hata, T.; Kawamoto, K.; Asai, A.; Tsunekuni, K.; et al. Oncometabolite d-2-hydroxyglurate directly induces epithelial-mesenchymal transition and is associated with distant metastasis in colorectal cancer. Sci. Rep. 2016, 6, 36289. [Google Scholar] [CrossRef]
- Toh, T.B.; Lim, J.J.; Chow, E.K.H. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29. [Google Scholar] [CrossRef]
- Turcan, S.; Makarov, V.; Taranda, J.; Wang, Y.; Fabius, A.W.M.; Wu, W.; Zheng, Y.; El-Amine, N.; Haddock, S.; Nanjangud, G.; et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 2018, 50, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Raineri, S.; Mellor, J. IDH1: Linking metabolism and epigenetics. Front. Genet. 2018, 9, 493. [Google Scholar] [CrossRef] [PubMed]
- Gagné, L.M.; Boulay, K.; Topisirovic, I.; Huot, M.É.; Mallette, F.A. Oncogenic activities of IDH1/2 mutations: From epigenetics to cellular signaling. Trends Cell Biol. 2017, 27, 738–752. [Google Scholar]
- Ye, D.; Guan, K.L.; Xiong, Y. Metabolism, activity, and targeting of d- and l-2-hydroxyglutarates. Trends Cancer 2018, 4, 151–165. [Google Scholar] [CrossRef]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 2009, 324, 261–265. [Google Scholar] [CrossRef]
- Jiang, B.; Zhao, W.; Shi, M.; Zhang, J.; Chen, A.; Ma, H.; Suleman, M.; Lin, F.; Zhou, L.; Wang, J.; et al. IDH1 Arg-132 mutant promotes tumor formation through down-regulating p53. J. Biol. Chem. 2018, 293, 9747–9758. [Google Scholar] [CrossRef]
- Tarhonskaya, H.; Rydzik, A.M.; Leung, I.K.H.; Loik, N.D.; Chan, M.C.; Kawamura, A.; McCullagh, J.S.O.; Claridge, T.D.W.; Flashman, E.; Schofield, C.J. Non-enzymatic chemistry enables 2-hydroxyglutarate-mediated activation of 2-oxoglutarate oxygenases. Nat. Commun. 2014, 5, 3423. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Itsumi, M.; Elia, A.J.; Harris, I.S.; Chio, I.I.C.; Cairns, R.A.; Mccracken, S.; Wakeham, A.; Haight, J.; et al. d-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012, 26, 2038–2049. [Google Scholar] [CrossRef]
- Abdollahi, A.; Hahnfeldt, P.; Maercker, C.; Gröne, H.J.; Debus, J.; Ansorge, W.; Folkman, J.; Hlatky, L.; Huber, P.E. Endostatin’s antiangiogenic signaling network. Mol. Cell 2004, 13, 649–663. [Google Scholar] [CrossRef]
- Kalluri, R. Basement membranes: Structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 2003, 3, 422–433. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Carbonneau, M.; Gagne, L.M.; Lalonde, M.E.; Germain, M.A.; Motorina, A.; Guiot, M.C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7, 12700. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef] [PubMed]
- Grassian, A.R.; Parker, S.J.; Davidson, S.M.; Divakaruni, A.S.; Green, C.R.; Zhang, X.; Slocum, K.L.; Pu, M.; Lin, F.; Vickers, C.; et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014, 74, 3317–3331. [Google Scholar] [CrossRef]
- Parker, S.J.; Metallo, C.M. Metabolic consequences of oncogenic IDH mutations. Pharmacol. Ther. 2015, 152, 54–62. [Google Scholar] [CrossRef]
- Li, F.; He, X.; Ye, D.; Lin, Y.; Yu, H.; Yao, C.; Huang, L.; Zhang, J.; Wang, F.; Xu, S.; et al. NADP+-IDH mutations promote hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Mol. Cell 2015, 60, 661–675. [Google Scholar] [CrossRef]
- Izquierdo-Garcia, J.L.; Viswanath, P.; Eriksson, P.; Cai, L.; Radoul, M.; Chaumeil, M.M.; Blough, M.; Luchman, H.A.; Weiss, S.; Cairncross, J.G.; et al. IDH1 mutation induces reprogramming of pyruvate metabolism. Cancer Res. 2015, 75, 2999–3009. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell 2018, 175, 101–116.e25. [Google Scholar] [CrossRef]
- Chaumeil, M.M.; Radoul, M.; Najac, C.; Eriksson, P.; Viswanath, P.; Blough, M.D.; Chesnelong, C.; Luchman, H.A.; Cairncross, J.G.; Ronen, S.M. Hyperpolarized 13C MR imaging detects no lactate production in mutant IDH1 gliomas: Implications for diagnosis and response monitoring. Neuroimage Clin. 2016, 12, 180–189. [Google Scholar] [CrossRef]
- Viswanath, P.; Chaumeil, M.M.; Ronen, S.M. Molecular imaging of metabolic reprograming in mutant IDH cells. Front. Oncol. 2016, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Chesnelong, C.; Chaumeil, M.M.; Blough, M.D.; Al-Najjar, M.; Stechishin, O.D.; Chan, J.A.; Pieper, R.O.; Ronen, S.M.; Weiss, S.; Luchman, H.A.; et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro. Oncol. 2014, 16, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Goyal, L.; Yau, T.; Poon, R.T.; Ancukiewicz, M.; Deshpande, V.; Christiani, D.C.; Liebman, H.M.; Yang, H.; Kim, H.; et al. Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin. Cancer Res. 2014, 20, 1884–1890. [Google Scholar] [CrossRef]
- Delahousse, J.; Verlingue, L.; Broutin, S.; Legoupil, C.; Touat, M.; Doucet, L.; Ammari, S.; Lacroix, L.; Ducreux, M.; Scoazec, J.Y.; et al. Circulating oncometabolite d-2-hydroxyglutarate enantiomer is a surrogate marker of isocitrate dehydrogenase–mutated intrahepatic cholangiocarcinomas. Eur. J. Cancer 2018, 90, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Dong, Q.; Zhang, C.; Kuan, P.F.; Liu, Y.; Jeck, W.R.; Andersen, J.B.; Jiang, W.; Savich, G.L.; Tan, T.X.; et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013, 32, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Parachoniak, C.A.; Ghanta, K.S.; Fitamant, J.; Ross, K.N.; Najem, M.S.; Gurumurthy, S.; Akbay, E.A.; Sia, D.; Cornella, H.; et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014, 513, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Miao, R.; Liu, T.; Xiang, X.; Gu, J.; Jia, Y.; Li, Z.; Fu, Y.; He, Y.; Zhang, Y.; et al. IDH1 as a frequently mutated gene has potential effect on exosomes releasement by epigenetically regulating P2RX7 in intrahepatic cholangiocarcinoma. Biomed. Pharmacother. 2019, 113, 108774. [Google Scholar] [CrossRef]
- Nepal, C.; O’Rourke, C.J.; Oliveira, D.V.N.P.; Taranta, A.; Shema, S.; Gautam, P.; Calderaro, J.; Barbour, A.; Raggi, C.; Wennerberg, K.; et al. Genomic perturbations reveal distinct regulatory networks in intrahepatic cholangiocarcinoma. Hepatology 2018, 68, 949–963. [Google Scholar] [CrossRef]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 2017, 18, 2780–2794. [Google Scholar] [CrossRef]
- Fujiwara, H.; Tateishi, K.; Misumi, K.; Hayashi, A.; Igarashi, K.; Kato, H.; Nakatsuka, T.; Suzuki, N.; Yamamoto, K.; Kudo, Y.; et al. Mutant IDH1 confers resistance to energy stress in normal biliary cells through PFKP-induced aerobic glycolysis and AMPK activation. Sci. Rep. 2019, 9, 18859. [Google Scholar] [CrossRef]
- Ding, N.; Che, L.; Li, X.L.; Liu, Y.; Jiang, L.J.; Fan, B.; Tao, J.Y.; Chen, X.; Ji, J.F. Oncogenic potential of IDH1 R132C mutant in cholangiocarcinoma development in mice. World J. Gastroenterol. 2016, 22, 2071–2080. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, K.J.; Luo, L.; Hsu, V.; Gudi, R.; Dorff, S.E.; Przepiorka, D.; Deisseroth, A.; Shen, Y.L.; Sheth, C.M.; Charlab, R.; et al. FDA approval summary: Ivosidenib for relapsed or refractory acute myeloid leukemia with an isocitrate dehydrogenase-1 mutation. Clin. Cancer Res. 2019, 25, 3205–3209. [Google Scholar] [CrossRef] [PubMed]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Cho, Y.S.; Levell, J.R.; Liu, G.; Caferro, T.; Sutton, J.; Shafer, C.M.; Costales, A.; Manning, J.R.; Zhao, Q.; Sendzik, M.; et al. Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor. ACS Med. Chem. Lett. 2017, 8, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Caravella, J.A.; Lin, J.; Diebold, R.B.; Campbell, A.M.; Ericsson, A.; Gustafson, G.; Wang, Z.; Castro, J.; Clarke, A.; Gotur, D.; et al. Structure-based design and identification of FT-2102 (Olutasidenib), a potent mutant-selective IDH1 inhibitor. J. Med. Chem. 2020, 63, 1612–1623. [Google Scholar]
- Pusch, S.; Krausert, S.; Fischer, V.; Balss, J.; Ott, M.; Schrimpf, D.; Capper, D.; Sahm, F.; Eisel, J.; Beck, A.C.; et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 2017, 133, 629–644. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Herbst, L.; Pusch, S.; Klett, L.; Goparaju, R.; Stichel, D.; Kaulfuss, S.; Panknin, O.; Zimmermann, K.; Toschi, L.; et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia 2017, 31, 2020–2028. [Google Scholar] [CrossRef]
- Deng, G.; Shen, J.; Yin, M.; McManus, J.; Mathieu, M.; Gee, P.; He, T.; Shi, C.; Bedel, O.; McLean, L.R.; et al. Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. J. Biol. Chem. 2015, 290, 762–774. [Google Scholar] [CrossRef]
- Fan, B.; Mellinghoff, I.K.; Wen, P.Y.; Lowery, M.A.; Goyal, L.; Tap, W.D.; Pandya, S.S.; Manyak, E.; Jiang, L.; Liu, G.; et al. Clinical pharmacokinetics and pharmacodynamics of ivosidenib, an oral, targeted inhibitor of mutant IDH1, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 433–444. [Google Scholar] [CrossRef]
- Lowery, M.A.; Burris, H.A.; Janku, F.; Shroff, R.T.; Cleary, J.M.; Azad, N.S.; Goyal, L.; Maher, E.A.; Gore, L.; Hollebecque, A.; et al. Safety and activity of ivosidenib in patients with IDH1-mutant advanced cholangiocarcinoma: A phase 1 study. Lancet Gastroenterol. Hepatol. 2019, 4, 711–720. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Crispo, F.; Condelli, V.; Lepore, S.; Notarangelo, T.; Sgambato, A.; Esposito, F.; Maddalena, F.; Landriscina, M. Metabolic dysregulations and epigenetics: A bidirectional interplay that drives tumor progression. Cells 2019, 8, 798. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Gordan, J.D.; Kleinstiver, B.P.; Vu, P.; Najem, M.S.; Yeo, J.C.; Shi, L.; Kato, Y.; Levin, R.S.; Webber, J.T.; et al. Isocitrate dehydrogenase mutations confer dasatinib hypersensitivity and SRC dependence in intrahepatic cholangiocarcinoma. Cancer Discov. 2016, 6, 727–739. [Google Scholar] [CrossRef]
- Fujiwara, H.; Tateishi, K.; Kato, H.; Nakatsuka, T.; Yamamoto, K.; Tanaka, Y.; Ijichi, H.; Takahara, N.; Mizuno, S.; Kogure, H.; et al. Isocitrate dehydrogenase 1 mutation sensitizes intrahepatic cholangiocarcinoma to the BET inhibitor JQ1. Cancer Sci. 2018, 109, 3602–3610. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wild, A.T.; Turcan, S.; Wu, W.H.; Sigel, C.; Klimstra, D.S.; Ma, X.; Gong, Y.; Holland, E.C.; Huse, J.T.; et al. Targeting therapeutic vulnerabilities with PARP inhibition and radiation in IDH-mutant gliomas and cholangiocarcinomas. Sci. Adv. 2020, 6, eaaz3221. [Google Scholar] [CrossRef]
- Johannessen, T.C.A.; Mukherjee, J.; Viswanath, P.; Ohba, S.; Ronen, S.M.; Bjerkvig, R.; Pieper, R.O. Rapid conversion of mutant IDH1 from driver to passenger in a model of human gliomagenesis. Mol. Cancer Res. 2016, 14, 976–983. [Google Scholar] [CrossRef]
- Su, L.; Zhang, X.; Zheng, L.; Wang, M.; Zhu, Z.; Li, P. Mutation of isocitrate dehydrogenase 1 in cholangiocarcinoma impairs tumor progression by inhibiting isocitrate metabolism. Front. Endocrinol. (Lausanne) 2020, 11, 189. [Google Scholar] [CrossRef]
- Tan, F.; Jiang, Y.; Sun, N.; Chen, Z.; Lv, Y.; Shao, K.; Li, N.; Qiu, B.; Gao, Y.; Li, B.; et al. Identification of isocitrate dehydrogenase 1 as a potential diagnostic and prognostic biomarker for non-small cell lung cancer by proteomic analysis. Mol. Cell. Proteom. 2012, 11, M111.008821. [Google Scholar] [CrossRef]
- Sun, N.; Chen, Z.; Tan, F.; Zhang, B.; Yao, R.; Zhou, C.; Li, J.; Gao, Y.; Liu, Z.; Tan, X.; et al. Isocitrate dehydrogenase 1 is a novel plasma biomarker for the diagnosis of non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 5136–5145. [Google Scholar] [CrossRef]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-associated IDH1 promotes growth and resistance to targeted therapies in the absence of mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef]
- Zarei, M.; Lal, S.; Parker, S.J.; Nevler, A.; Vaziri-Gohar, A.; Dukleska, K.; Mambelli-Lisboa, N.C.; Moffat, C.; Blanco, F.F.; Chand, S.N.; et al. Posttranscriptional upregulation of IDH1 by HuR establishes a powerful survival phenotype in pancreatic cancer cells. Cancer Res. 2017, 77, 4460–4471. [Google Scholar] [CrossRef] [PubMed]
- Al-Amodi, H.S.A.B.; Nabih, E.S.; Kamel, H.F.M.; El Sayed, M.A.; Dwedar, I.A.M. Wild-type isocitrate dehydrogenase 1 over-expression is related to cancer stem cells survival in lung adenocarcinoma. Cancer Investig. 2018, 36, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, F.; Liu, X.; Zhang, T.; Liu, F.; Ge, X.; Mao, Y.; Hua, D. Correlation of IDH1 and B7–H3 expression with prognosis of CRC patients. Eur. J. Surg. Oncol. 2018, 44, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Xiao, L.; Zhao, Y.; Shi, J.; Yuan, Y.; Gu, Y.; Zhang, F.; Gao, X.; Yang, Y.; Yang, R.; et al. Wild-type IDH1 and mutant IDH1 opposingly regulate podoplanin expression in glioma. Transl. Oncol. 2020, 13, 100758. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).