
An Optimization of Liquid–Liquid Extraction of Urinary Volatile and Semi-Volatile Compounds and Its Application for Gas Chromatography-Mass Spectrometry and Proton Nuclear Magnetic Resonance Spectroscopy

 ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Results and Discussion
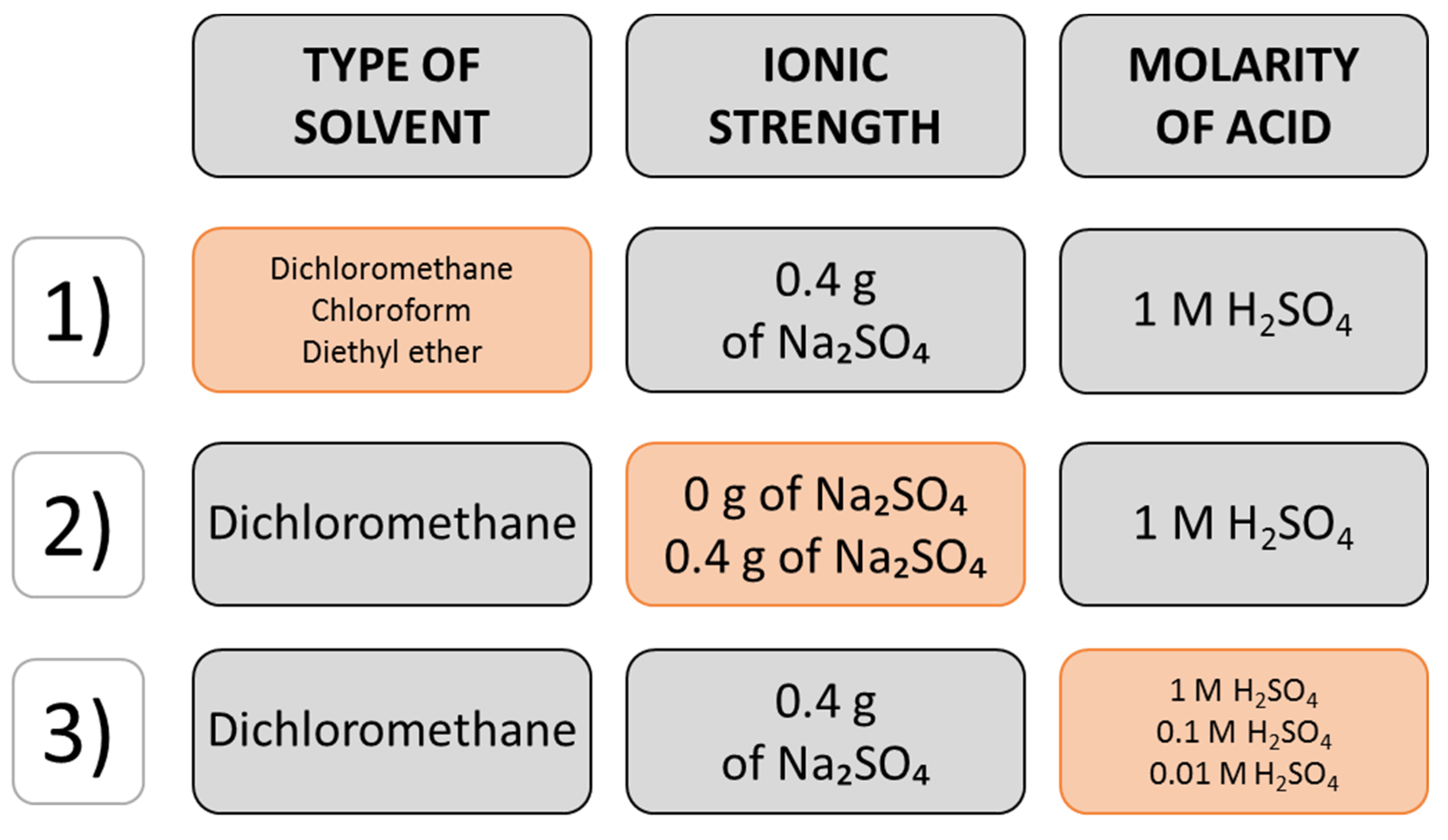
2.1. Optimization of the Extraction Parameters
2.2. Analytical Performance
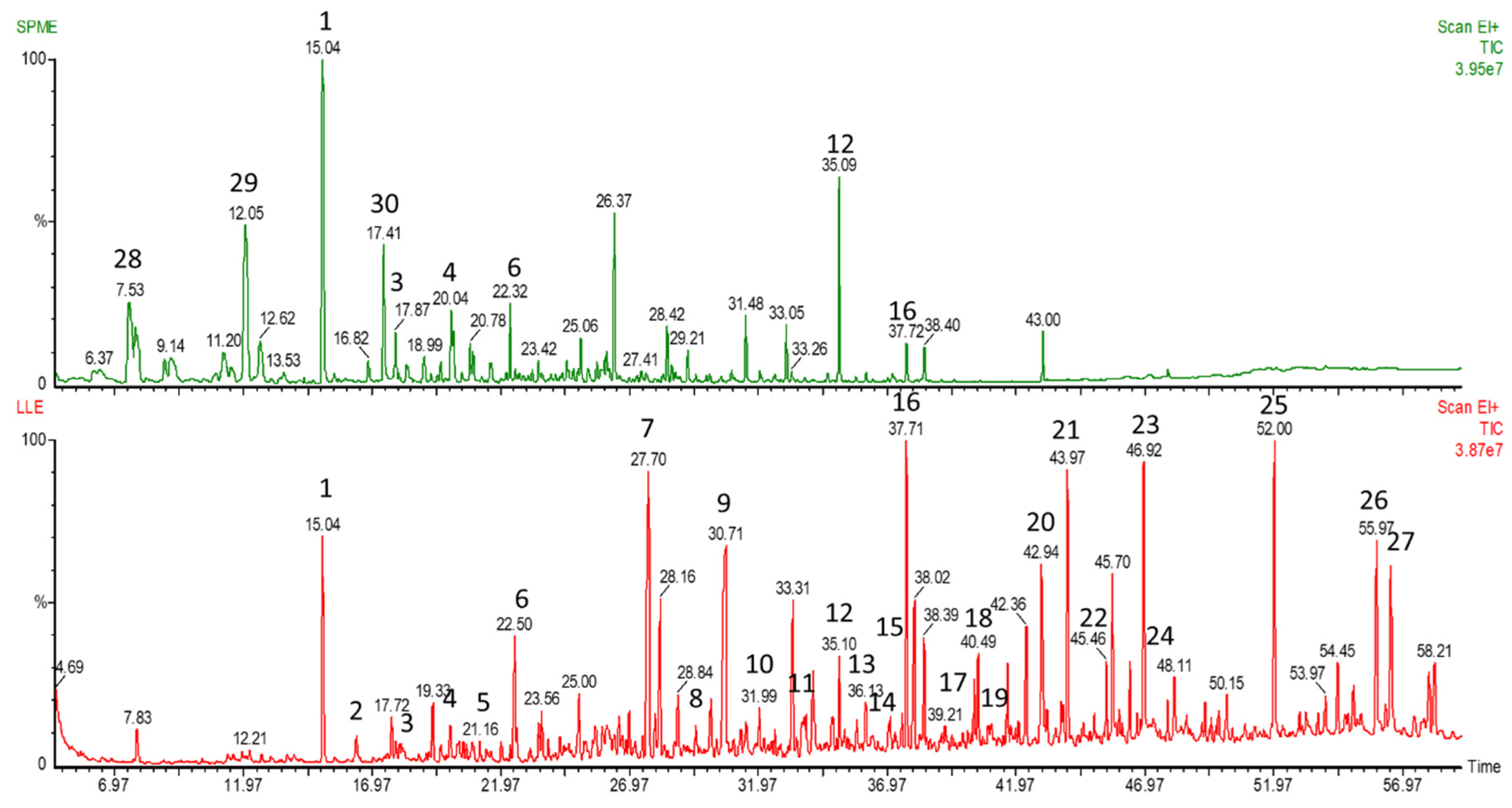
2.3. Method Application—GC-MS
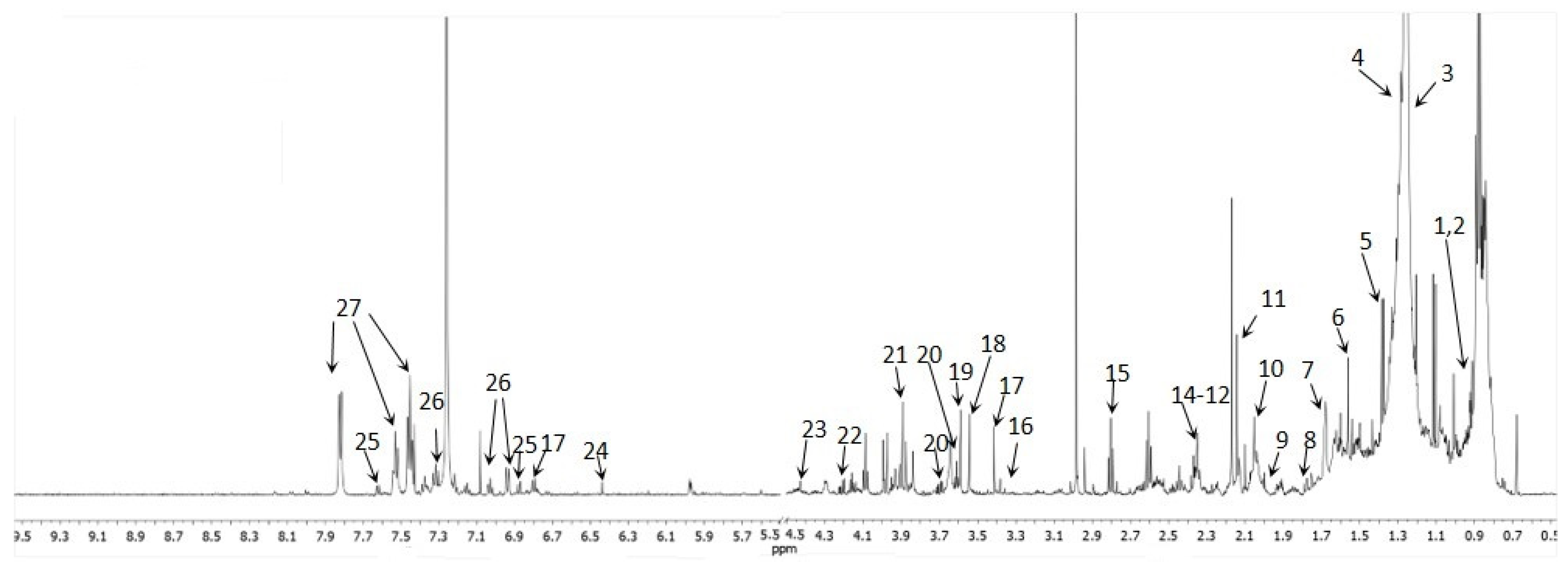
2.4. Method Application—1H-NMR
3. Materials and Methods
3.1. Chemicals and Preparation of Calibration Solutions
3.2. Urine Samples
3.3. Extraction Optimization
3.4. Analytical Performance
3.5. SPME
3.6. Gas Chromatography-Mass Spectrometry
3.7. 1H-NMR
3.8. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Lacy Costello, B.; Amann, A.; Al-Kateb, H.; Flynn, C.; Filipiak, W.; Khalid, T.; Osborne, D.; Ratcliffe, N.M. A review of the volatiles from the healthy human body. J. Breath Res. 2014, 8, 014001. [Google Scholar] [CrossRef] [PubMed]
- Živković Semren, T.; Brčić Karačonji, I.; Safner, T.; Brajenović, N.; Tariba Lovaković, B.; Pizent, A. Gas chromatographic-mass spectrometric analysis of urinary volatile organic metabolites: Optimization of the HS-SPME procedure and sample storage conditions. Talanta 2018, 176, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Bouatra, S.; Aziat, F.; Mandal, R.; Guo, A.C.; Wilson, M.R.; Knox, C.; Bjorndahl, T.C.; Krishnamurthy, R.; Saleem, F.; Liu, P.; et al. The Human Urine Metabolome. PLoS ONE 2013, 8, e73076. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, M.; Carvalho, M.; Henrique, R.; Jerónimo, C.; Moreira, N.; De Lourdes Bastos, M.; De Pinho, P.G. Analysis of volatile human urinary metabolome by solid-phase microextraction in combination with gas chromatography-mass spectrometry for biomarker discovery: Application in a pilot study to discriminate patients with renal cell carcinoma. Eur. J. Cancer 2014, 50, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Li, X.H.; Chen, W.N. An untargeted fecal and urine metabolomics analysis of the interplay between the gut microbiome, diet and human metabolism in Indian and Chinese adults. Sci. Rep. 2019, 9, 9191. [Google Scholar] [CrossRef]
- Zheng, L.; Wang, J.; Gao, W.; Hu, C.; Wang, S.; Rong, R.; Guo, Y.; Zhu, T.; Zhu, D. GC/MS-based urine metabolomics analysis of renal allograft recipients with acute rejection. J. Transl. Med. 2018, 16, 202. [Google Scholar] [CrossRef]
- Zabek, A.; Paslawski, R.; Paslawska, U.; Wojtowicz, W.; Drozdz, K.; Polakof, S.; Podhorska, M.; Dziegiel, P.; Mlynarz, P.; Szuba, A. The influence of different diets on metabolism and atherosclerosis processes—A porcine model: Blood serum, urine and tissues 1H-NMR metabolomics targeted analysis. PLoS ONE 2017, 12, e0184798. [Google Scholar] [CrossRef]
- Taunk, K.; Taware, R.; More, T.H.; Porto-Figueira, P.; Pereira, J.A.M.; Mohapatra, R.; Soneji, D.; Câmara, J.S.; Nagarajaram, H.A.; Rapole, S. A non-invasive approach to explore the discriminatory potential of the urinary volatilome of invasive ductal carcinoma of the breast. RSC Adv. 2018, 8, 25040–25050. [Google Scholar] [CrossRef]
- Taware, R.; Taunk, K.; Pereira, J.A.M.; Dhakne, R.; Kannan, N.; Soneji, D.; Câmara, J.S.; Nagarajaram, H.A.; Rapole, S. Investigation of urinary volatomic alterations in head and neck cancer: A non-invasive approach towards diagnosis and prognosis. Metabolomics 2017, 13, 111. [Google Scholar] [CrossRef]
- Khalid, T.; Aggio, R.; White, P.; De Lacy Costello, B.; Persad, R.; Al-Kateb, H.; Jones, P.; Probert, C.S.; Ratcliffe, N. Urinary volatile organic compounds for the detection of prostate cancer. PLoS ONE 2015, 10, e0143283. [Google Scholar] [CrossRef]
- Antón, A.P.; Ferreira, A.M.C.; Pinto, C.G.; Cordero, B.M.; Pavón, J.L.P. Headspace generation coupled to gas chromatography-mass spectrometry for the automated determination and quantification of endogenous compounds in urine. Aldehydes as possible markers of oxidative stress. J. Chromatogr. A 2014, 1367, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Arasaradnam, R.P.; Westenbrink, E.; McFarlane, M.J.; Harbord, R.; Chambers, S.; O’Connell, N.; Bailey, C.; Nwokolo, C.U.; Bardhan, K.D.; Savage, R.; et al. Differentiating coeliac disease from irritable bowel syndrome by urinary volatile organic compound analysis—A pilot study. PLoS ONE 2014, 9, e107312. [Google Scholar] [CrossRef] [PubMed]
- Arasaradnam, R.P.; Ouaret, N.; Thomas, M.G.; Quraishi, N.; Heatherington, E.; Nwokolo, C.U.; Bardhan, K.D.; Covington, J.A. A novel tool for noninvasive diagnosis and tracking of patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2013, 19, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Drabińska, N.; Azeem, H.A.; Krupa-Kozak, U. A targeted metabolomic protocol for quantitative analysis of volatile organic compounds in urine of children with celiac disease. RSC Adv. 2018, 8, 36534–36541. [Google Scholar] [CrossRef]
- Wang, M.; Xie, R.; Jia, X.; Liu, R. Urinary Volatile Organic Compounds as Potential Biomarkers in Idiopathic Membranous Nephropathy. Med. Princ. Pract. 2017, 26, 375–380. [Google Scholar] [CrossRef]
- Zhu, S.; Corsetti, S.; Wang, Q.; Li, C.; Huang, Z.; Nabi, G. Optical sensory arrays for the detection of urinary bladder cancer-related volatile organic compounds. J. Biophotonics 2019, 12, c201800165. [Google Scholar] [CrossRef]
- Wang, Y.; Hua, L.; Jiang, J.; Xie, Y.; Hou, K.; Li, Q.; Wu, C.; Li, H. High-pressure photon ionization time-of-flight mass spectrometry combined with dynamic purge-injection for rapid analysis of volatile metabolites in urine. Anal. Chim. Acta 2018, 1008, 74–81. [Google Scholar] [CrossRef]
- Zou, X.; Lu, Y.; Xia, L.; Zhang, Y.; Li, A.; Wang, H.; Huang, C.; Shen, C.; Chu, Y. Detection of Volatile Organic Compounds in a Drop of Urine by Ultrasonic Nebulization Extraction Proton Transfer Reaction Mass Spectrometry. Anal. Chem. 2018, 90, 2210–2215. [Google Scholar] [CrossRef]
- Batty, C.A.; Cauchi, M.; Hunter, J.O.; Woolner, J.; Baglin, T.; Turner, C. Differences in microbial metabolites in urine headspace of subjects with Immune Thrombocytopenia (ITP) detected by volatile organic compound (VOC) analysis and metabolomics. Clin. Chim. Acta 2016, 461, 61–68. [Google Scholar] [CrossRef]
- Drabinska, N.; Jarocka-Cyrta, E.; Ratcliffe, N.M.; Krupa-Kozak, U. The profile of urinary headspace volatile organic compounds after 12-week intake of oligofructose-enriched inulin by children and adolescents with celiac disease on a gluten-free diet: Results of a pilot, randomized, placebo-controlled clinical trial. Molecules 2019, 24, 1341. [Google Scholar] [CrossRef]
- Aggio, R.B.M.; Mayor, A.; Coyle, S.; Reade, S.; Khalid, T.; Ratcliffe, N.M.; Probert, C.S.J. Freeze-drying: An alternative method for the analysis of volatile organic compounds in the headspace of urine samples using solid phase micro-extraction coupled to gas chromatography—Mass spectrometry. Chem. Cent. J. 2016, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Burden, H.; Persad, R.; Whittington, K.; De Lacy Costello, B.; Ratcliffe, N.M.; Probert, C.S. A comparative study of the analysis of human urine headspace using gas chromatography-mass spectrometry. J. Breath Res. 2008, 2, 037022. [Google Scholar] [CrossRef] [PubMed]
- O’Lenick, C.R.; Pleil, J.D.; Stiegel, M.A.; Sobus, J.R.; Wallace, M.A.G. Detection and analysis of endogenous polar volatile organic compounds (PVOCs) in urine for human exposome research. Biomarkers 2018, 24, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.A.R.; Barros, E.B.P.; Rezende, C.M. Chapter 78—Method Development and Optimization of Liquid–Liquid Extraction for the Quantitative Analysis of Volatile Compounds from Brazilian Grape Juices. In Flavour Science Proceedings from XIII Weurman Flavour Research Symposium; Ferreira, V., Lopez, R.B.T.-F.S., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 417–421. ISBN 978-0-12-398549-1. [Google Scholar]
- Orak, H.H.; Bahrisefit, I.S.; Sabudak, T. Antioxidant Activity of Extracts of Soursop (Annona muricata L.) Leaves, Fruit Pulps, Peels, and Seeds. Polish J. Food Nutr. Sci. 2019, 69, 359–366. [Google Scholar] [CrossRef]
- Soliman, M.A.; Pedersen, J.A.; Suffet, I.M. Rapid gas chromatography–mass spectrometry screening method for human pharmaceuticals, hormones, antioxidants and plasticizers in water. J. Chromatogr. A 2004, 1029, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H. New trends in sample preparation for clinical and pharmaceutical analysis. TrAC Trends Anal. Chem. 2003, 22, 232–244. [Google Scholar] [CrossRef]
- Zlatkis, A.; Liebich, H.M. Profile of Volatile Metabolites in Human Urine. Clin. Chem. 1971, 17, 592–594. [Google Scholar] [CrossRef]
- Seyler, T.H.; Kim, J.G.; Hodgson, J.A.; Cowan, E.A.; Blount, B.C.; Wang, L. Quantitation of Urinary Volatile Nitrosamines from Exposure to Tobacco Smoke*. J. Anal. Toxicol. 2013, 37, 195–202. [Google Scholar] [CrossRef]
- Cozzolino, R.; De Magistris, L.; Saggese, P.; Stocchero, M.; Martignetti, A.; Di Stasio, M.; Malorni, A.; Marotta, R.; Boscaino, F.; Malorni, L. Use of solid-phase microextraction coupled to gas chromatography-mass spectrometry for determination of urinary volatile organic compounds in autistic children compared with healthy controls. Anal. Bioanal. Chem. 2014, 406, 4649–4662. [Google Scholar] [CrossRef]
- Cequier-Sánchez, E.; Rodríguez, C.; Ravelo, Á.G.; Zárate, R. Dichloromethane as a Solvent for Lipid Extraction and Assessment of Lipid Classes and Fatty Acids from Samples of Different Natures. J. Agric. Food Chem. 2008, 56, 4297–4303. [Google Scholar] [CrossRef]
- Ciska, E.; Drabińska, N.; Honke, J.; Narwojsz, A. Boiled Brussels sprouts: A rich source of glucosinolates and the corresponding nitriles. J. Funct. Foods 2015, 19, 91–99. [Google Scholar] [CrossRef]
- Risticevic, S.; Lord, H.; Górecki, T.; Arthur, C.L.; Pawliszyn, J. Protocol for solid-phase microextraction method development. Nat. Protoc. 2010, 5, 122–139. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Vallejo, M.; García, A.; Barbas, C. Method validation strategies involved in non-targeted metabolomics. J. Chromatogr. A 2014, 1353, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Kordalewska, M.; Macioszek, S.; Wawrzyniak, R.; Sikorska-Wiśniewska, M.; Śledziński, T.; Chmielewski, M.; Mika, A.; Markuszewski, M.J. Multiplatform metabolomics provides insight into the molecular basis of chronic kidney disease. J. Chromatogr. B 2019, 1117, 49–57. [Google Scholar] [CrossRef]
- Rosen Vollmar, A.K.; Rattray, N.J.W.; Cai, Y.; Santos-Neto, Á.J.; Deziel, N.C.; Jukic, A.M.Z.; Johnson, C.H. Normalizing Untargeted Periconceptional Urinary Metabolomics Data: A Comparison of Approaches. Metabolites 2019, 9, 198. [Google Scholar] [CrossRef]
- Rocha, S.M.; Caldeira, M.; Carrola, J.; Santos, M.; Cruz, N.; Duarte, I.F. Exploring the human urine metabolomic potentialities by comprehensive two-dimensional gas chromatography coupled to time of flight mass spectrometry. J. Chromatogr. A 2012, 1252, 155–163. [Google Scholar] [CrossRef]
- Görling, B.; Bräse, S.; Luy, B. NMR chemical shift ranges of urine metabolites in various organic solvents. Metabolites 2016, 6, 27. [Google Scholar] [CrossRef]
- Fathi, F.; Brun, A.; Rott, K.H.; Cobra, P.F.; Tonelli, M.; Eghbalnia, H.R.; Caviedes-Vidal, E.; Karasov, W.H.; Markley, J.L. NMR-based identification of metabolites in polar and non-polar extracts of avian liver. Metabolites 2017, 7, 61. [Google Scholar] [CrossRef]
- Silva, C.L.; Passos, M.; Cmara, J.S. Investigation of urinary volatile organic metabolites as potential cancer biomarkers by solid-phase microextraction in combination with gas chromatography-mass spectrometry. Br. J. Cancer 2011, 105, 1894–1904. [Google Scholar] [CrossRef]
Sample Availability: Not available |




{kind=link}
{kind=link}
{kind=link}
| Factor Analyzed | Number of Peaks Detected | Constant Conditions |
|---|---|---|
| Type of solvent (4 mL added) | ||
| DCM * | 205.0 ± 46.1 a,** | 1 M acid, salt addition |
| Chloroform | 121.0 ± 47.6 b | |
| Diethyl ether | 20.7 ± 4.0 c | |
| Acid molarity (2 mL added) | ||
| 0.01 M | 137.0 ± 2.7 b | solvent: DCM, salt addition |
| 0.1 M | 120.0 ± 21.1 b | |
| 1 M | 205.0 ± 46.1 a | |
| Ionic strength | ||
| Salt addition (0.2 g) | 205.0 ± 46.1 a | solvent: DCM, 1 M acid |
| No salt | 156.0 ± 44.7 a | |
| Retention Time | Compound | Intraday Precision [RSD*%] | Interday Precision [RSD%] | Linear Range [μmol/L] | R2 | LOD ** [μmol/L] | LOQ *** [μmol/L] |
|---|---|---|---|---|---|---|---|
| 9.78 | heptanal | 9 | 9 | 8.857–70.853 | 0.984 | 4.4 | 14.8 |
| 13.11 | octanal | 9 | 19 | 8.004–64.035 | 0.986 | 4.0 | 13.3 |
| 16.15 | nonanal | 13 | 17 | 7.268–58.142 | 0.976 | 3.6 | 12.1 |
| 19.84 | decanal | 6 | 16 | 6.642–53.137 | 0.983 | 3.3 | 11.1 |
| 29.70 | hexanoic acid | 15 | 15 | 9.997–79.977 | 0.985 | 10.0 | 33.3 |
| 32.51 | heptanoic acid | 13 | 26 | 8.814–70.515 | 0.988 | 8.8 | 29.4 |
| 35.05 | octanoic acid | 11 | 15 | 3.944–63.102 | 0.990 | 3.9 | 13.1 |
| Urine Sample | Number of Compounds Detected Using GC-MS |
|---|---|
| 1 | 336 |
| 2 | 326 |
| 3 | 330 |
| 4 | 337 |
| 5 | 338 |
| 6 | 272 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drabińska, N.; Młynarz, P.; de Lacy Costello, B.; Jones, P.; Mielko, K.; Mielnik, J.; Persad, R.; Ratcliffe, N.M. An Optimization of Liquid–Liquid Extraction of Urinary Volatile and Semi-Volatile Compounds and Its Application for Gas Chromatography-Mass Spectrometry and Proton Nuclear Magnetic Resonance Spectroscopy. Molecules 2020, 25, 3651. https://doi.org/10.3390/molecules25163651
Drabińska N, Młynarz P, de Lacy Costello B, Jones P, Mielko K, Mielnik J, Persad R, Ratcliffe NM. An Optimization of Liquid–Liquid Extraction of Urinary Volatile and Semi-Volatile Compounds and Its Application for Gas Chromatography-Mass Spectrometry and Proton Nuclear Magnetic Resonance Spectroscopy. Molecules. 2020; 25(16):3651. https://doi.org/10.3390/molecules25163651
Chicago/Turabian StyleDrabińska, Natalia, Piotr Młynarz, Ben de Lacy Costello, Peter Jones, Karolina Mielko, Justyna Mielnik, Raj Persad, and Norman Mark Ratcliffe. 2020. "An Optimization of Liquid–Liquid Extraction of Urinary Volatile and Semi-Volatile Compounds and Its Application for Gas Chromatography-Mass Spectrometry and Proton Nuclear Magnetic Resonance Spectroscopy" Molecules 25, no. 16: 3651. https://doi.org/10.3390/molecules25163651
APA StyleDrabińska, N., Młynarz, P., de Lacy Costello, B., Jones, P., Mielko, K., Mielnik, J., Persad, R., & Ratcliffe, N. M. (2020). An Optimization of Liquid–Liquid Extraction of Urinary Volatile and Semi-Volatile Compounds and Its Application for Gas Chromatography-Mass Spectrometry and Proton Nuclear Magnetic Resonance Spectroscopy. Molecules, 25(16), 3651. https://doi.org/10.3390/molecules25163651