
1,4-Disubstituted 1,2,3-Triazoles as Amide Bond Surrogates for the Stabilisation of Linear Peptides with Biological Activity



Abstract
1. Introduction
2. 1,2,3-Triazole as Amide Bond Isostere
3. Synthesis of Triazolo-Peptidomimetics
4. Applications
4.1. PACE4 Inhibitors
4.2. Leu-Enkephalin
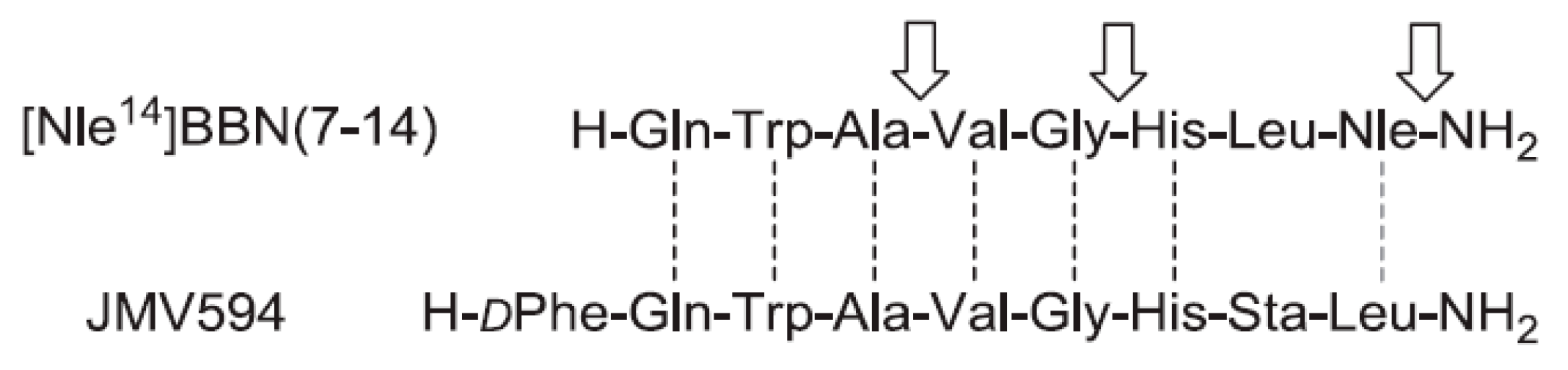
4.3. Bombesin
4.4. Kisspeptin
4.5. Neurotensin
4.6. Cathepsin K & S Inhibitors
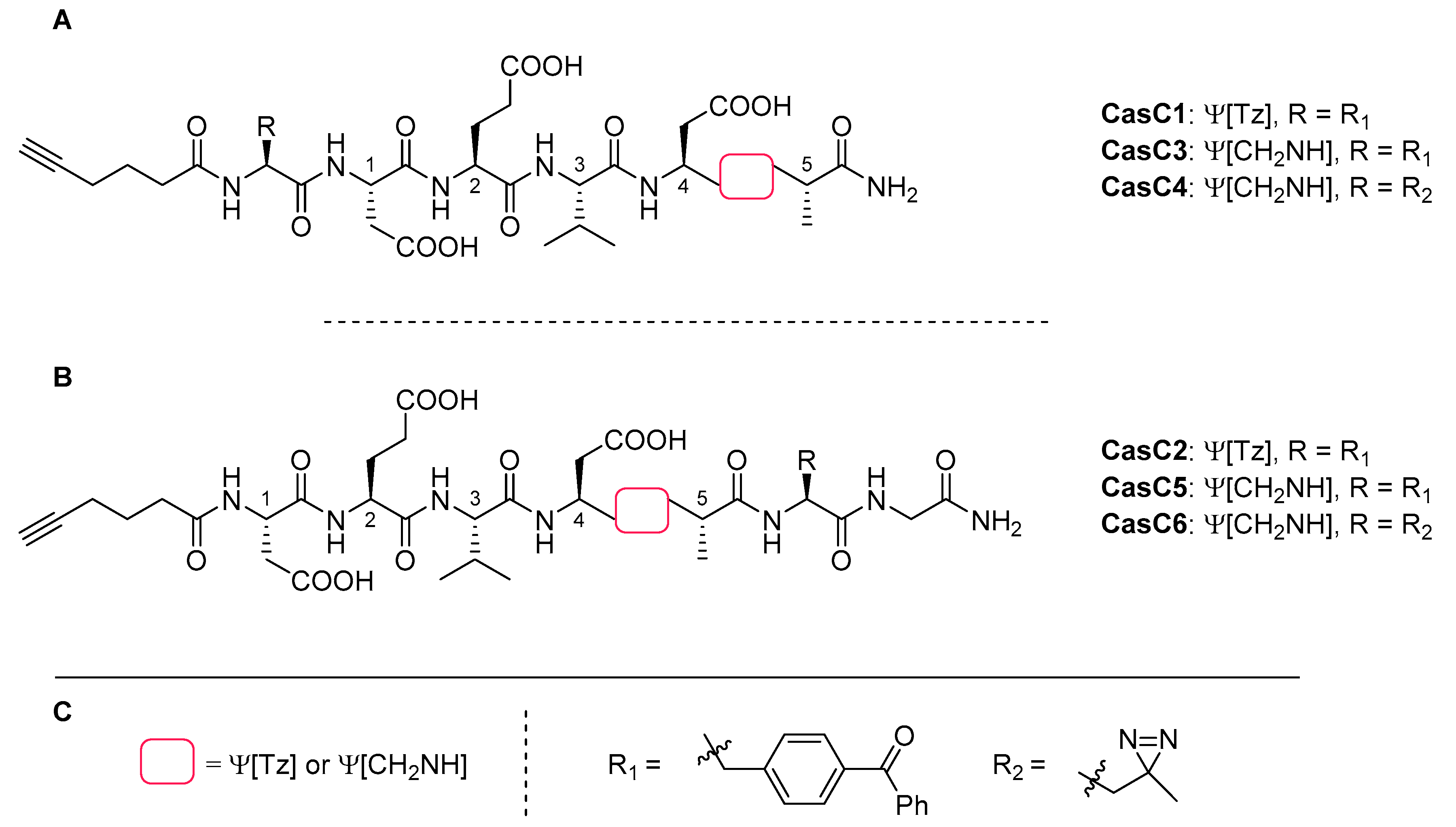
4.7. Caspase-3 Inhibitors
4.8. NRP-1/VEGF165 Inhibitors
4.9. Minigastrins
4.10. Angiotensin
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Di, L. Strategic Approaches to Optimizing Peptide ADME Properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, M.J. The Oral Bioavailability of Peptides and Related Drugs. In Delivery Systems for Peptide Drugs; Davis, S.S., Illum, L., Tomlinson, E., Eds.; NATO ASI Series; Springer US: Boston, MA, USA, 1986; pp. 139–151. ISBN 978-1-4757-9960-6. [Google Scholar]
- Werle, M.; Bernkop-Schnürch, A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.-F.; Yang, H.; Zhao, Y.-Z.; Xue, M. Metabolism of Peptide Drugs and Strategies to Improve their Metabolic Stability. Curr. Drug Metab. 2018, 19, 892–901. [Google Scholar] [CrossRef]
- Goodman, M.; Felix, A. Synthesis of Peptides and Peptidomimetics; Georg Thieme Verlag: Stuttgart, Germany, 2002; ISBN 978-1-58890-023-4. [Google Scholar]
- Nock, B.A.; Maina, T.; Krenning, E.P.; Jong, M. de “To Serve and Protect”: Enzyme Inhibitors as Radiopeptide Escorts Promote Tumor Targeting. J. Nucl. Med. 2014, 55, 121–127. [Google Scholar] [CrossRef]
- Choudhary, A.; Raines, R.T. An Evaluation of Peptide-Bond Isosteres. ChemBioChem 2011, 12, 1801–1807. [Google Scholar] [CrossRef]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical Modifications Designed to Improve Peptide Stability: Incorporation of Non-Natural Amino Acids, Pseudo-Peptide Bonds, and Cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef]
- Cour, T.F.M.L.; Hansen, H.A.S.; Clausen, K.; Lawesson, S.-O. The geometry of the thiopeptide unit. Int. J. Pept. Protein Res. 1983, 22, 509–512. [Google Scholar] [CrossRef]
- Chen, X.; Mietlicki-Baase, E.G.; Barrett, T.M.; McGrath, L.E.; Koch-Laskowski, K.; Ferrie, J.J.; Hayes, M.R.; Petersson, E.J. Thioamide Substitution Selectively Modulates Proteolysis and Receptor Activity of Therapeutic Peptide Hormones. J. Am. Chem. Soc. 2017, 139, 16688–16695. [Google Scholar] [CrossRef]
- Allmendinger, T.; Furet, P.; Hungerbühler, E. Fluoroolefin dipeptide isosteres—I.: The synthesis of Glyψ(CF=CH)Gly and racemic Pheψy(CF=CH)Gly. Tetrahedron Lett. 1990, 31, 7297–7300. [Google Scholar] [CrossRef]
- Malde, A.K.; Khedkar, S.A.; Coutinho, E.C. The B(OH)−NH Analog Is a Surrogate for the Amide Bond (CO−NH) in Peptides: An ab Initio Study. J. Chem. Theory Comput. 2007, 3, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, S.; Trinquier, G. The −BF–NH– Link as a Peptide-Bond Surrogate. J. Phys. Chem. B 2012, 116, 8863–8872. [Google Scholar] [CrossRef] [PubMed]
- Freidinger, R.M.; Veber, D.F.; Perlow, D.S.; Brooks; Saperstein, R. Bioactive conformation of luteinizing hormone-releasing hormone: Evidence from a conformationally constrained analog. Science 1980, 210, 656–658. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, J.; Belleau, B. Synthesis of chiral piperazin-2-ones as model peptidomimetics. J. Chem. Soc. Perkin 1 1989, 9, 1687–1689. [Google Scholar] [CrossRef]
- Jones, R.C.F.; Ward, G.J. Amide bond isosteres: Imidazolines in pseudopeptide chemistry. Tetrahedron Lett. 1988, 29, 3853–3856. [Google Scholar] [CrossRef]
- De Luca, L.; Falorni, M.; Giacomelli, G.; Porcheddu, A. New pyrazole containing bicarboxylic α-amino acids: Mimics of the cis amide bond. Tetrahedron Lett. 1999, 40, 8701–8704. [Google Scholar] [CrossRef]
- May, B.C.H.; Abell, A.D. The synthesis and crystal structure of alpha-keto tetrazole-based dipeptide mimics. Tetrahedron Lett. 2001, 42, 5641–5644. [Google Scholar] [CrossRef]
- Hitotsuyanagi, Y.; Motegi, S.; Fukaya, H.; Takeya, K. A cis Amide Bond Surrogate Incorporating 1,2,4-Triazole. J. Org. Chem. 2002, 67, 3266–3271. [Google Scholar] [CrossRef]
- Valverde, I.E.; Mindt, T.L. 1,2,3-Triazoles as Amide-bond Surrogates in Peptidomimetics. Chimia 2013, 67, 262–266. [Google Scholar] [CrossRef]
- Hou, J.; Liu, X.; Shen, J.; Zhao, G.; Wang, P.G. The impact of click chemistry in medicinal chemistry. Expert Opin. Drug Discov. 2012, 7, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Horne, W.S.; Stout, C.D.; Ghadiri, M.R. A Heterocyclic Peptide Nanotube. J. Am. Chem. Soc. 2003, 125, 9372–9376. [Google Scholar] [CrossRef]
- Horne, W.S.; Yadav, M.K.; Stout, C.D.; Ghadiri, M.R. Heterocyclic Peptide Backbone Modifications in an α-Helical Coiled Coil. J. Am. Chem. Soc. 2004, 126, 15366–15367. [Google Scholar] [CrossRef] [PubMed]
- Angelo, N.G.; Arora, P.S. Nonpeptidic Foldamers from Amino Acids: Synthesis and Characterization of 1,3-Substituted Triazole Oligomers. J. Am. Chem. Soc. 2005, 127, 17134–17135. [Google Scholar] [CrossRef] [PubMed]
- van Maarseveen, J.H.; Horne, W.S.; Ghadiri, M.R. Efficient Route to C2 Symmetric Heterocyclic Backbone Modified Cyclic Peptides. Org. Lett. 2005, 7, 4503–4506. [Google Scholar] [CrossRef]
- Angelo, N.G.; Arora, P.S. Solution- and Solid-Phase Synthesis of Triazole Oligomers That Display Protein-Like Functionality. J. Org. Chem. 2007, 72, 7963–7967. [Google Scholar] [CrossRef]
- Boddaert, T.; Solà, J.; Helliwell, M.; Clayden, J. Chemical communication: Conductors and insulators of screw-sense preference between helical oligo(aminoisobutyric acid) domains. Chem. Commun. 2012, 48, 3397–3399. [Google Scholar] [CrossRef]
- Salah, K.B.H.; Legrand, B.; Das, S.; Martinez, J.; Inguimbert, N. Straightforward strategy to substitute amide bonds by 1,2,3-triazoles in peptaibols analogs using Aibψ[Tz]-Xaa dipeptides. Pept. Sci. 2015, 104, 611–621. [Google Scholar] [CrossRef]
- Salah, K.B.H.; Das, S.; Ruiz, N.; Andreu, V.; Martinez, J.; Wenger, E.; Amblard, M.; Didierjean, C.; Legrand, B.; Inguimbert, N. How are 1,2,3-triazoles accommodated in helical secondary structures? Org. Biomol. Chem. 2018, 16, 3576–3583. [Google Scholar] [CrossRef]
- Schröder, D.C.; Kracker, O.; Fröhr, T.; Góra, J.; Jewginski, M.; Nieß, A.; Antes, I.; Latajka, R.; Marion, A.; Sewald, N. 1,4-Disubstituted 1H-1,2,3-Triazole Containing Peptidotriazolamers: A New Class of Peptidomimetics With Interesting Foldamer Properties. Front. Chem. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Bock, V.D.; Perciaccante, R.; Jansen, T.P.; Hiemstra, H.; van Maarseveen, J.H. Click Chemistry as a Route to Cyclic Tetrapeptide Analogues: Synthesis of cyclo-[Pro-Val-ψ(triazole)-Pro-Tyr]. Org. Lett. 2006, 8, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Bock, V.D.; Speijer, D.; Hiemstra, H.; Maarseveen, J.H. van 1,2,3-Triazoles as peptide bond isosteres: Synthesis and biological evaluation of cyclotetrapeptide mimics. Org. Biomol. Chem. 2007, 5, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Springer, J.; de Cuba, K.R.; Calvet-Vitale, S.; Geenevasen, J.A.J.; Hermkens, P.H.H.; Hiemstra, H.; van Maarseveen, J.H. Backbone Amide Linker Strategy for the Synthesis of 1,4-Triazole-Containing Cyclic Tetra- and Pentapeptides. Eur. J. Org. Chem. 2008, 2008, 2592–2600. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, L.; Wan, J.; Li, Y.; Xu, Y.; Pan, Y. Design and synthesis of cyclo[-Arg-Gly-Asp-Ψ(triazole)-Gly-Xaa-] peptide analogues by click chemistry. Tetrahedron 2008, 64, 10728–10734. [Google Scholar] [CrossRef]
- Horne, W.S.; Olsen, C.A.; Beierle, J.M.; Montero, A.; Ghadiri, M.R. Probing the Bioactive Conformation of an Archetypal Natural Product HDAC Inhibitor with Conformationally Homogeneous Triazole-Modified Cyclic Tetrapeptides. Angew. Chem. Int. Ed. 2009, 48, 4718–4724. [Google Scholar] [CrossRef]
- Beierle, J.M.; Horne, W.S.; van Maarseveen, J.H.; Waser, B.; Reubi, J.C.; Ghadiri, M.R. Conformationally Homogeneous Heterocyclic Pseudotetrapeptides as Three-Dimensional Scaffolds for Rational Drug Design: Receptor-Selective Somatostatin Analogues. Angew. Chem. Int. Ed. 2009, 48, 4725–4729. [Google Scholar] [CrossRef]
- Singh, E.K.; Nazarova, L.A.; Lapera, S.A.; Alexander, L.D.; McAlpine, S.R. Histone deacetylase inhibitors: Synthesis of cyclic tetrapeptides and their triazole analogs. Tetrahedron Lett. 2010, 51, 4357–4360. [Google Scholar] [CrossRef]
- Davis, M.R.; Singh, E.K.; Wahyudi, H.; Alexander, L.D.; Kunicki, J.B.; Nazarova, L.A.; Fairweather, K.A.; Giltrap, A.M.; Jolliffe, K.A.; McAlpine, S.R. Synthesis of sansalvamide A peptidomimetics: Triazole, oxazole, thiazole, and pseudoproline containing compounds. Tetrahedron 2012, 68, 1029–1051. [Google Scholar] [CrossRef]
- Tischler, M.; Nasu, D.; Empting, M.; Schmelz, S.; Heinz, D.W.; Rottmann, P.; Kolmar, H.; Buntkowsky, G.; Tietze, D.; Avrutina, O. Braces for the Peptide Backbone: Insights into Structure–Activity Relationships of Protease Inhibitor Mimics with Locked Amide Conformations. Angew. Chem. Int. Ed. 2012, 51, 3708–3712. [Google Scholar] [CrossRef]
- Oueis, E.; Jaspars, M.; Westwood, N.J.; Naismith, J.H. Enzymatic Macrocyclization of 1,2,3-Triazole Peptide Mimetics. Angew. Chem. Int. Ed. 2016, 55, 5842–5845. [Google Scholar] [CrossRef] [PubMed]
- Valverde, I.E.; Lecaille, F.; Lalmanach, G.; Aucagne, V.; Delmas, A.F. Synthesis of a Biologically Active Triazole-Containing Analogue of Cystatin A Through Successive Peptidomimetic Alkyne–Azide Ligations. Angew. Chem. Int. Ed. 2012, 51, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Bittermann, H.; Gmeiner, P. Triazolopeptides: Chirospecific synthesis and cis/trans prolyl ratios of structural isomers. Tetrahedron 2006, 62, 8919–8927. [Google Scholar] [CrossRef]
- Hartwig, S.; Hecht, S. Polypseudopeptides with Variable Stereochemistry: Synthesis via Click-Chemistry, Postfunctionalization, and Conformational Behavior in Solution. Macromolecules 2010, 43, 242–248. [Google Scholar] [CrossRef]
- Kann, N.; Johansson, J.R.; Beke-Somfai, T. Conformational properties of 1,4- and 1,5-substituted 1,2,3-triazole amino acids–building units for peptidic foldamers. Org. Biomol. Chem. 2015, 13, 2776–2785. [Google Scholar] [CrossRef]
- Charron, C.L.; Hickey, J.L.; Nsiama, T.K.; Cruickshank, D.R.; Turnbull, W.L.; Luyt, L.G. Molecular imaging probes derived from natural peptides. Nat. Prod. Rep. 2016, 33, 761–800. [Google Scholar] [CrossRef]
- Fani, M.; Maecke, H.R.; Okarvi, S.M. Radiolabeled Peptides: Valuable Tools for the Detection and Treatment of Cancer. Theranostics 2012, 2, 481–501. [Google Scholar] [CrossRef]
- Reubi, J.C.; Maecke, H.R. Peptide-Based Probes for Cancer Imaging. J. Nucl. Med. 2008, 49, 1735–1738. [Google Scholar] [CrossRef]
- Reilly, R.M.; Sandhu, J.; Alvarez-Diez, T.M.; Gallinger, S.; Kirsh, J.; Stern, H. Problems of Delivery of Monoclonal Antibodies. Clin. Pharmacokinet. 1995, 28, 126–142. [Google Scholar] [CrossRef]
- Tam, A.; Arnold, U.; Soellner, M.B.; Raines, R.T. Protein Prosthesis: 1,5-Disubstituted[1,2,3]triazoles as cis-Peptide Bond Surrogates. J. Am. Chem. Soc. 2007, 129, 12670–12671. [Google Scholar] [CrossRef]
- Pokorski, J.K.; Miller Jenkins, L.M.; Feng, H.; Durell, S.R.; Bai, Y.; Appella, D.H. Introduction of a Triazole Amino Acid into a Peptoid Oligomer Induces Turn Formation in Aqueous Solution. Org. Lett. 2007, 9, 2381–2383. [Google Scholar] [CrossRef] [PubMed]
- Ahsanullah; Schmieder, P.; Kühne, R.; Rademann, J. Metal-Free, Regioselective Triazole Ligations that Deliver Locked cis Peptide Mimetics. Angew. Chem. Int. Ed. 2009, 48, 5042–5045. [Google Scholar] [CrossRef] [PubMed]
- Tietze, D.; Tischler, M.; Voigt, S.; Imhof, D.; Ohlenschläger, O.; Görlach, M.; Buntkowsky, G. Development of a Functional cis-Prolyl Bond Biomimetic and Mechanistic Implications for Nickel Superoxide Dismutase. Chem.–Eur. J. 2010, 16, 7572–7578. [Google Scholar] [CrossRef] [PubMed]
- Kracker, O.; Góra, J.; Krzciuk-Gula, J.; Marion, A.; Neumann, B.; Stammler, H.-G.; Nieß, A.; Antes, I.; Latajka, R.; Sewald, N. 1,5-Disubstituted 1,2,3-Triazole-Containing Peptidotriazolamers: Design Principles for a Class of Versatile Peptidomimetics. Chem.–Eur. J. 2018, 24, 953–961. [Google Scholar] [CrossRef]
- Palmer, M.H.; Findlay, R.H.; Gaskell, A.J. Electronic charge distribution and moments of five- and six-membered heterocycles. J. Chem. Soc. Perkin Trans. 2 1974, 420–428. [Google Scholar] [CrossRef]
- Bates, W.W.; Hobbs, M.E. The Dipole Moments of Some Acid Amides and the Structure of the Amide Group 1. J. Am. Chem. Soc. 1951, 73, 2151–2156. [Google Scholar] [CrossRef]
- Bourne, Y.; Kolb, H.C.; Radić, Z.; Sharpless, K.B.; Taylor, P.; Marchot, P. Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Proc. Natl. Acad. Sci. USA 2004, 101, 1449–1454. [Google Scholar] [CrossRef]
- Brik, A.; Alexandratos, J.; Lin, Y.-C.; Elder, J.H.; Olson, A.J.; Wlodawer, A.; Goodsell, D.S.; Wong, C.-H. 1,2,3-Triazole as a Peptide Surrogate in the Rapid Synthesis of HIV-1 Protease Inhibitors. ChemBioChem 2005, 6, 1167–1169. [Google Scholar] [CrossRef]
- Massarotti, A.; Aprile, S.; Mercalli, V.; Del Grosso, E.; Grosa, G.; Sorba, G.; Tron, G.C. Are 1,4- and 1,5-Disubstituted 1,2,3-Triazoles Good Pharmacophoric Groups? ChemMedChem 2014, 9, 2497–2508. [Google Scholar] [CrossRef]
- Schulze, B.; Schubert, U.S. Beyond click chemistry–supramolecular interactions of 1,2,3-triazoles. Chem. Soc. Rev. 2014, 43, 2522–2571. [Google Scholar] [CrossRef]
- Seebach, D.; Matthews, J.L. β-Peptides: A surprise at every turn. Chem. Commun. 1997, 2015–2022. [Google Scholar] [CrossRef]
- Prasher, P.; Sharma, M. Tailored therapeutics based on 1,2,3-1H-triazoles: A mini review. MedChemComm 2019, 10, 1302–1328. [Google Scholar] [CrossRef] [PubMed]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Meghani, N.M.; Amin, H.H.; Lee, B.-J. Mechanistic applications of click chemistry for pharmaceutical drug discovery and drug delivery. Drug Discov. Today 2017, 22, 1604–1619. [Google Scholar] [CrossRef]
- Jiang, X.; Hao, X.; Jing, L.; Wu, G.; Kang, D.; Liu, X.; Zhan, P. Recent applications of click chemistry in drug discovery. Expert Opin. Drug Discov. 2019, 14, 779–789. [Google Scholar] [CrossRef]
- Rani, A.; Singh, G.; Singh, A.; Maqbool, U.; Kaur, G.; Singh, J. CuAAC-ensembled 1,2,3-triazole-linked isosteres as pharmacophores in drug discovery: Review. RSC Adv. 2020, 10, 5610–5635. [Google Scholar] [CrossRef]
- Xi, W.; Scott, T.F.; Kloxin, C.J.; Bowman, C.N. Click Chemistry in Materials Science. Adv. Funct. Mater. 2014, 24, 2572–2590. [Google Scholar] [CrossRef]
- Döhler, D.; Michael, P.; Binder, W.H. CuAAC-Based Click Chemistry in Self-Healing Polymers. Acc. Chem. Res. 2017, 50, 2610–2620. [Google Scholar] [CrossRef]
- Li, H.; Aneja, R.; Chaiken, I. Click Chemistry in Peptide-Based Drug Design. Molecules 2013, 18, 9797–9817. [Google Scholar] [CrossRef] [PubMed]
- Ahmad Fuaad, A.A.H.; Azmi, F.; Skwarczynski, M.; Toth, I. Peptide Conjugation via CuAAC ‘Click’ Chemistry. Molecules 2013, 18, 13148–13174. [Google Scholar] [CrossRef] [PubMed]
- Goddard-Borger, E.D.; Stick, R.V. An Efficient, Inexpensive, and Shelf-Stable Diazotransfer Reagent: Imidazole-1-sulfonyl Azide Hydrochloride. Org. Lett. 2007, 9, 3797–3800. [Google Scholar] [CrossRef] [PubMed]
- Proteau-Gagné, A.; Rochon, K.; Roy, M.; Albert, P.-J.; Guérin, B.; Gendron, L.; Dory, Y.L. Systematic replacement of amides by 1,4-disubstituted[1,2,3]triazoles in Leu-enkephalin and the impact on the delta opioid receptor activity. Bioorg. Med. Chem. Lett. 2013, 23, 5267–5269. [Google Scholar] [CrossRef] [PubMed]
- Valverde, I.E.; Bauman, A.; Kluba, C.A.; Vomstein, S.; Walter, M.A.; Mindt, T.L. 1,2,3-Triazoles as Amide Bond Mimics: Triazole Scan Yields Protease-Resistant Peptidomimetics for Tumor Targeting. Angew. Chem. Int. Ed. 2013, 52, 8957–8960. [Google Scholar] [CrossRef]
- Beltramo, M.; Robert, V.; Galibert, M.; Madinier, J.-B.; Marceau, P.; Dardente, H.; Decourt, C.; De Roux, N.; Lomet, D.; Delmas, A.F.; et al. Rational Design of Triazololipopeptides Analogs of Kisspeptin Inducing a Long-Lasting Increase of Gonadotropins. J. Med. Chem. 2015, 58, 3459–3470. [Google Scholar] [CrossRef]
- Fedorczyk, B.; Lipiński, P.F.J.; Puszko, A.K.; Tymecka, D.; Wilenska, B.; Dudka, W.; Perret, G.Y.; Wieczorek, R.; Misicka, A. Triazolopeptides Inhibiting the Interaction between Neuropilin-1 and Vascular Endothelial Growth Factor-165. Molecules 2019, 24, 1756. [Google Scholar] [CrossRef]
- Dickson, H.D.; Smith, S.C.; Hinkle, K.W. A convenient scalable one-pot conversion of esters and Weinreb amides to terminal alkynes. Tetrahedron Lett. 2004, 45, 5597–5599. [Google Scholar] [CrossRef]
- Reginato, G.; Mordini, A.; Messina, F.; Degl’Innocenti, A.; Poli, G. A new stereoselective synthesis of chiral γ-functionalized (E)-allylic amines. Tetrahedron 1996, 52, 10985–10996. [Google Scholar] [CrossRef]
- Grob, N.M.; Häussinger, D.; Deupi, X.; Schibli, R.; Behe, M.; Mindt, T.L. Triazolo-Peptidomimetics: Novel Radiolabeled Minigastrin Analogs for Improved Tumor Targeting. J. Med. Chem. 2020, 63, 4484–4495. [Google Scholar] [CrossRef]
- Punna, S.; Finn, M.G. A Convenient Colorimetric Test for Aliphatic Azides. Synlett 2004, 99–100. [Google Scholar] [CrossRef]
- Day, R.; Neugebauer, W.A.; Dory, Y. Stable Peptide-Based Pace4 Inhibitors. WO2013/029180A1, 7 March 2013. [Google Scholar]
- Proteau-Gagné, A.; Bournival, V.; Rochon, K.; Dory, Y.L.; Gendron, L. Exploring the Backbone of Enkephalins To Adjust Their Pharmacological Profile for the δ-Opioid Receptor. ACS Chem. Neurosci. 2010, 1, 757–769. [Google Scholar] [CrossRef]
- Vigna, S.R.; Giraud, A.S.; Reeve, J.R.; Walsh, J.H. Biological activity of oxidized and reduced iodinated bombesins. Peptides 1988, 9, 923–926. [Google Scholar] [CrossRef]
- Valverde, I.E.; Vomstein, S.; Fischer, C.A.; Mascarin, A.; Mindt, T.L. Probing the Backbone Function of Tumor Targeting Peptides by an Amide-to-Triazole Substitution Strategy. J. Med. Chem. 2015, 58, 7475–7484. [Google Scholar] [CrossRef]
- Llinares, M.; Devin, C.; Chaloin, O.; Azay, J.; Noel-Artis, A.M.; Bernad, N.; Fehrentz, J.A.; Martinez, J. Syntheses and biological activities of potent bombesin receptor antagonists. J. Pept. Res. 1999, 53, 275–283. [Google Scholar] [CrossRef]
- Valverde, I.E.; Huxol, E.; Mindt, T.L. Radiolabeled antagonistic bombesin peptidomimetics for tumor targeting. J. Label. Compd. Radiopharm. 2014, 57, 275–278. [Google Scholar] [CrossRef]
- de Roux, N.; Genin, E.; Carel, J.-C.; Matsuda, F.; Chaussain, J.-L.; Milgrom, E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc. Natl. Acad. Sci. USA 2003, 100, 10972–10976. [Google Scholar] [CrossRef]
- Seminara, S.B.; Messager, S.; Chatzidaki, E.E.; Thresher, R.R.; Acierno, J.S.; Shagoury, J.K.; Bo-Abbas, Y.; Kuohung, W.; Schwinof, K.M.; Hendrick, A.G.; et al. The GPR54 Gene as a Regulator of Puberty. N. Engl. J. Med. 2003, 349, 1614–1627. [Google Scholar] [CrossRef]
- Caraty, A.; Smith, J.T.; Lomet, D.; Ben Saïd, S.; Morrissey, A.; Cognie, J.; Doughton, B.; Baril, G.; Briant, C.; Clarke, I.J. Kisspeptin Synchronizes Preovulatory Surges in Cyclical Ewes and Causes Ovulation in Seasonally Acyclic Ewes. Endocrinology 2007, 148, 5258–5267. [Google Scholar] [CrossRef]
- Mascarin, A.; Valverde, I.E.; Vomstein, S.; Mindt, T.L. 1,2,3-Triazole Stabilized Neurotensin-Based Radiopeptidomimetics for Improved Tumor Targeting. Bioconjug. Chem. 2015, 26, 2143–2152. [Google Scholar] [CrossRef]
- Souazé, F.; Dupouy, S.; Viardot-Foucault, V.; Bruyneel, E.; Attoub, S.; Gespach, C.; Gompel, A.; Forgez, P. Expression of Neurotensin and NT1 Receptor in Human Breast Cancer: A Potential Role in Tumor Progression. Cancer Res. 2006, 66, 6243–6249. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Waser, B.; Friess, H.; Büchler, M.; Laissue, J. Neurotensin receptors: A new marker for human ductal pancreatic adenocarcinoma. Gut 1998, 42, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, R.; Scheunemann, M.; Heichert, C.; Mäding, P.; Wittrisch, H.; Kretzschmar, M.; Rodig, H.; Tourwé, D.; Iterbeke, K.; Chavatte, K.; et al. Biodistribution and catabolism of 18F-labeled neurotensin(8–13) analogs. Nucl. Med. Biol. 2002, 29, 61–72. [Google Scholar] [CrossRef]
- Bruehlmeier, M.; Garayoa, E.G.; Blanc, A.; Holzer, B.; Gergely, S.; Tourwé, D.; Schubiger, P.A.; Bläuenstein, P. Stabilization of neurotensin analogues: Effect on peptide catabolism, biodistribution and tumor binding. Nucl. Med. Biol. 2002, 29, 321–327. [Google Scholar] [CrossRef]
- Mascarin, A.; Valverde, I.E.; Mindt, T.L. Radiolabeled analogs of neurotensin (8–13) containing multiple 1,2,3-triazoles as stable amide bond mimics in the backbone. MedChemComm 2016, 7, 1640–1646. [Google Scholar] [CrossRef]
- Galibert, M.; Wartenberg, M.; Lecaille, F.; Saidi, A.; Mavel, S.; Joulin-Giet, A.; Korkmaz, B.; Brömme, D.; Aucagne, V.; Delmas, A.F.; et al. Substrate-derived triazolo- and azapeptides as inhibitors of cathepsins K and S. Eur. J. Med. Chem. 2018, 144, 201–210. [Google Scholar] [CrossRef]
- Lecaille, F.; Brömme, D.; Lalmanach, G. Biochemical properties and regulation of cathepsin K activity. Biochimie 2008, 90, 208–226. [Google Scholar] [CrossRef]
- Wilkinson, R.D.A.; Williams, R.; Scott, C.J.; Burden, R.E. Cathepsin S: Therapeutic, diagnostic, and prognostic potential. Biol. Chem. 2015, 396, 867–882. [Google Scholar] [CrossRef]
- Lützner, N.; Kalbacher, H. Quantifying Cathepsin S Activity in Antigen Presenting Cells Using a Novel Specific Substrate. J. Biol. Chem. 2008, 283, 36185–36194. [Google Scholar] [CrossRef]
- Lecaille, F.; Vandier, C.; Godat, E.; Hervé-Grépinet, V.; Brömme, D.; Lalmanach, G. Modulation of hypotensive effects of kinins by cathepsin K. Arch. Biochem. Biophys. 2007, 459, 129–136. [Google Scholar] [CrossRef]
- Van Kersavond, T.; Konopatzki, R.; Chakrabarty, S.; Blank-Landeshammer, B.; Sickmann, A.; Verhelst, S.H.L. Short Peptides with Uncleavable Peptide Bond Mimetics as Photoactivatable Caspase-3 Inhibitors. Molecules 2019, 24, 206. [Google Scholar] [CrossRef]
- Jubb, A.M.; Strickland, L.A.; Liu, S.D.; Mak, J.; Schmidt, M.; Koeppen, H. Neuropilin-1 expression in cancer and development. J. Pathol. 2012, 226, 50–60. [Google Scholar] [CrossRef]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 Is Expressed by Endothelial and Tumor Cells as an Isoform-Specific Receptor for Vascular Endothelial Growth Factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef]
- Naik, A.; Al-Zeheimi, N.; Bakheit, C.S.; Al Riyami, M.; Al Jarrah, A.; Al Moundhri, M.S.; Al Habsi, Z.; Basheer, M.; Adham, S.A. Neuropilin-1 Associated Molecules in the Blood Distinguish Poor Prognosis Breast Cancer: A Cross-Sectional Study. Sci. Rep. 2017, 7, 3301. [Google Scholar] [CrossRef]
- Binétruy-Tournaire, R.; Demangel, C.; Malavaud, B.; Vassy, R.; Rouyre, S.; Kraemer, M.; Plouët, J.; Derbin, C.; Perret, G.; Mazié, J.C. Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis. EMBO J. 2000, 19, 1525–1533. [Google Scholar] [CrossRef]
- Tymecka, D.; Puszko, A.K.; Lipiński, P.F.J.; Fedorczyk, B.; Wilenska, B.; Sura, K.; Perret, G.Y.; Misicka, A. Branched pentapeptides as potent inhibitors of the vascular endothelial growth factor 165 binding to Neuropilin-1: Design, synthesis and biological activity. Eur. J. Med. Chem. 2018, 158, 453–462. [Google Scholar] [CrossRef]
- Reubi, J.C.; Schaer, J.-C.; Waser, B. Cholecystokinin(CCK)-A and CCK-B/Gastrin Receptors in Human Tumors. Cancer Res. 1997, 57, 1377–1386. [Google Scholar]
- Laverman, P.; Joosten, L.; Eek, A.; Roosenburg, S.; Peitl, P.K.; Maina, T.; Mäcke, H.; Aloj, L.; von Guggenberg, E.; Sosabowski, J.K.; et al. Comparative biodistribution of 12 111In-labelled gastrin/CCK2 receptor-targeting peptides. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1410–1416. [Google Scholar] [CrossRef]
- Grob, N.M.; Schmid, S.; Schibli, R.; Behe, M.; Mindt, T.L. Design of Radiolabeled Analogs of Minigastrin by Multiple Amide-to-Triazole Substitutions. J. Med. Chem. 2020, 63, 4496–4505. [Google Scholar] [CrossRef]
- Renziehausen, A.; Wang, H.; Rao, B.; Weir, L.; Nigro, C.L.; Lattanzio, L.; Merlano, M.; Vega-Rioja, A.; del Carmen Fernandez-Carranco, M.; Hajji, N.; et al. The renin angiotensin system (RAS) mediates bifunctional growth regulation in melanoma and is a novel target for therapeutic intervention. Oncogene 2019, 38, 2320–2336. [Google Scholar] [CrossRef]
- Magnani, F.; Pappas, C.G.; Crook, T.; Magafa, V.; Cordopatis, P.; Ishiguro, S.; Ohta, N.; Selent, J.; Bosnyak, S.; Jones, E.S.; et al. Electronic Sculpting of Ligand-GPCR Subtype Selectivity: The Case of Angiotensin II. ACS Chem. Biol. 2014, 9, 1420–1425. [Google Scholar] [CrossRef] [PubMed]
- Vrettos, E.I.; Valverde, I.E.; Mascarin, A.; Pallier, P.N.; Cerofolini, L.; Fragai, M.; Parigi, G.; Hirmiz, B.; Bekas, N.; Grob, N.M.; et al. Single peptide backbone surrogate mutations to regulate angiotensin GPCR subtype selectivity. Chem.–Eur. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Coy, D.H.; Heinz-Erian, P.; Jiang, N.Y.; Sasaki, Y.; Taylor, J.; Moreau, J.P.; Wolfrey, W.T.; Gardner, J.D.; Jensen, R.T. Probing peptide backbone function in bombesin. A reduced peptide bond analogue with potent and specific receptor antagonist activity. J. Biol. Chem. 1988, 263, 5056–5060. [Google Scholar]
- Horwell, D.C.; Howson, W.; Naylor, D.; Osborne, S.; Pinnock, R.D.; Ratcliffe, G.S.; Suman-Chauhan, N. Alanine scan and N-methyl amide derivatives of Ac-bombesin[7-14]. Development of a proposed binding conformation at the neuromedin B (NMB) and gastrin releasing peptide (GRP) receptors. Int. J. Pept. Protein Res. 1996, 48, 522–531. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Sequence | t1/2 [h] b | Ki [nM] |
|---|---|---|---|
| Multi-Leu a | Ac-Leu-Leu-Leu-Leu-Arg-Val-Lys-Arg-NH2 | 2.1 ± 0.2 | 38 |
| P1 | Ac-Leu-LeuΨ[Tz]Leu-Leu-Arg-Val-Lys-Arg-NH2 | 1.0 ± 0.2 | 600 |
| P2 | Ac-LeuΨ[Tz]Leu-Leu-Leu-Arg-Val-Lys-Arg-NH2 | 4.0 ± 0.5 | 37 |
| Compound | Sequence | Ki [nM] b |
|---|---|---|
| Leu-Enk a | Tyr-Gly-Gly-Phe-Leu | 6.3 ± 0.9 |
| Enk1 | Try-Gly-Gly-PheΨ[Tz]Leu | 89 ± 12 |
| Enk2 | Try-Gly-GlyΨ[Tz]Phe-Leu | 460 ± 250 |
| Enk3 | Try-GlyΨ[Tz]Gly-Phe-Leu | >1000 |
| Enk4 | TryΨ[Tz]Gly-Gly-Phe-Leu | >1000 |
| Comp. | Sequence | t1/2 [h] b | Uptake after 4 h [%] c | KD [nM] d |
|---|---|---|---|---|
| BBN1 a | [177Lu]Lu-DOTA-PEG4-Gln-Trp-Ala-Val-Gly-His-Leu-Nle-NH2 | 5 | 27.7 | 2.0 ± 0.6 |
| BBN2 | [177Lu]Lu-DOTA-PEG4-Gln-Trp-Ala-Val-Gly-His-Leu-NleΨ[Tz]H | 6 | 29.1 | 3.0 ± 0.5 |
| BBN3 | [177Lu]Lu-DOTA-PEG4-Gln-Trp-Ala-Val-Gly-His-LeuΨ[Tz]Nle-NH2 | 60 | 0.2 | n.d. |
| BBN4 | [177Lu]Lu-DOTA-PEG4-Gln-Trp-Ala-Val-Gly-HisΨ[Tz]Leu-Nle-NH2 | >100 | n.o. | n.d. |
| BBN5 | [177Lu]Lu-DOTA-PEG4-Gln-Trp-Ala-Val-GlyΨ[Tz]His-Leu-Nle-NH2 | 17 | 28.3 | 3.1 ± 1.0 |
| BBN6 | [177Lu]Lu-DOTA-PEG4-Gln-Trp-Ala-ValΨ[Tz]Gly-His-Leu-Nle-NH2 | 25 | 8.4 | 48.6 ± 11.5 |
| BBN7 | [177Lu]Lu-DOTA-PEG4-Gln-Trp-AlaΨ[Tz]Val-Gly-His-Leu-Nle-NH2 | 16 | 24.5 | 5.9 ± 1.8 |
| BBN8 | [177Lu]Lu-DOTA-PEG4-Gln-TrpΨ[Tz]Ala-Val-Gly-His-Leu-Nle-NH2 | 8 | n.o. | n.d. |
| BBN9 | [177Lu]Lu-DOTA-PEG4-GlnΨ[Tz]Trp-Ala-Val-Gly-His-Leu-Nle-NH2 | 14 | n.o. | n.d. |
| BBN10 | [177Lu]Lu-DOTA-PEG4Ψ[Tz]Gln-Trp-Ala-Val-Gly-His-Leu-Nle-NH2 | 5 | 0.5 | n.d. |
| BBN11 | [177Lu]Lu-DOTA-PEG4-Gln-Trp-AlaΨ[Tz]Val-GlyΨ[Tz]His-Leu-Nle-NH2 | 27 | 21.7 ± 0.2 | 25.6 ± 6.9 |
| BBN12 | [177Lu]Lu-DOTA-PEG4-Gln-Trp-Ala-ValΨ[Tz]GlyΨ[Tz]His-Leu-Nle-NH2 | 40 | 3.5 ± 0.6 | >1000 |
| BBN13 | [177Lu]Lu-DOTA-PEG4-Gln-Trp-AlaΨ[Tz]ValΨ[Tz]Gly-His-Leu-Nle-NH2 | 66 | 0.1 ± 0.1 | >1000 |
| BBN14 | [177Lu]Lu-DOTA-PEG4-Gln-Trp-AlaΨ[Tz]ValΨ[Tz]GlyΨ[Tz]His-Leu-Nle-NH2 | 61 | 0.3 ± 0.1 | >1000 |
| Comp. | Sequence b | Stability after 48 h [%] c,d | Uptake after 4 h [%] d,e | KD [nM] f |
|---|---|---|---|---|
| BBN15 a | [177Lu]Lu-DOTA-PEG4-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu | 65 | 30 | 2.7 |
| BBN16 | [177Lu]Lu-DOTA-PEG4-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-LeuΨ[Tz]H | 75 | 13 | 8.1 |
| BBN17 | [177Lu]Lu-DOTA-PEG4-d-Phe-Gln-Trp-Ala-Val-GlyΨ[Tz]His-Sta-Leu | n.d. | n.d. | >1000 |
| BBN18 | [177Lu]Lu-DOTA-PEG4-d-Phe-Gln-Trp-AlaΨ[Tz]Val-Gly-His-Sta-Leu | n.d. | n.d. | >1000 |
| Comp. | Sequence | Stability after 6 h [%] b,c | EC50 [nM] d |
|---|---|---|---|
| KP1 a | Tyr-Asn-Trp-Asn-Ser-Phe-Gly-Leu-Arg-Tyr-NH2 | n.d. | 2.5 ± 2.2 |
| KP2 | Ac-Tyr-Asn-Trp-Asn-Ser-Phe-Gly-Leu-Arg-Tyr-NH2 | 2.6 ± 0.4 | 0.07 ± 0.06 |
| KP3 | Ac-Tyr-Asn-Trp-Asn-Ser-Phe-GlyΨ[Tz]Leu-Arg-Tyr-NH2 | 40.8 ± 2.9 | 0.07 ± 0.06 |
| KP4 | Ac-Tyr-Asn-Trp-Asn-Ser-PheΨ[Tz]Gly-Leu-Arg-Tyr-NH2 | 61.4 ± 6.4 | 0.6 ± 0.05 |
| KP5 | Ac-Tyr-Asn-Trp-Asn-Ser-PheΨ[Tz]GlyΨ[Tz]Leu-Arg-Tyr-NH2 | 50.7 ± 3.2 | 120 ± 87 |
| Comp. | Sequence | Stability after 4 h [%] b (t1/2 in min) c | Uptake after 4 h [%] d | KD [nM] e |
|---|---|---|---|---|
| NT1 a | [177Lu]Lu-DOTA-PEG4-Arg-Arg-Pro-Tyr-Ile-Leu | 0.9 ± 0.3 (39.4) | 7.3. ± 0.4 | 3.7 ± 0.8 |
| NT2 | [177Lu]Lu-DOTA-PEG4-Arg-Arg-Pro-Tyr-Ile-LeuΨ[Tz]H | 21.3 ± 1.8 (69.7) | n.o. | n.d. |
| NT3 | [177Lu]Lu-DOTA-PEG4-Arg-Arg-Pro-Tyr-IleΨ[Tz]Leu | 30.0 ± 3.6 (72.0) | n.o. | n.d. |
| NT4 | [177Lu]Lu-DOTA-PEG4-Arg-Arg-Pro-TyrΨ[Tz]Ile-Leu | 46.1. ± 3.8 (164.0) | n.o. | n.d. |
| NT5 | [177Lu]Lu-DOTA-PEG4-Arg-Arg-ProΨ[Tz]Tyr-Ile-Leu | 0 (13.0) | n.o. | n.d. |
| NT6 | [177Lu]Lu-DOTA-PEG4-ArgΨ[Tz]Arg-Pro-Tyr-Ile-Leu | 6.5 ± 4.6 (64.9) | 6.4 ± 1.2 | 8.8 ± 1.7 |
| NT7 | [177Lu]Lu-DOTA-PEG4Ψ[Tz]Arg-Arg-Pro-Tyr-Ile-Leu | 2.2. ± 1.2 (46.9) | 9.4 ± 0.5 | 4.5 ± 0.8 |
| NT8 a | [177Lu]Lu-DOTA-PEG4-Arg-Arg-Pro-Tyr-Tle-Leu | 70.6. ± 1.4 (n.d.) | 1.3 ± 0.2 | 507 ± 114 |
| NT9 | [177Lu]Lu-DOTA-PEG4-ArgΨ[Tz]Arg-Pro-Tyr-Tle-Leu | 97.7 ± 2.3 (n.d.) | 2.1 ± 0.1 | 214 ± 45 |
| NT10 | [177Lu]Lu-DOTA-PEG4Ψ[Tz]Arg-Arg-Pro-Tyr-Tle-Leu | 94.7 ± 4.2 (n.d.) | 1.2 ± 0.2 | >1000 |
| NT11 | [177Lu]Lu-DOTA-PEG4Ψ[Tz]ArgΨ[Tz]Arg-Pro-Tyr-Ile-Leu | 0.2 ± 0.2 (13) | 10.8 ± 0.4 | 4.6 ± 2.3 |
| NT12 | [177Lu]Lu-DOTA-PEG4Ψ[Tz]ArgΨ[Tz]Arg-Pro-Tyr-Tle-Leu | 97.2 ± 3.1 (n.d.) | 2.2 ± 0.1 | >1000 |
| Comp. | Sequence a | Ki for CatS, pH 5.5 [nM] b | Ki for CatS, pH 7.4 [nM] b | Ki for CatK, pH 5.5 [nM] b | Ki for CatL, pH 5.5 [nM] b |
|---|---|---|---|---|---|
| CatS1 | Ac-Gly-Arg-Trp-His-Pro-Met-GlyΨ[Tz]Ala-Pro-Trp-Glu-D-Ala-D-Arg-NH2 | 15,000 ± 5000 | 42,000 ± 8000 | 10,000 ± 3600 | 30,000 ± 3200 |
| CatS2 | Ac-Gly-Arg-Trp-His-Pro-Met-azaGly-Ala-Pro-Trp-Glu-D-Ala-D-Arg-NH2 | 26 ± 5 | 17 ± 3 | 3 ± 0.8 | 5 ± 0.5 |
| CatK1 | Abz-Arg-Pro-Pro-GlyΨ[Tz]Phe-Ser-Pro-Phe-Arg-Tyr(3-NO2)-NH2 | n.o. | n.o. | 800 ± 330 | 22,000 ± 2000 |
| CatK2 | Abz-Arg-Pro-Pro-azaGly-Phe-Ser-Pro-Phe-Arg-Tyr(3-NO2)-NH2 | n.o. | n.o. | 9 ± 0.7 | 2000 ± 400 |
| Compound | Sequence b | Inhibition at 10 μM [%] c,d | IC50 [μM] d |
|---|---|---|---|
| A7R a | Ala-Thr-Trp-Lys-Pro-Pro-Arg | 61.0 ± 0.4 | 5.86 |
| NV1 | Lys(Har)-Pro-Ala-Arg a | n.d. | 0.3 |
| NV2 | Lys(Har)-GlyΨ[Tz]GlyΨ[Tz]Arg | 58.1 ± 2.1 | 8.39 |
| NV3 | d-Lys(Har)-GlyΨ[Tz]GlyΨ[Tz]Arg | 52.6 ± 1.3 | 10.22 |
| Compound | Sequence | t1/2 [h] b,d | Uptake after 4 h [%] b,e | IC50 [nM] c,f |
|---|---|---|---|---|
| [Nle15]MG11 a | Lu-DOTA-d-Glu-Ala-Tyr-Gly-Trp-Nle-Asp-Phe-NH2 | 3.9 (3.8–4.1) | 32.2 ± 3.2 | 15.4 (11.0–21.1) |
| MGN1 | Lu-DOTA-d-Glu-Ala-Tyr-Gly-Trp-Nle-Asp-PheΨ[Tz]H | 34.9 (31.3–39.3) | 2.5 ± 2.9 | 200.5 (164–245) |
| MGN2 | Lu-DOTA-d-Glu-Ala-Tyr-Gly-Trp-Nle-AspΨ[Tz]Phe-NH2 | 35.9 (32.2–40.3) | 0.1 ± 0.08 | > 50,000 |
| MGN3 | Lu-DOTA-d-Glu-Ala-Tyr-Gly-Trp-NleΨ[Tz]Asp-Phe-NH2 | 114.3 (96.5–139.7) | 0.2 ± 0.13 | > 50,000 |
| MGN4 | Lu-DOTA-d-Glu-Ala-Tyr-Gly-TrpΨ[Tz]Nle-Asp-Phe-NH2 | 349.8 (263–520) | 33.1 ± 1.9 | 25.4 (18.3–34.6) |
| MGN5 | Lu-DOTA-d-Glu-Ala-Tyr-GlyΨ[Tz]Trp-Nle-Asp-Phe-NH2 | 3.8 (3.6–4.0) | 41.7 ± 3.9 | 15.6 (12.3–19.7) |
| MGN6 | Lu-DOTA-d-Glu-Ala-TyrΨ[Tz]Gly-Trp-Nle-Asp-Phe-NH2 | 2.6 (2.5–2.7) | 54.3 ± 5.1 | 1.7 (1.3–2.3) |
| MGN7 | Lu-DOTA-d-Glu-AlaΨ[Tz]Tyr-Gly-Trp-Nle-Asp-Phe-NH2 | 51.4 (46.2–57.7) | 29.6 ±2.7 | 20.9 (17.0–25.7) |
| MGN8 | Lu-DOTA-d-GluΨ[Tz]Ala-Tyr-Gly-Trp-Nle-Asp-Phe-NH2 | 7.7 (7.3–8.1) | 39.6 ± 2.7 | 8.0 (6.3–10.9) |
| MGN9 | Lu-DOTA-d-Glu-Ala-TyrΨ[Tz]Gly-TrpΨ[Tz]Nle-Asp-Phe-NH2 | 279.5 (206–431) | 28.2 ± 3.0 | 65.8 (53.2–80.9) |
| MGN10 | Lu-DOTA-d-Glu-Ala-TyrΨ[Tz]GlyΨ[Tz]Trp-Nle-Asp-Phe-NH2 | 1.9 (1.85–2.04) | 49.6 ± 3.0 | 12.4 (10.9–14.0) |
| MGN11 | Lu-DOTA-d-Glu-AlaΨ[Tz]Tyr-Gly-TrpΨ[Tz]Nle-Asp-Phe-NH2 | 386.1 (249–842) | 31.1 ± 3.9 | 91.0 (76.3–108.3) |
| MGN12 | Lu-DOTA-d-Glu-AlaΨ[Tz]TyrΨ[Tz]Gly-Trp-Nle-Asp-Phe-NH2 | 4.1 (3.8–4.4) | 48.3 ± 2.2 | 5.3 (4.5–6.1) |
| MGN13 | Lu-DOTA-d-GluΨ[Tz]Ala-TyrΨ[Tz]Gly-Trp-Nle-Asp-Phe-NH2 | 14.7 (14.0–15.5) | 58.4 ± 3.5 | 5.8 (5.3–6.4) |
| MGN14 | Lu-DOTA-d-GluΨ[Tz]AlaΨ[Tz]Tyr-Gly-Trp-Nle-Asp-Phe-NH2 | 6.4 (5.8–7.2) | 40.9 ± 2.5 | 15.6 (12.6–19.1) |
| MGN15 | Lu-DOTA-d-GluΨ[Tz]AlaΨ[Tz]TyrΨ[Tz]Gly-Trp-Nle-Asp-Phe-NH2 | 8.1 (7.7–8.6) | 47.1 ± 1.6 | 7.3 (6.0–8.7) |
| Compound | Sequence | IC50 for AT2R [nM] b | AT2R/AT1R Selectivity c |
|---|---|---|---|
| All a | Asp-Arg-Val-Tyr-Ile-His-Pro-Phe | 0.12 ± 0.01 | 13.7 |
| [Tyr6]All | Asp-Arg-Val-Tyr-Ile-Tyr-Pro-Phe | 4.0 d | 18,000 d |
| All1 | Asp-Arg-Val-Tyr-IleΨ[Tz]Tyr-Pro-Phe | 1990 ± 88 | >5 |
| All2 | Asp-Arg-Val-TyrΨ[Tz]Ile-Tyr-Pro-Phe | 2.8 ± 0.08 | >3611 |
| All3 | Asp-Arg-ValΨ[Tz]Tyr-Ile-Tyr-Pro-Phe | 155 ± 6 | >64 |
| All4 | Asp-ArgΨ[Tz]Val-Tyr-Ile-Tyr-Pro-Phe | 7.5 ± 0.05 | >1336 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rečnik, L.-M.; Kandioller, W.; Mindt, T.L. 1,4-Disubstituted 1,2,3-Triazoles as Amide Bond Surrogates for the Stabilisation of Linear Peptides with Biological Activity. Molecules 2020, 25, 3576. https://doi.org/10.3390/molecules25163576
Rečnik L-M, Kandioller W, Mindt TL. 1,4-Disubstituted 1,2,3-Triazoles as Amide Bond Surrogates for the Stabilisation of Linear Peptides with Biological Activity. Molecules. 2020; 25(16):3576. https://doi.org/10.3390/molecules25163576
Chicago/Turabian StyleRečnik, Lisa-Maria, Wolfgang Kandioller, and Thomas L. Mindt. 2020. "1,4-Disubstituted 1,2,3-Triazoles as Amide Bond Surrogates for the Stabilisation of Linear Peptides with Biological Activity" Molecules 25, no. 16: 3576. https://doi.org/10.3390/molecules25163576
APA StyleRečnik, L.-M., Kandioller, W., & Mindt, T. L. (2020). 1,4-Disubstituted 1,2,3-Triazoles as Amide Bond Surrogates for the Stabilisation of Linear Peptides with Biological Activity. Molecules, 25(16), 3576. https://doi.org/10.3390/molecules25163576