
Mass Spectrometry and 1H-NMR Study of Schinopsis lorentzii (Quebracho) Tannins as a Source of Hypoglycemic and Antioxidant Principles


Abstract
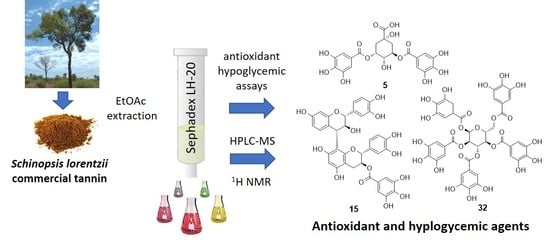
1. Introduction
2. Results and Discussion
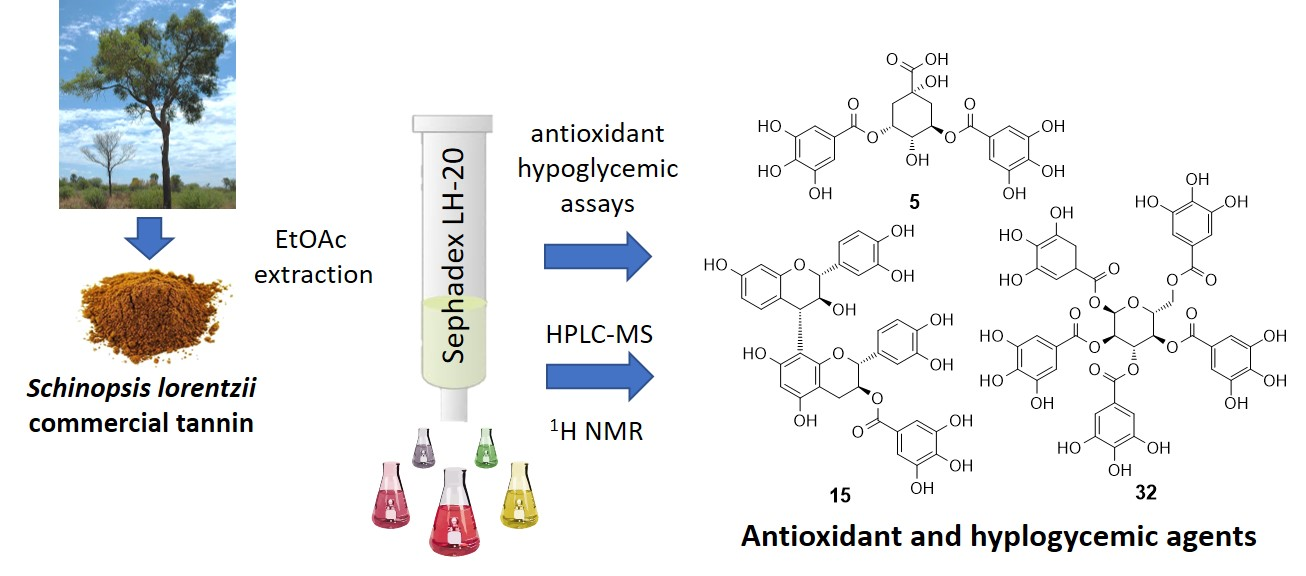
2.1. Extraction and Fractionation of Tan’Activ QS-SOL
2.2. Antioxidant Activity and α-Glucosidase and α-Amylase Inhibition for SL-T, SL-A and A-1–A-9 Fractions
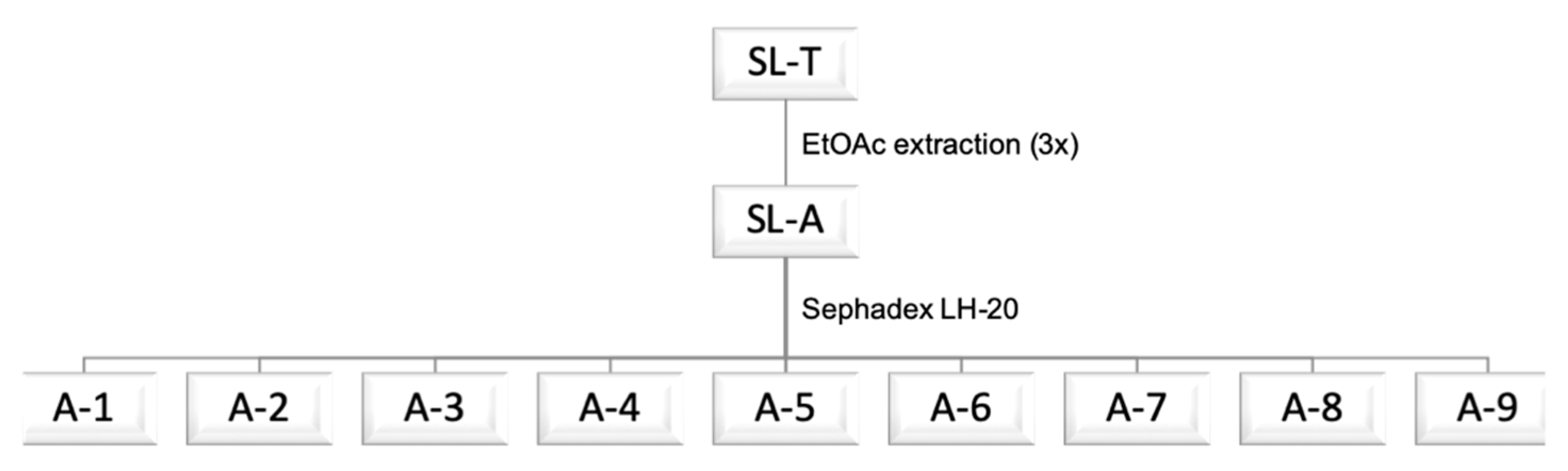
2.3. Correlation Analysis
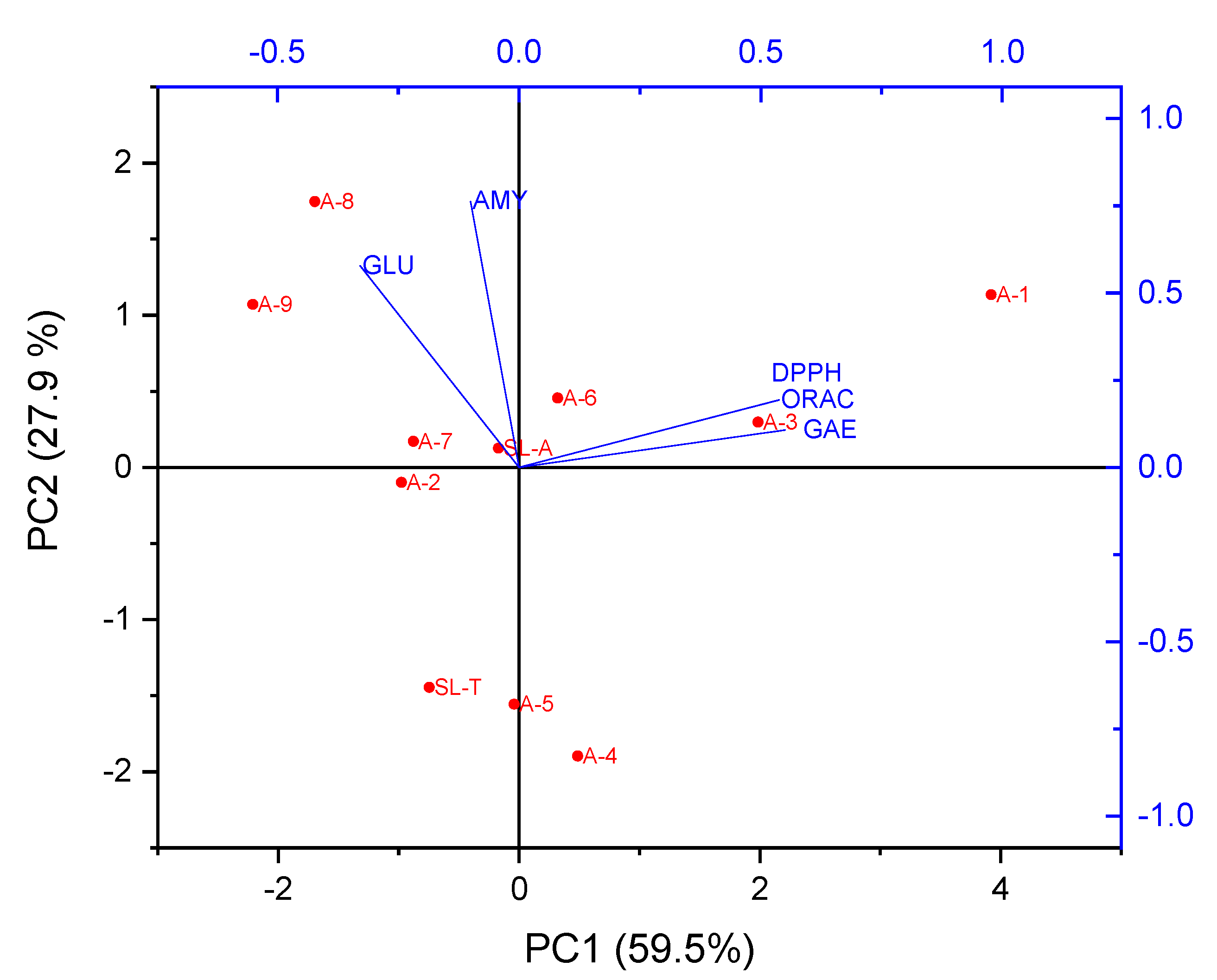
2.4. Principal Component Analysis (PCA)
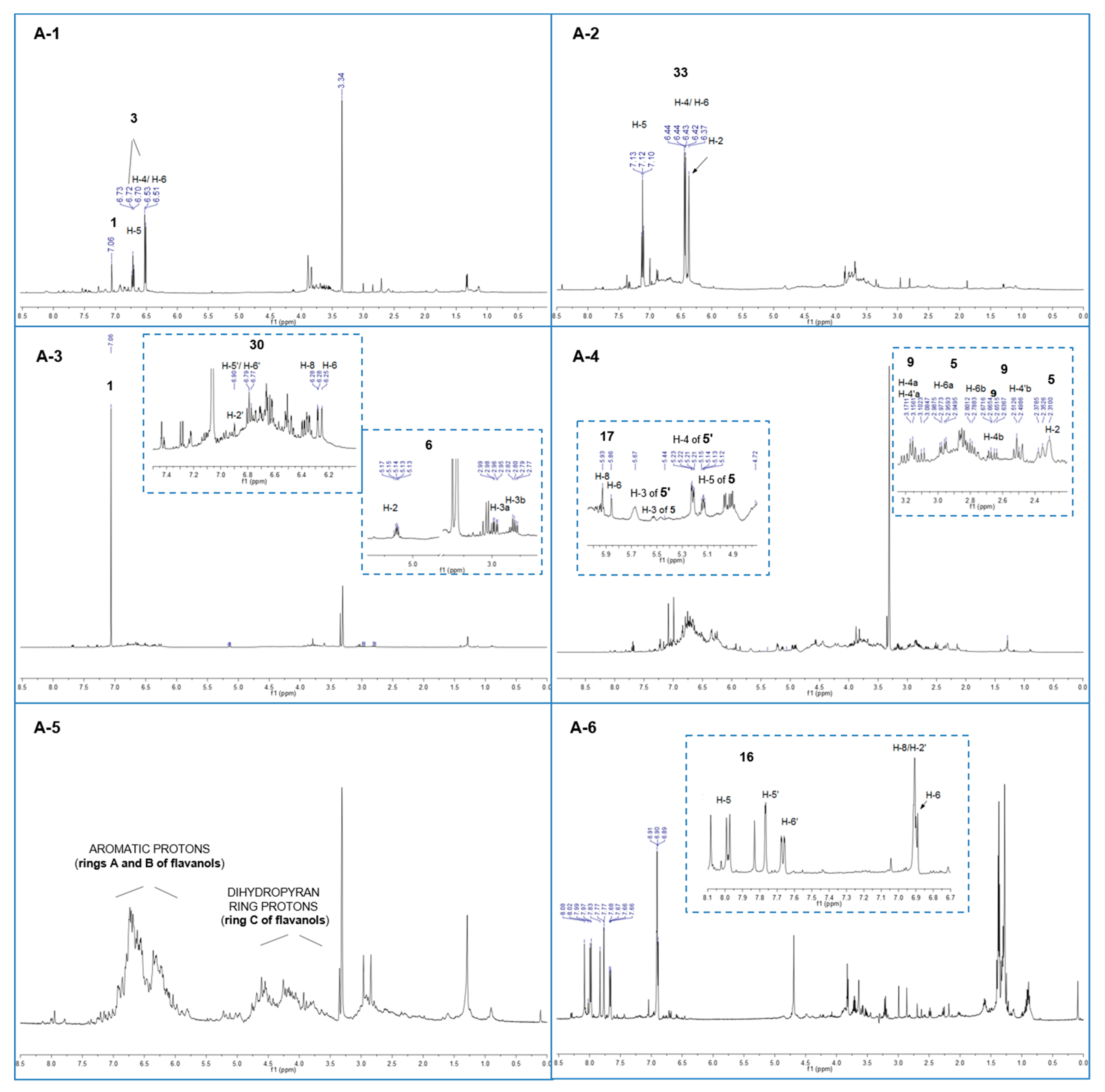
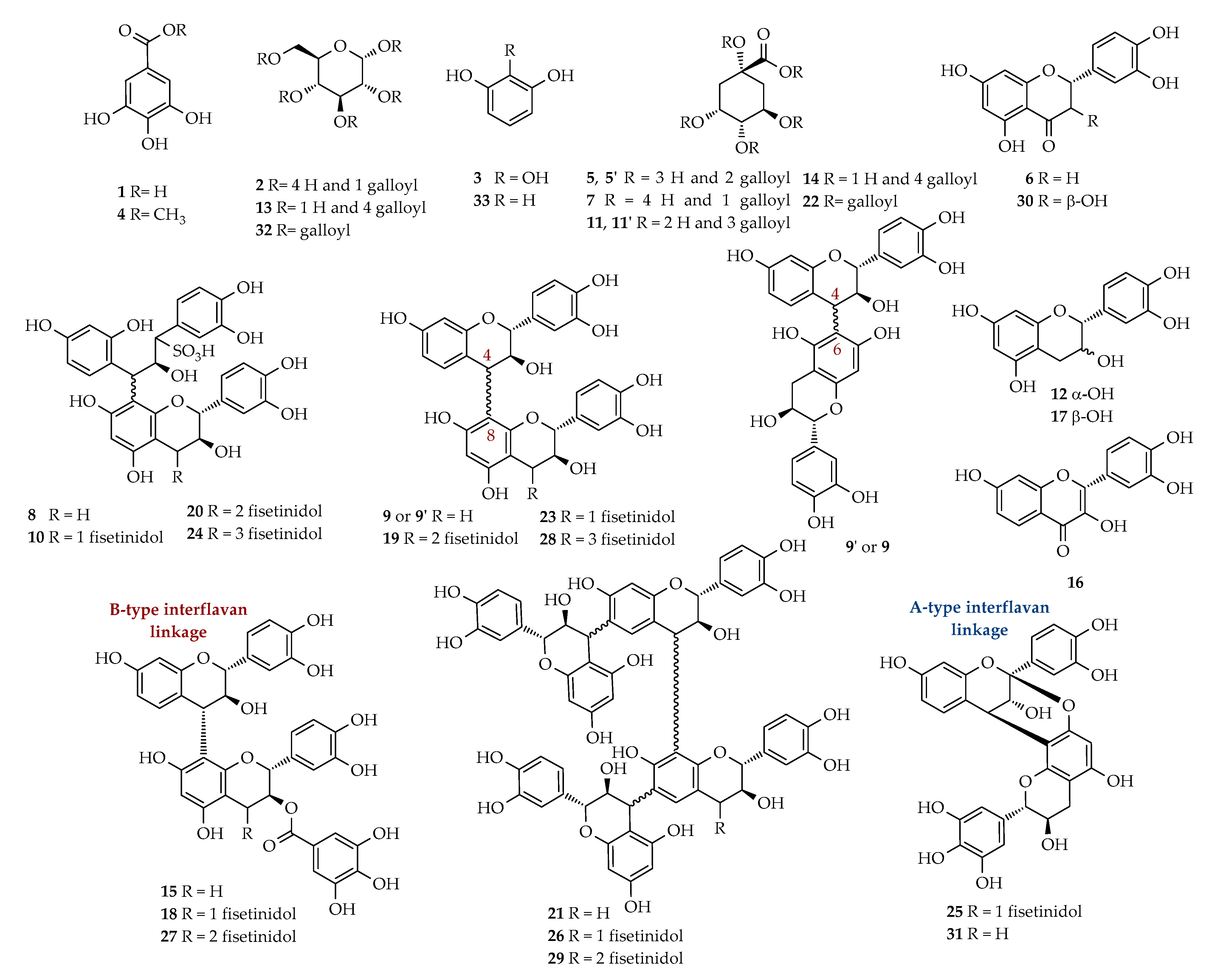
2.5. Mass Spectrometric and 1H-NMR Analysis of A-1–A-9 Fractions
3. Materials and Methods
3.1. Chemicals
3.2. HPLC/ESI-MS/MS Analysis
3.3. NMR Analysis
3.4. Preparation of Tannin Extract (CSE)
3.5. Sephadex LH-20 Fractionation
3.6. Determination of Total Phenols (GAE)
3.7. DPPH Radical Scavenging Activity Assay
3.8. Determination of Oxygen Radical Absorbance Capacity (ORAC)
3.9. Measurements of α-Glucosidase Inhibition
3.10. Measurements of α-Amylase Inhibition
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, T.V.; Prioletta, A.; Zuo, P.; Folli, F. Hyperglycemia-induced Oxidative Stress and its Role in Diabetes Mellitus Related Cardiovascular Diseases. Curr. Pharm. Des. 2013, 19, 5695–5703. [Google Scholar] [CrossRef]
- Chaudhury, A.; Duvoor, C.; Dendi, V.S.R.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical Review of Antidiabetic Drugs: Implications for Type 2 Diabetes Mellitus Management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef]
- Derosa, G.; Maffioli, P. alpha-Glucosidase inhibitors and their use in clinical practice. Arch. Med. Sci. 2012, 8, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Godbout, A.; Chiasson, J.L. Who should benefit from the use of alpha-glucosidase inhibitors? Curr. Diab. Rep. 2007, 7, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Tundis, R.; Loizzo, M.R.; Menichini, F. Natural Products as alpha-Amylase and alpha-Glucosidase Inhibitors and their Hypoglycaemic Potential in the Treatment of Diabetes: An Update. Mini-Rev. Med. Chem. 2010, 10, 315–331. [Google Scholar] [CrossRef]
- Qi, L.W.; Liu, E.H.; Chu, C.; Peng, Y.B.; Cai, H.X.; Li, P. Anti-Diabetic Agents from Natural Products-An Update from 2004 to 2009. Curr. Top. Med. Chem. 2010, 10, 434–457. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.Y.; Qian, K.D.; Morris-Natschke, S.L.; Hsu, C.S.; Lee, K.H. Recent discovery of plant-derived anti-diabetic natural products. Nat. Prod. Rep. 2012, 29, 580–606. [Google Scholar] [CrossRef]
- Cardullo, N.; Spatafora, C.; Musso, N.; Barresi, V.; Condorelli, D.; Tringali, C. Resveratrol-Related Polymethoxystilbene Glycosides: Synthesis, Antiproliferative Activity, and Glycosidase Inhibition. J. Nat. Prod. 2015, 78, 2675–2683. [Google Scholar] [CrossRef] [PubMed]
- Pulvirenti, L.; Muccilli, V.; Cardullo, N.; Spatafora, C.; Tringali, C. Chemoenzymatic Synthesis and alpha-Glucosidase Inhibitory Activity of Dimeric Neolignans Inspired by Magnolol. J. Nat. Prod. 2017, 80, 1648–1657. [Google Scholar] [CrossRef]
- Cardullo, N.; Catinella, G.; Floresta, G.; Muccilli, V.; Rosselli, S.; Rescifina, A.; Bruno, M.; Tringali, C. Synthesis of Rosmarinic Acid Amides as Antioxidative and Hypoglycemic Agents. J. Nat. Prod. 2019, 82, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Kasturi, S.; Surarapu, S.; Uppalanchi, S.; Anireddy, J.S.; Dwivedi, S.; Anantaraju, H.S.; Perumal, Y.; Sigalapalli, D.K.; Babu, B.N.; Ethiraj, K.S. Synthesis and alpha-glucosidase inhibition activity of dihydroxy pyrrolidines. Bioorg. Med. Chem. Lett. 2017, 27, 2818–2823. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.M.; Rahim, F.; Wadood, A.; Kosar, N.; Taha, M.; Lalani, S.; Khan, A.; Fakhri, M.I.; Junaid, M.; Rehman, W.; et al. Synthesis and molecular docking studies of potent alpha-glucosidase inhibitors based on biscoumarin skeleton. Eur. J. Med. Chem. 2014, 81, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Ajebli, M.; Eddouks, M. The Promising Role of Plant Tannins as Bioactive Antidiabetie Agents. Curr. Med. Chem. 2019, 26, 4852–4884. [Google Scholar] [CrossRef]
- Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouysegu, L. Plant Polyphenols: Chemical Properties, Biological Activities, and Synthesis. Angew. Chem. Int. Ed. 2011, 50, 586–621. [Google Scholar] [CrossRef] [PubMed]
- Cardullo, N.; Muccilli, V.; Pulvirenti, L.; Cornu, A.; Pouysegu, L.; Deffieux, D.; Quideau, S.; Tringali, C. C-glucosidic ellagitannins and galloylated glucoses as potential functional food ingredients with anti-diabetic properties: A study of alpha-glucosidase and alpha-amylase inhibition. Food Chem. 2020, 313. [Google Scholar] [CrossRef] [PubMed]
- Muccilli, V.; Cardullo, N.; Spatafora, C.; Cunsolo, V.; Tringali, C. alpha-Glucosidase inhibition and antioxidant activity of an oenological commercial tannin. Extraction, fractionation and analysis by HPLC/ESI-MS/MS and H-1 NMR. Food Chem. 2017, 215, 50–60. [Google Scholar] [CrossRef]
- Cardullo, N.; Muccilli, V.; Saletti, R.; Giovando, S.; Tringali, C. A mass spectrometry and H-1 NMR study of hypoglycemic and antioxidant principles from a Castanea sativa tannin employed in oenology. Food Chem. 2018, 268, 585–593. [Google Scholar] [CrossRef]
- European Union Law. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/animal-feed-eu-reg-comm_register_feed_additives_1831-03.pdf (accessed on 16 July 2020).
- Yanagida, A.; Shoji, T.; Shibusawa, Y. Separation of proanthocyanidins by degree of polymerization by means of size-exclusion chromatography and related techniques. J. Biochem. Biophys. Methods 2003, 56, 311–322. [Google Scholar] [CrossRef]
- Guyot, S.; Marnet, N.; Drilleau, J.F. Thiolysis-HPLC characterization of apple procyanidins covering a large range of polymerization states. J. Agric. Food Chem. 2001, 49, 14–20. [Google Scholar] [CrossRef]
- Costamagna, M.S.; Zampini, I.C.; Alberto, M.R.; Cuello, S.; Torres, S.; Perez, J.; Quispe, C.; Schmeda-Hirschmann, G.; Isla, M.I. Polyphenols rich fraction from Geoffroea decorticans fruits flour affects key enzymes involved in metabolic syndrome, oxidative stress and inflammatory process. Food Chem. 2016, 190, 392–402. [Google Scholar] [CrossRef]
- Venter, P.B.; Senekal, N.D.; Kemp, G.; Amra-Jordaan, M.; Khan, P.; Bonnet, S.L.; van der Westhuizen, J.H. Analysis of commercial proanthocyanidins. Part 3: The chemical composition of wattle (Acacia mearnsii) bark extract. Phytochemistry 2012, 83, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Boulekbache-Makhlouf, L.; Meudec, E.; Chibane, M.; Mazauric, J.P.; Slimani, S.; Henry, M.; Cheynier, V.; Madani, K. Analysis by High-Performance Liquid Chromatography Diode Array Detection Mass Spectrometry of Phenolic Compounds in Fruit of Eucalyptus globulus Cultivated in Algeria. J. Agric. Food Chem. 2010, 58, 12615–12624. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kwon, Y.S.; Chun, W.J.; Kim, T.Y.; Sun, J.H.; Yu, C.Y.; Kim, M.J. Rhus verniciflua Stokes flavonoid extracts have anti-oxidant, anti-microbial and alpha-glucosidase inhibitory effect. Food Chem. 2010, 120, 539–543. [Google Scholar] [CrossRef]
- Meyers, K.J.; Swiecki, T.J.; Mitchell, A.E. Understanding the native Californian diet: Identification of condensed and hydrolyzable Tannins in tanoak acorns (Lithocarpus densiflorus). J. Agric. Food Chem. 2006, 54, 7686–7691. [Google Scholar] [CrossRef] [PubMed]
- Falcao, L.; Araujo, M.E.M. Vegetable Tannins Used in the Manufacture of Historic Leathers. Molecules 2018, 23, 1081. [Google Scholar] [CrossRef]
- Venter, P.B.; Sisa, M.; van der Merwe, M.J.; Bonnet, S.L.; van der Westhuizen, J.H. Analysis of commercial proanthocyanidins. Part 1: The chemical composition of quebracho (Schinopsis lorentzii and Schinopsis balansae) heartwood extract. Phytochemistry 2012, 73, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.W.; Kelm, M.A.; Hammerstone, J.F.; Zhang, Z.; Beecher, G.; Holden, J.; Haytowitz, D.; Prior, R.L. Liquid chromatographic/electrospray ionization mass spectrometric studies of proanthocyanidins in foods. J. Mass Spectrom. 2003, 38, 1272–1280. [Google Scholar] [CrossRef]
- Hamed, A.I.; Al-Ayed, A.S.; Moldoch, J.; Piacente, S.; Oleszek, W.; Stochmal, A. Profiles analysis of proanthocyanidins in the argun nut (Medemia argun-an ancient Egyptian palm) by LC-ESI-MS/MS. J. Mass Spectrom. 2014, 49, 306–315. [Google Scholar] [CrossRef]
- Venter, P.B.; Senekal, N.D.; Amra-Jordaan, M.; Bonnet, S.L.; Van der Westhuizen, J.H. Analysis of commercial proanthocyanidins. Part 2: An electrospray mass spectrometry investigation into the chemical composition of sulfited quebracho (Schinopsis lorentzii and Schinopsis balansae) heartwood extract. Phytochemistry 2012, 78, 156–169. [Google Scholar] [CrossRef]
- Abraham, R.J.; Mobli, M. Modelling 1H-NMR Spectra of Organic Compounds:Theory, Applications and NMR Prediction Software; Wiley: Chichester, UK, 2008. [Google Scholar]
- Pizzi, A.; Daling, G.M.E. Laminating Wood Adhesives by Generation of Resorcinol From Tannin Extracts. J. Appl. Polym. Sci. 1980, 25, 1039–1048. [Google Scholar] [CrossRef]
- Li, H.J.; Deinzer, M.L. Tandem mass spectrometry for sequencing proanthocyanidins. Anal. Chem. 2007, 79, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.L.; Li, X.; Saleri, F.; Guo, M.Q. Analysis of Flavonoids in Rhamnus davurica and Its Antiproliferative Activities. Molecules 2016, 21, 1275. [Google Scholar] [CrossRef] [PubMed]
- Rossato, M.F.; Trevisan, G.; Walker, C.I.B.; Klafke, J.Z.; de Oliveira, A.P.; Villarinho, J.G.; Zanon, R.B.; Royes, L.F.F.; Athayde, M.L.; Gomez, M.V.; et al. Eriodictyol: A flavonoid antagonist of the TRPV1 receptor with antioxidant activity. Biochem. Pharmacol. 2011, 81, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Gottumukkala, R.V.; Nadimpalli, N.; Sukala, K.; Subbaraju, G.V. Determination of Catechin and Epicatechin Content in Chocolates by High-Performance Liquid Chromatography. Int. Sch. Res. Notices. 2014, 2014, 628196. [Google Scholar] [CrossRef]
- Naeimi, A.F.; Alizadeh, M. Antioxidant properties of the flavonoid fisetin: An updated review of in vivo and in vitro studies. Trends Food Sci. Tech. 2017, 70, 34–44. [Google Scholar] [CrossRef]
- Goncalves, R.; Mateus, N.; de Freitas, V. Inhibition of alpha-amylase activity by condensed tannins. Food Chem. 2011, 125, 665–672. [Google Scholar] [CrossRef]
- Ma, C.M.; Sato, N.; Li, X.Y.; Nakamura, N.; Hattori, M. Flavan-3-ol contents, anti-oxidative and alpha-glucosidase inhibitory activities of Cynomorium songaricum. Food Chem. 2010, 118, 116–119. [Google Scholar] [CrossRef]
- Spatafora, C.; Barbagallo, E.; Amico, V.; Tringali, C. Grape stems from Sicilian Vitis vinifera cultivars as a source of polyphenol-enriched fractions with enhanced antioxidant activity. Lwt-Food Sci. Technol. 2013, 54, 542–548. [Google Scholar] [CrossRef]
- Amico, V.; Chillemi, R.; Mangiafico, S.; Spatafora, C.; Tringali, C. Polyphenol-enriched fractions from Sicilian grape pomace: HPLC-DAD analysis and antioxidant activity. Bioresour. Technol. 2008, 99, 5960–5966. [Google Scholar] [CrossRef]
- Gao, H.; Huang, Y.N.; Gao, B.; Xu, P.Y.; Inagaki, C.; Kawabata, J. alpha-Glucosidase inhibitory effect by the flower buds of Tussilago farfara L. Food Chemistry 2008, 106, 1195–1201. [Google Scholar] [CrossRef]
- de Sales, P.M.; de Souza, P.M.; Simeoni, L.A.; Magalhaes, P.D.; Silveira, D. alpha-Amylase Inhibitors: A Review of Raw Material and Isolated Compounds from Plant Source. J. Pharm. Pharm. Sci. 2012, 15, 141–183. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lian, G.Y.; Yu, B. Naturally Occurring Polyphenolic Glucosidase Inhibitors. Isr. J. Chem. 2015, 55, 268–284. [Google Scholar] [CrossRef]
Sample Availability: Samples of the fractions A-1–A-9 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Weight % | GAE (mg/g) 1 | DPPH (SC50 ± SD) 2 | ORAC (TE ± SD) 3 | α-GLU (IC50 ± SD) 4 | α-AMY (IC50 ± SD) 4 |
|---|---|---|---|---|---|---|
| SL-T | - | 303.5 ± 8.8 | 7.4 ± 0.8 | 1075.4 ± 71.6 | 48.9 ± 2.2 | 129.3 ± 13.0 |
| SL-A | 34.7 5 | 316.3 ± 10.4 | 5.5 ± 0.6 | 1410.7 ± 60.8 | 6.3 ± 0.3 | 86.1 ± 11.3 |
| A-1 | 3.9 6 | 867.5 ± 12.9 | 4.0 ± 0.6 | 3345.4 ± 36.3 | 14.5 ± 1.2 | 79.5 ± 10.4 |
| A-2 | 3.2 6 | 357.3 ± 16.4 | 7.7 ± 0.7 | 896.5 ± 11.6 | 47.2 ± 0.1 | 66.5 ± 8.9 |
| A-3 | 6.4 6 | 756.4 ± 15.3 | 4.9 ± 0.4 | 1895.1 ± 23.2 | 24.9 ± 1.6 | 81.8 ± 9.3 |
| A-4 | 4.7 6 | 467.3 ± 7.2 | 6.1 ± 1.8 | 1440.2 ± 16.9 | 31.6 ± 1.9 | 294.7 ± 15.9 |
| A-5 | 21.1 6 | 475.1 ± 8.5 | 6.0 ± 0.5 | 719.0 ± 36.7 | 22.4 ± 0.3 | 172.8 ± 13.9 |
| A-6 | 15.2 6 | 483.1 ± 19.5 | 5.3 ± 0.6 | 1224.9 ± 32.9 | 8.9 ± 1.0 | 72.5 ± 6.6 |
| A-7 | 31.1 6 | 388.2 ± 5.5 | 6.1 ± 0.3 | 793.5 ± 13.7 | 3.6 ± 0.2 | 93.6 ± 11.3 |
| A-8 | 5.6 6 | 316.7 ± 3.7 | 6.6 ± 0.5 | 936.2 ± 28.2 | 2.1 ± 0.8 | 64.2 ± 8.4 |
| A-9 | 8.8 6 | 279.0 ± 9.1 | 8.1 ± 0.6 | 653.6 ± 19.8 | 2.6 ± 0.3 | 66.6 ± 7.4 |
| Que | - | - | 3.9 ± 0.5 | 4.8 ± 0.3 7 | 5.5 ± 0.7 | 47.6 ± 6.1 |
| Aca | - | - | - | - | 97.2 ± 4.8 | 36.8 ± 9.3 |
| Coefficients of PC1 | Coefficients of PC2 | |
|---|---|---|
| GAE | 0.55081 | 0.10679 |
| DPPH | 0.53842 | 0.19330 |
| ORAC | 0.53726 | 0.19248 |
| α-GLU | −0.32863 | 0.57787 |
| α-AMY | −0.10031 | 0.76174 |
| tR (min) | Identification | Calcd mass | [M − H]− | MS/MS Fragments m/z (Relative intensity) | 1H-NMR Signals δ (multiplicity, J = Hz, assignment) | Fraction |
|---|---|---|---|---|---|---|
| 3.1 | Gallic acid (1) | 170 | 169 | 125 (100) | 7.06 (s, H-2/H-6) | A-1–A-3 |
| 3.2 | Monogalloylglucose isomer (2) | 332 | 331 | 271 (100); 241 (30); 169 (10) | A-1 | |
| 3.5 | Pyrogallol (3) 1 | 126 | 125 | - | 6.72 (t, J = 8.1, H-5), 6.52 J = 8.1, H-4/H-6) | A-1 |
| 20.0 | Gallic acid methyl ester (4) | 184 | 183 | A-9 | ||
| 21.1 | 3,5-digalloylquinic acid (5) 2 | 496 | 495 | 343 (100); 325 (50) | 5.53 (m, H-3), 5.14 (bdd, J = 7.6, 5.0, H-5); 2.95 (dd, J = 14.0, 5.0, H- 6a), 2.79 (dd, J = 14.0, 7.6, H-6b) | A-4 |
| 21.1 | 3,4-digalloylquinic acid (5′) 2 | 496 | 495 | 343 (100); 325 (50) | 5.68 (m, H-3), 5.22 (dd, J = 8.4, 2.8, H-4) 4.43 (m, H-5), 2.32 (m, H-2) | A-4 |
| 27.7 | Eriodictyol (6) | 288 | 287 | 269 (100). 163 (20) | 5.14 (dd, J = 6.7, 5.0, H-2), 2.97 (dd, J = 13.9, 6.7, H-3a), 2.80 (dd, J = 13.9, 5.0, H-3b) | A-3 |
| 28.0 | Monogalloylquinic acid isomer (7) | 344 | 343 | 325 (100). 191(40) | A-1 | |
| 28.3 | Dimer isomer (C-SF) (8) 3 | 644 | 643 | 561 (100;) 409(20;) 289(10) | A-1–A-3 | |
| 29.3 | Dimer isomer (C-F) (9) 2,3 | 562 | 561 | 543 (20); 541 (40); 409 (100); 289 (60); 271 (30) | 4.57 (d, 5.7 Hz, H-2 F), 4.45 (m, H-3 C), 4.00 (m, H-3 F), 3.09 (dd, J = 15.2, 8.7, H-4a C), 2.66 (dd, J = 15.2, 6.5, H-4b C) | A-1; A-4 |
| 29.7 | Trimer isomer (C-F-SF) (10) 3 | 916 | 915 | 833 (100); 681 (20); 561 (20); 289 (20) | A-1–A-3 | |
| 32.4 | Trigalloylquinic acid isomer (11) | 648 | 647 | 495 (100); 477 (20) | A-8 | |
| 37.0 | Dimer isomer (C-F) (9′) 2,3 | 562 | 561 | 543 (20); 541 (40); 409 (100); 289 (60) | A-5–A-8 | |
| 37.3 | Catechin (12) | 290 | 289 | 271(100); 245(10); 179 (30); 167 (90); 151 (5); 137 (5); 109 (5) | A-3 | |
| 37.5 | Tetragalloylglucose isomer (13) | 788 | 787 | 635 (20); 617 (100) | A-9 | |
| 38.2 | Tetragalloylquinic acid isomer (14) | 800 | 799 | 647 (100); 601 (10); 477 (5); 495 (5) | A-8; A-9 | |
| 39.2 | Trigalloylquinic acid isomer (11′) 2 | 648 | 647 | 495 (100); 477 (20) | A-5–A-7; A-9 | |
| 39.3 | Dimer isomer (CG-F) (15) 3 | 714 | 713 | 695 (10); 603 (10); 561 (100); 573 (40); 441 (100) | A-8; A-9 | |
| 40.3 | Fisetin (16) | 286 | 285 | 163 (100); 241 (20); 267 (30) | 7.98 (d, J = 9.4, H-5), 7.77 (d, J = 1.9, H-5′), 7.67 (dd, J = 8.5, 1.9, H-6′), 6.90 (bs, H-2′, H-8), 6.89 (d, J = 9.4, H-6) | A-6 |
| 40.4 | Epi-catechin (17) | 290 | 289 | 271(70); 245(100); 179 (30); 167 (60); 151 (5); 137 (5); 109 (5) | 5.93 (d, J = 1.9, H-8), 5.86 (d, J = 1.9, H-6), 4.67 (d, J = 5.6, H-2), 3.97 (m, H- 3) | A-4 |
| 40.5 | Trimer isomer (CG-F-F) (18) 3 | 986 | 985 | 831 (20); 749 (40); 697 (80); 679 (80); 577 (90); 561 (70); 529 (100) | A-8; A-9 | |
| 41.1 | Tetramer isomer (C-F-F-F) (19) 3 | 1106 | 1105 | 1086 (20); 995 (40); 953 (100); 935 (60); 833 (70); 561 (20) | A-5–A-9 | |
| 41.3 | Tetramer isomer (C-F-F-SF) (20) 3 | 1188 | 1187 | 1185 (100); 953 (30); 833 (10) | A-2 | |
| 41.4 | Tetramer isomer (C-C-F-F) (21) 1,3 | 1123 | 1122 | - | A-5; A-7; A-8 | |
| 42.0 | Pentagalloylquinic acid isomer (22) | 952 | 951 | 799 (100); 647 (10) | A-8 | |
| 42.1 | Trimer isomer (C-F-F) (23) 3 | 834 | 833 | 723 (10); 681 (95); 663 (50); 561 (100); 529 (75);289 (10) | A-5–A-9 | |
| 42.5 | Pentamer isomer (C-F-F-F-SF) (24) 2 | 1460 | 1459 | 1377 (100); 1225 (30); 1105 (30) | A-3 | |
| 43.3 | A-type trimer isomer (GC-F-F) (25) 2 | 848 | 847 | 737 (40); 695 (100); 575 (60) | A-5 | |
| 43.3 | Pentamer isomer (C-C-F-F-F) (26) 1,3 | 1395 | 1394 | - | A-5 | |
| 43.7 | Tetramer isomer (GC-F-F-F) (27) 1,3 | 1122 | 1121 | - | A-8 | |
| 45.0 | Pentamer isomer (C-F-F-F-F) (28) 3 | 1378 | 1377 | 1225 (100); 1207(50);1105 (50); 995 (30); 833 (20) | A-7 | |
| 45.1 | Hexamer isomer (C-C-F-F-F-F) (29) 1,3 | 1668 | 1667 | - | A-5 | |
| 46.2 | Taxifolin (30) | 304 | 303 | 285 (100); 259 (40);175 (70) | 6.90 (d, J = 1.9, H-2′), 6.79 (m, H-5′, H-6′), 6.28 (d, J = 1.9, H-8), 6.25 (d, J = 1.9, H-6) | A-3 |
| 48.1 | A-type bond dimer isomer (GC-F) (31) 3 | 576 | 575 | 533 (20); 467 (30); 437 (25); 425 (40); 409 (50); 289 (100) | A-5 | |
| 50.0 | Pentagalloylglucose isomer (32) | 940 | 939 | 787 (100); 769 (10) | A-9 | |
| - | Resorcinol (33) 4 | 7.12 (t, J = 8.2, H-5), 6.43 (dd, J = 8.2, 2.1, H-4/H-6), 6.37 (bt, J = 2.1, H-2) | A-2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardullo, N.; Muccilli, V.; Cunsolo, V.; Tringali, C. Mass Spectrometry and 1H-NMR Study of Schinopsis lorentzii (Quebracho) Tannins as a Source of Hypoglycemic and Antioxidant Principles. Molecules 2020, 25, 3257. https://doi.org/10.3390/molecules25143257
Cardullo N, Muccilli V, Cunsolo V, Tringali C. Mass Spectrometry and 1H-NMR Study of Schinopsis lorentzii (Quebracho) Tannins as a Source of Hypoglycemic and Antioxidant Principles. Molecules. 2020; 25(14):3257. https://doi.org/10.3390/molecules25143257
Chicago/Turabian StyleCardullo, Nunzio, Vera Muccilli, Vincenzo Cunsolo, and Corrado Tringali. 2020. "Mass Spectrometry and 1H-NMR Study of Schinopsis lorentzii (Quebracho) Tannins as a Source of Hypoglycemic and Antioxidant Principles" Molecules 25, no. 14: 3257. https://doi.org/10.3390/molecules25143257
APA StyleCardullo, N., Muccilli, V., Cunsolo, V., & Tringali, C. (2020). Mass Spectrometry and 1H-NMR Study of Schinopsis lorentzii (Quebracho) Tannins as a Source of Hypoglycemic and Antioxidant Principles. Molecules, 25(14), 3257. https://doi.org/10.3390/molecules25143257