
Pd(II)-Catalyzed C-H Acylation of (Hetero)arenes—Recent Advances



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
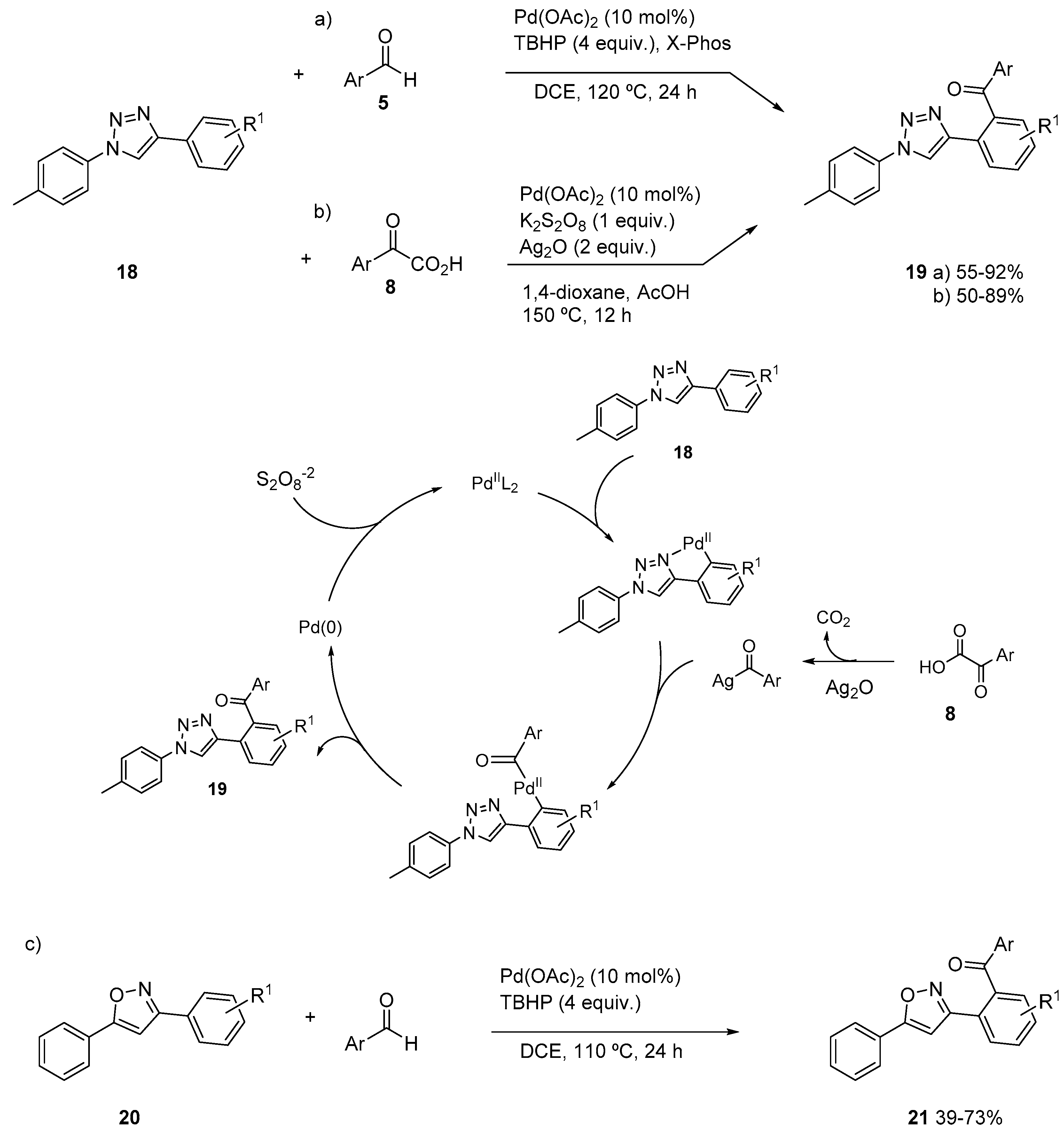{kind=link}
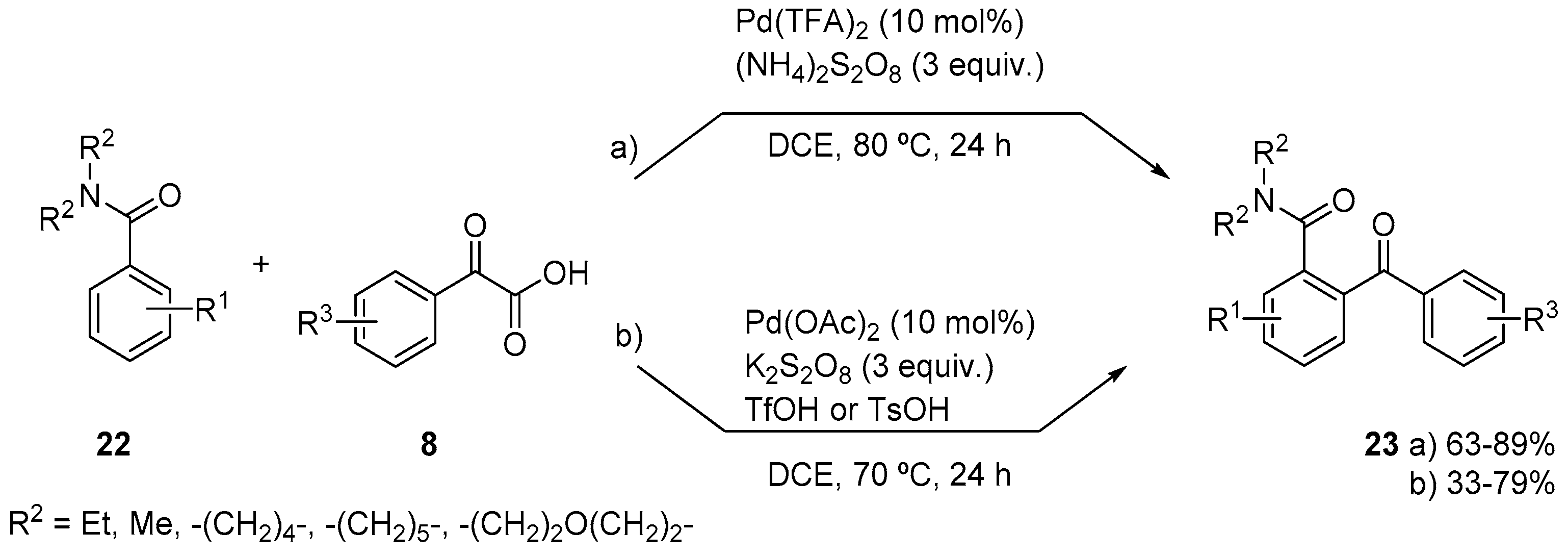{kind=link}
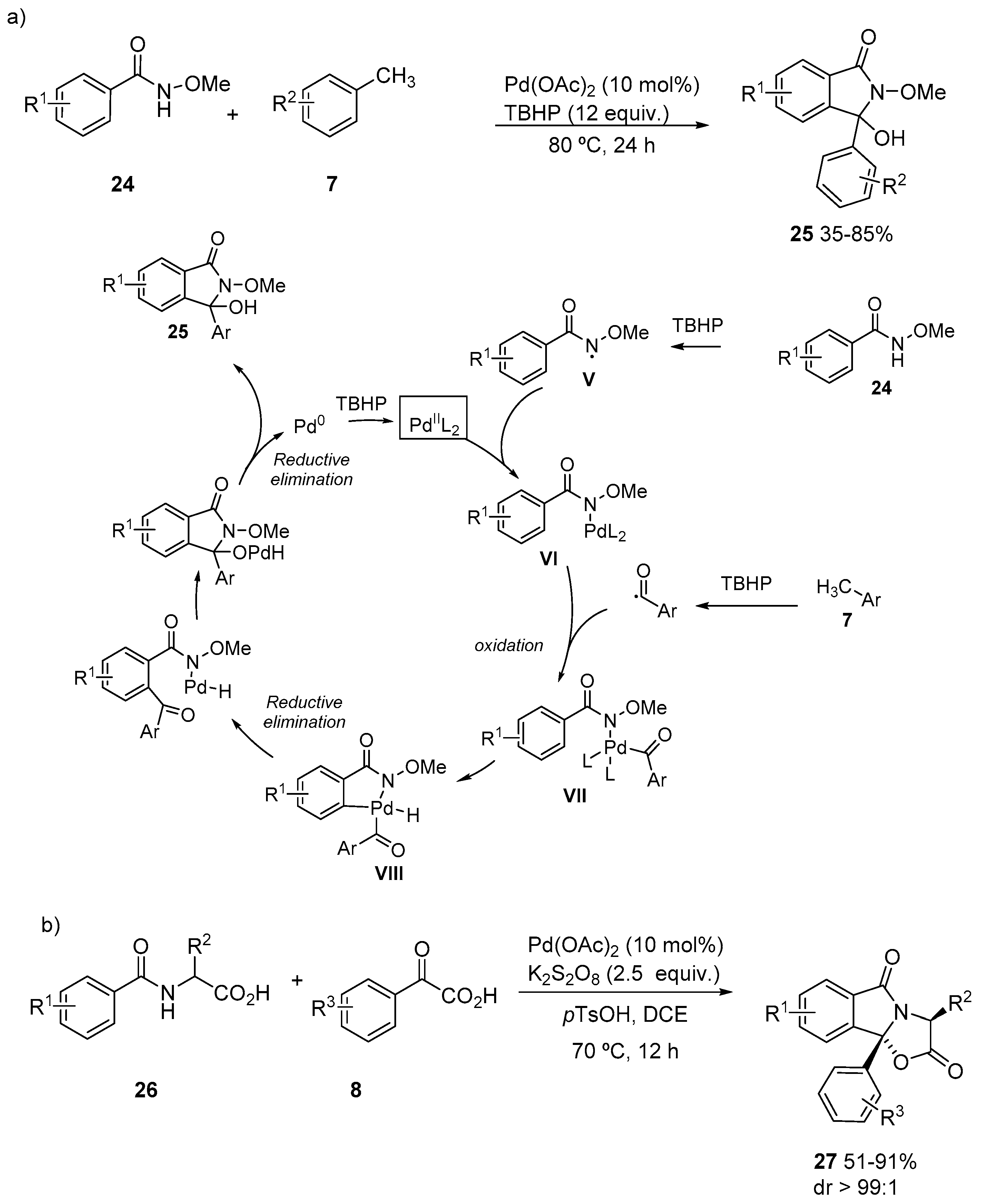{kind=link}
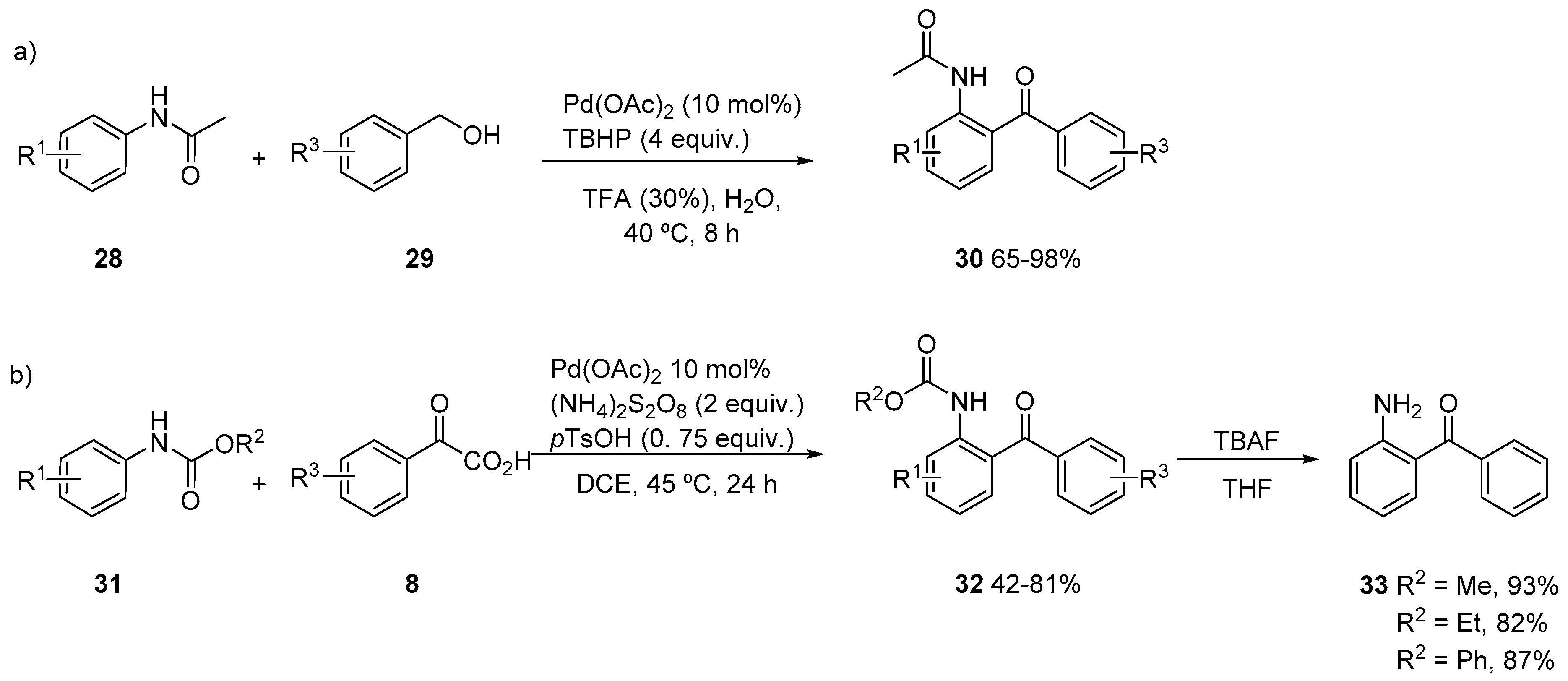{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
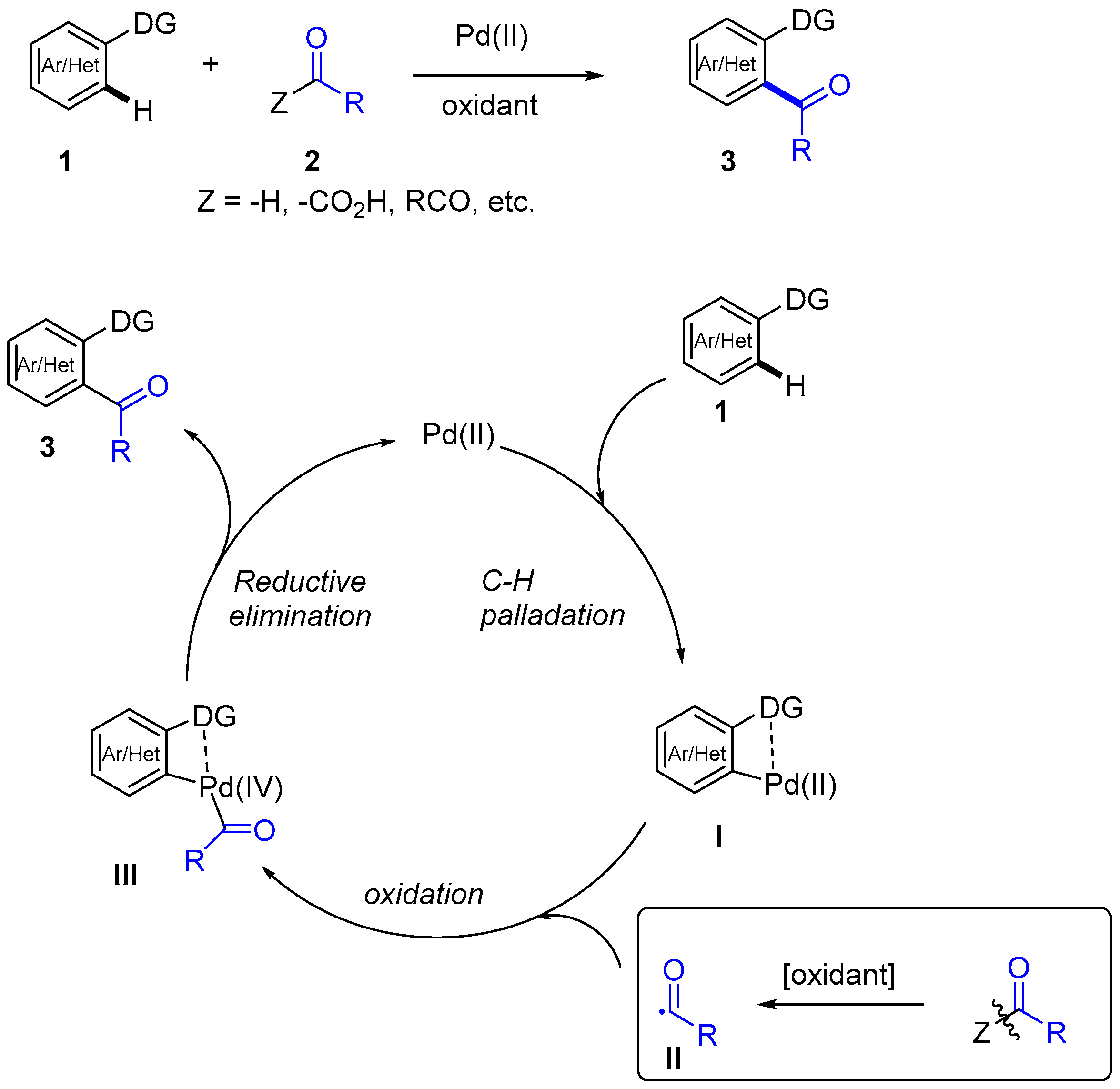
1. Introduction
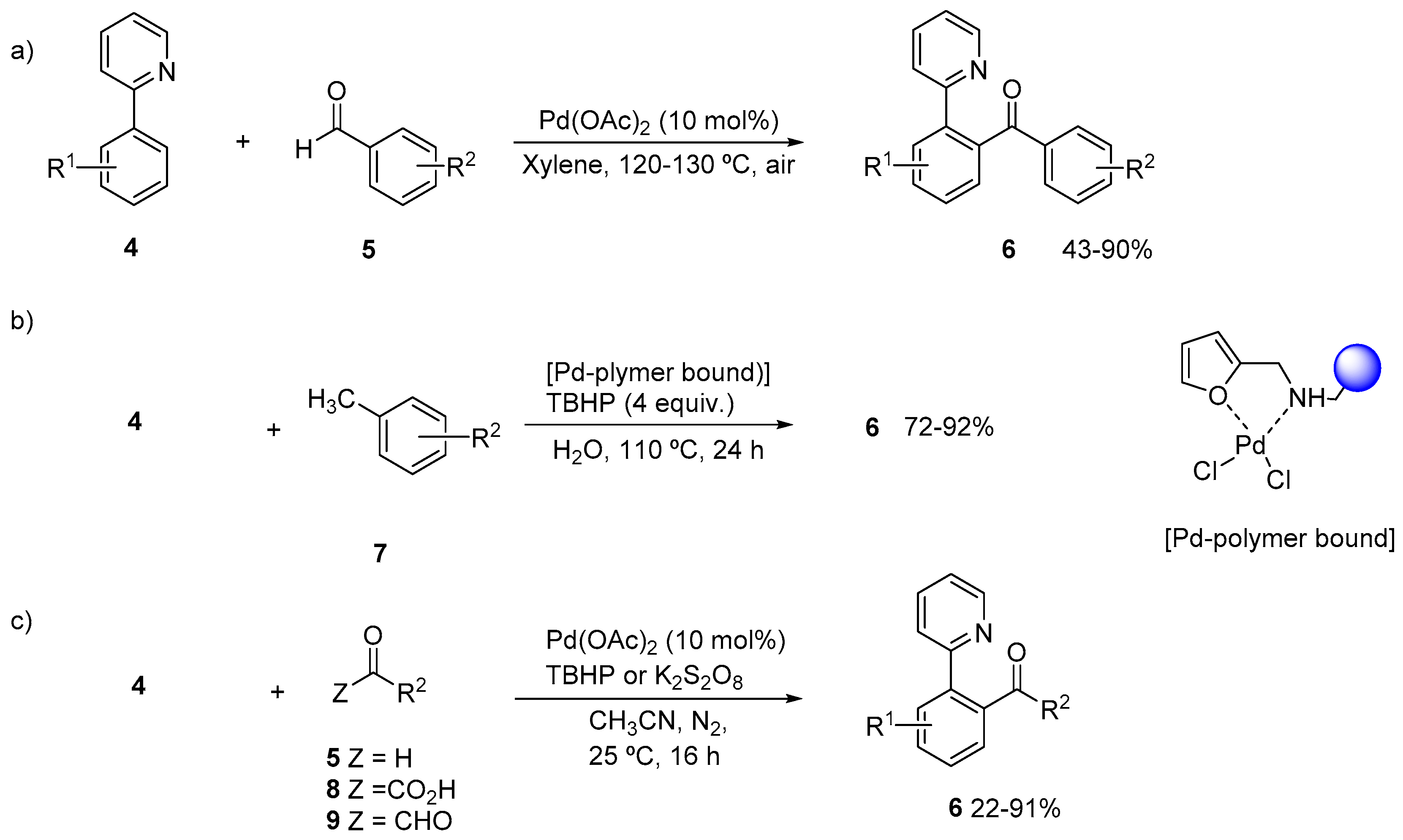
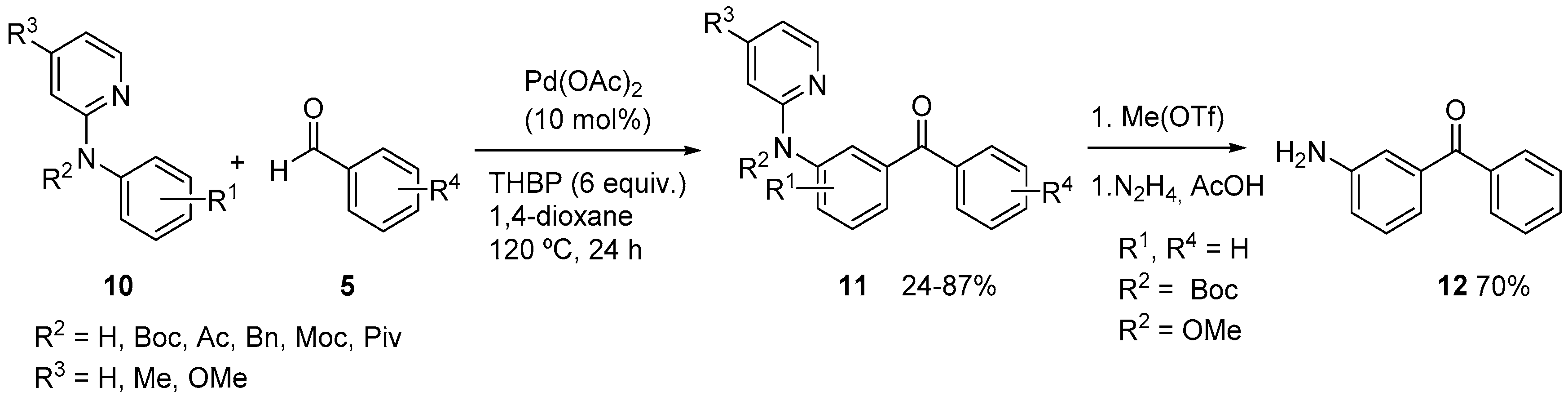
2. Acylation of Arenes
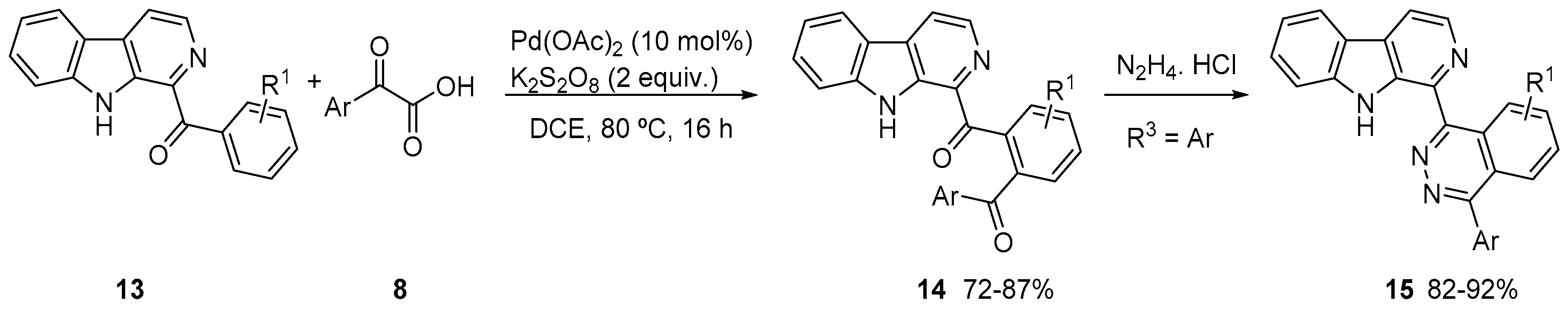
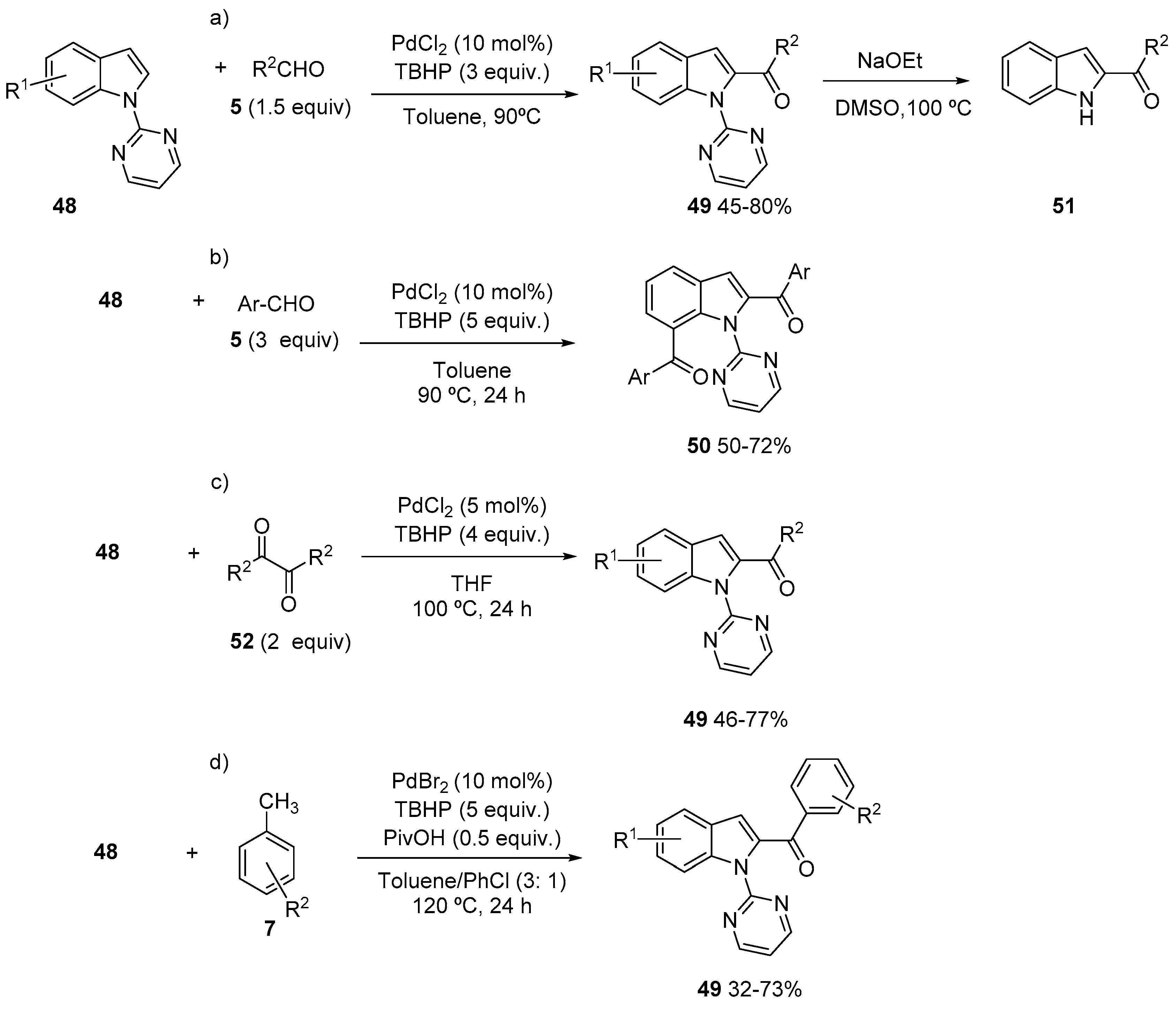
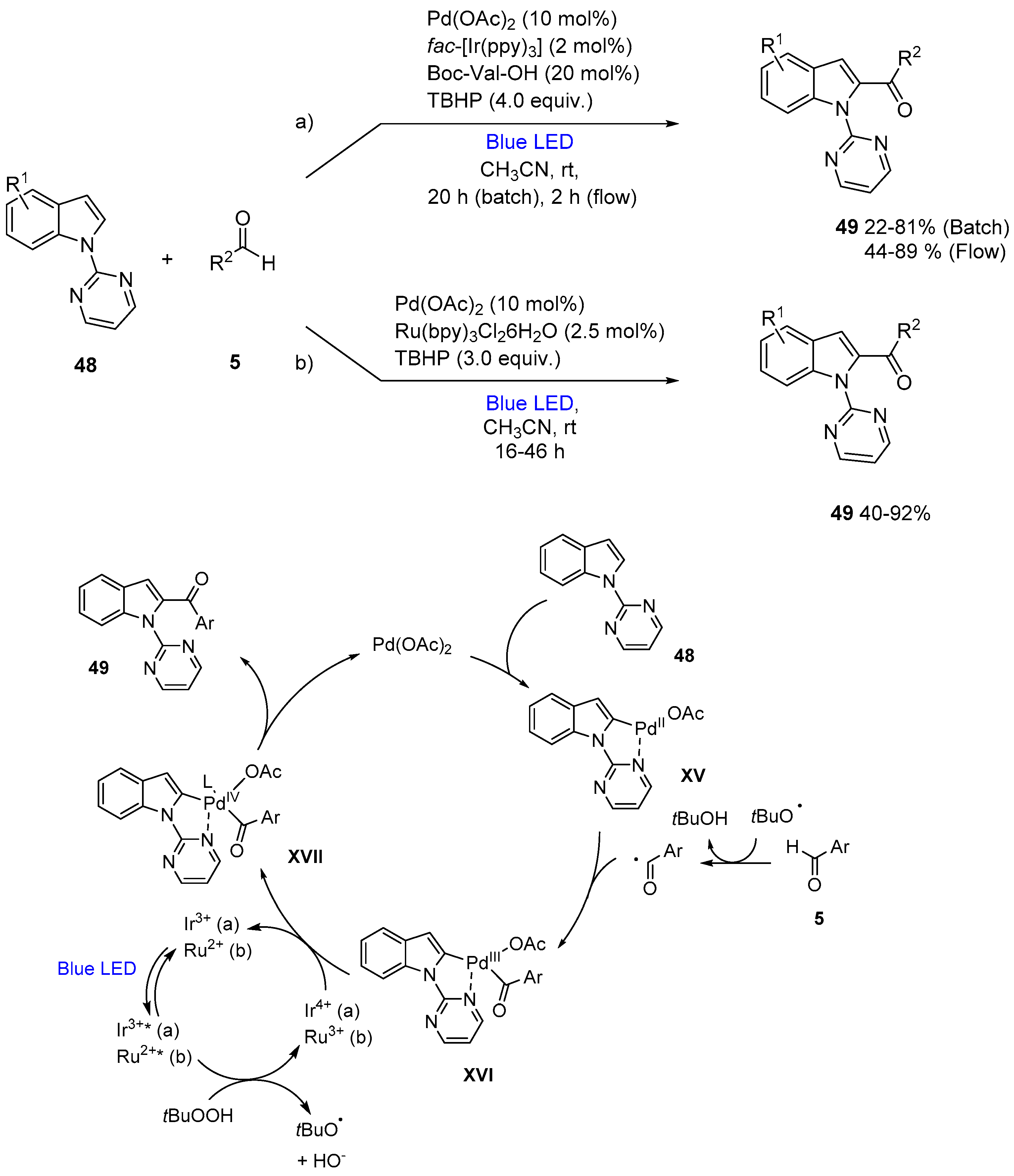
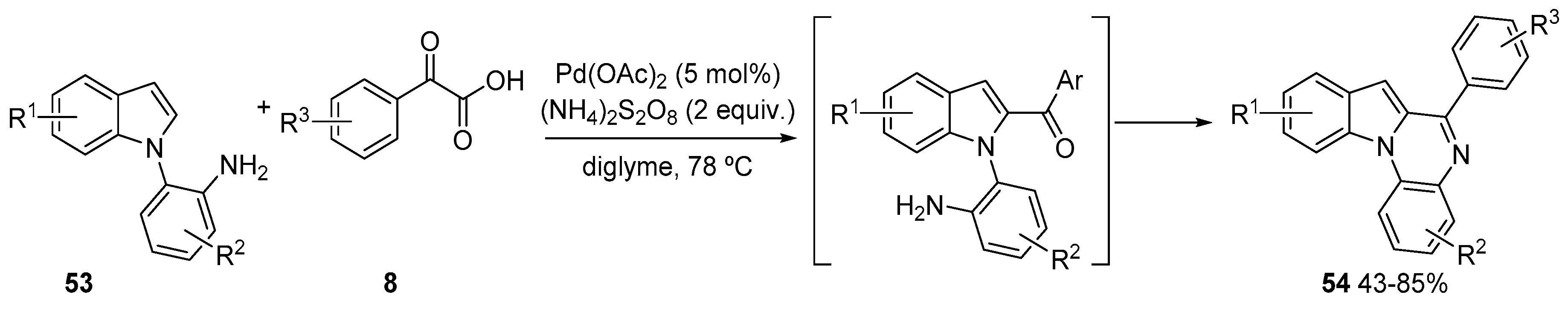
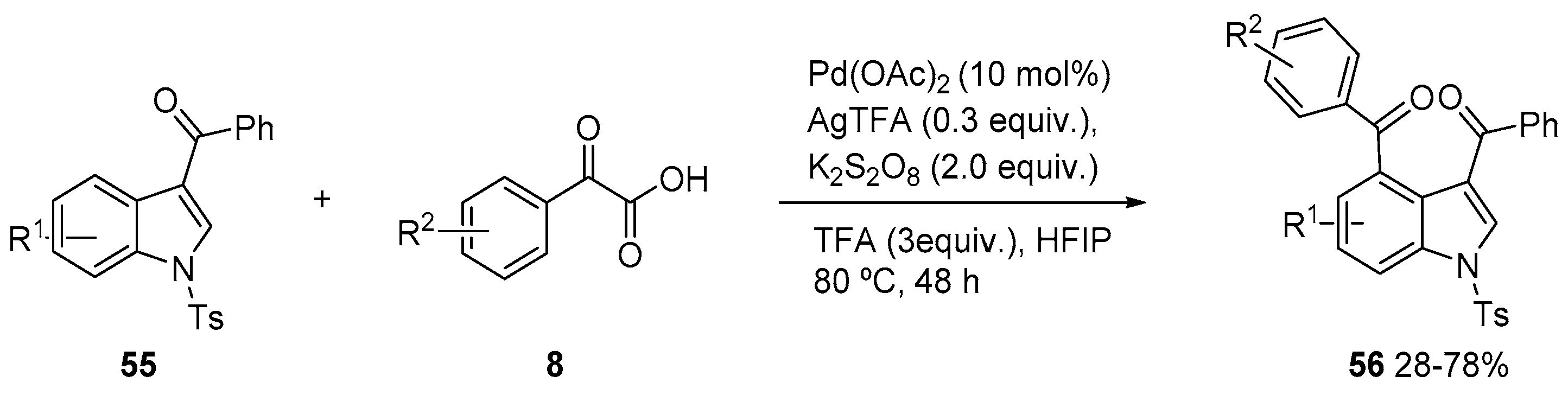
3. Acylation of Heteroarenes
4. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lei, A.; Shi, W.; Liu, W.; Zhang, H.; He, C. Oxidative Cross-Coupling Reactions; Wiley: Weinheim, Germany, 2017. [Google Scholar]
- Gensch, T.; Hopkinson, M.N.; Glorius, F.; Wencel-Delord, J. Mild Metal-catalyzed C–H Activation: Examples and Concepts. Chem. Soc. Rev. 2016, 45, 2900–2936. [Google Scholar] [CrossRef] [PubMed]
- Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia, M.F.; Wencel-Delord, J.; Besset, T.; et al. Comprehensive Overview of Directing Groups Applied in Metal-Catalysed C–H Functionalization Chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Zhang, S.; Wang, W.; Luo, F.; Cheng, J. Palladium-Catalyzed Acylation of sp2 C-H bond: Direct Access to Ketones from Aldehydes. Org. Lett. 2009, 11, 3120–3123. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Lei, Z.; Ngai, M.Y. Acyl Radical Chemistry via Visible-Light Photoredox Catalysis. Synthesis 2019, 51, 303–333. [Google Scholar] [CrossRef]
- Duan, P.; Yang, Y.; Ben, R.; Yan, Y.; Dai, l.; Hong, M.; Wu, Y.D.; Wang, D.; Zhang, X.; Zhao, J. Palladium-catalyzed Benzo[d]isoxazole Synthesis by C-H Activation/[4+1] Annulation. Chem. Sci. 2014, 5, 1574–1578. [Google Scholar] [CrossRef]
- Chu, J.H.; Chen, S.T.; Chiang, M.F.; Wu, M.J. Palladium Catalyzed Direct Ortho Aroylation of 2-Phenoxypyridines with Aldehydes and Catalytic Mechanistic Investigation. Organometallics 2015, 34, 953–966. [Google Scholar] [CrossRef]
- Wu, X.-F. Acylation of (Hetero)Arenes through C-H Activation with Aroyl Surrogates. Chem. Eur. J. 2015, 21, 12252–12265. [Google Scholar] [CrossRef]
- Hummel, J.R.; Boerth, J.A.; Ellman, J.A. Transition-Metal-Catalyzed C−H Bond Addition to Carbonyls, Imines and Related Polarized π Bonds. Chem. Rev. 2017, 117, 9163–9227. [Google Scholar] [CrossRef]
- Baslé, O.; Bidange, J.; Shuai, Q.; Li, C.J. Palladium-Catalyzed Oxidative sp2 CH Bond Acylation with Aldehydes. Adv. Synth. Catal. 2010, 352, 1145–1149. [Google Scholar] [CrossRef]
- Perumgani, C.P.; Parvathaneni, S.P.; Keesara, S.; Mandapati, M.R. Recyclable Pd(II) Complex Catalyzed Oxidative sp2 C-H Bond Acylation of 2-Aryl Pyridines with Toluene Derivatives. J. Organomet. Chem. 2016, 822, 189–195. [Google Scholar] [CrossRef]
- Li, M.; Ge, H. Decarboxylative Acylation of Arenes with α-Oxocarboxylic Acids via Palladium-Catalyzed C-H Activation. Org. Lett. 2010, 12, 3464–3467. [Google Scholar] [CrossRef] [PubMed]
- Hossian, A.; Manna, M.K.; Manna, K.; Jana, R. Palladium-catalyzed Decarboxylative, Decarbonylative and Dehydrogenative C(sp2)–H Acylation at Room Temperature. Org. Biomol. Chem. 2017, 15, 6592–6603. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.H.; Chiang, M.F.; Li, C.W.; Su, Z.H.; Lo, S.C.; Wu, M.J. Palladium Catalyzed Late Stage ortho-C-H Bond Aroylation of anilines using 4-Methoxy-2-pyridinyl as a Removable Directing Group. Organometallics 2019, 38, 2105–2119. [Google Scholar] [CrossRef]
- Kolle, S.; Batra, S. β-Carboline-directed Decarboxylative Acylation of Ortho-C(sp2)–H of the Aryl Ring of Aryl(β-carbolin-1-yl)methanones with α-Ketoacids under Palladium Catalysis. RSC Adv. 2016, 6, 50658–50665. [Google Scholar] [CrossRef]
- San Segundo, M.; Correa, A. Pd-catalyzed Site-selective C(sp2)–H Radical Acylation of Phenylalanine Containing Peptides with Aldehydes. Chem. Sci. 2019, 10, 8872–8879. [Google Scholar] [CrossRef]
- Wang, Z.; Tian, Q.; Yu, X.; Kuang, C. Palladium-Catalyzed Acylation of 2-Aryl-1,2,3-triazoles with Aldehydes. Adv. Synth. Catal. 2014, 356, 961–966. [Google Scholar] [CrossRef]
- Zhao, F.; Chen, Z.; Liu, Y.; Xie, K.; Jiang, Y. Palladium-Catalyzed Acylation of Arenes by 1,2,3-Triazole-Directed C–H Activation. Eur. J. Org. Chem. 2016, 5971–5979. [Google Scholar] [CrossRef]
- Ma, X.; Huang, H.; Yang, J.; Feng, X.; Xie, K. Palladium-Catalyzed Decarboxylative N-3-ortho-C–H Acylation of 1,4-Disubstituted 1,2,3-Triazoles with α-Oxocarboxylic Acids. Synthesis 2018, 50, 2567–2576. [Google Scholar] [CrossRef]
- Banerjee, A.; Bera, A.; Santra, S.K.; Guin, S.; Patel, B.K. Palladium-catalysed Regioselective Aroylation and Acetoxylation of 3,5-Diarylisoxazole via Ortho C–H Functionalisations. RSC Adv. 2014, 4, 8558–8566. [Google Scholar] [CrossRef]
- Snieckus, V. Directed Ortho Metalation. Tertiary Amide and O-carbamate Directors in Synthetic Strategies for Polysubstituted Aromatics. Chem. Rev. 1990, 90, 879–933. [Google Scholar] [CrossRef]
- Whisler, M.C.; MacNeil, S.; Snieckus, V.; Beak, P. Beyond Thermodynamic Acidity: A Perspective on the Complex-Induced Proximity Effect (CIPE) in Deprotonation Reactions. Angew. Chem. Int. Ed. 2004, 43, 2206–2225. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Park, E.; Kim, A.; Lee, Y.; Chi, K.W.; Kwak, J.H.; Jung, Y.H.; Kim, I.S. Rhodium-Catalyzed Oxidative ortho-Acylation of Benzamides with Aldehydes: Direct Functionalization of the sp2 C–H Bond. Org. Lett. 2011, 13, 4390–4393. [Google Scholar] [CrossRef]
- Meng, G.; Shi, S.; Szostak, M. Cross-Coupling of Amides by N–C Bond Activation. Synlett 2016, 27, 2530–2540. [Google Scholar] [CrossRef]
- Laha, J.K.; Patel, K.V.; Sharma, S. Palladium-Catalyzed Decarboxylative Ortho-Acylation of Tertiary Benzamides with Arylglyoxylic Acids. ACS Omega. 2017, 2, 3806–3815. [Google Scholar] [CrossRef] [PubMed]
- Jing, K.; Yao, J.-P.; Li, Z.-Y.; Li, Q.-L.; Lin, H.-S.; Wang, G.-W. Palladium-Catalyzed Decarboxylative Ortho-Acylation of Benzamides with α-Oxocarboxylic Acids. J. Org. Chem. 2017, 82, 12715–12725. [Google Scholar] [CrossRef]
- Yu, Q.; Zhang, N.; Huang, J.; Lu, S.; Zhu, Y.; Yu, X.; Zhao, K. Efficient Synthesis of Hydroxyl Isoindolones by a Pd-Mediated C-H Activation/Annulation Reaction. Chem. Eur. J. 2013, 19, 11184–11188. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Han, L.; Xu, B.; Zhao, L.; Zhou, J.; Zhang, H. Palladium-Catalyzed C-H Bond Ortho Acylation/Annulation with Toluene Derivatives. Asian J. Org. Chem. 2016, 5, 62–65. [Google Scholar] [CrossRef]
- Jing, K.; Wang, X.-N.; Wang, G.-W. Diastereoselective Synthesis of Oxazoloisoindolinones via Cascade Pd-Catalyzed Ortho-Acylation of N-Benzoyl α-Amino Acid Derivatives and Subsequent Double Intramolecular Cyclizations. J. Org. Chem. 2019, 84, 161–172. [Google Scholar] [CrossRef]
- Chan, C.W.; Zhou, Z.; Yu, W.Y. Palladium(II)-Catalyzed Direct Ortho-C−H Acylation of Anilides by Oxidative Cross-Coupling with Aldehydes using Tert-Butyl Hydroperoxide as Oxidant. Adv. Synth. Catal. 2011, 353, 2999–3006. [Google Scholar] [CrossRef]
- Wu, Y.; Li, B.; Mao, F.; Li, X.; Kwong, F.Y. Palladium-Catalyzed Oxidative C−H Bond Coupling of Steered Acetanilides and Aldehydes: A Facile Access to ortho-Acylacetanilides. Org. Lett. 2011, 13, 3258–3261. [Google Scholar] [CrossRef]
- Li, C.; Wang, L.; Li, P.; Zhou, W. Palladium(II)-Catalyzed Direct Ortho-C−H Acylation of Anilides by Oxidative Cross-Coupling with Aldehydes using Tert-Butyl Hydroperoxide as Oxidant. Chem. Eur. J. 2011, 17, 10208–10212. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Sharma, S.; Mishra, N.K.; Han, S.; Park, J.; Oh, H.; Ha, J.; Yoo, H.; Jung, H.-Y.; Kim, I.S. Direct and Site-Selective Palladium-Catalyzed C-7 Acylation of Indolines with Aldehydes. Adv. Synth. Catal. 2015, 357, 594–600. [Google Scholar] [CrossRef]
- Luo, F.; Yang, J.; Li, Z.; Xiang, H.; Zhou, X. Palladium-Catalyzed C–H Bond Acylation of Acetanilides with Benzylic Alcohols under Aqueous Conditions. Eur. J. Org. Chem. 2015, 2463–2469. [Google Scholar] [CrossRef]
- Li, Q.-L.; Li, Z.-Y.; Wang, G.-W. Palladium-Catalyzed Decarboxylative Ortho-Acylation of Anilines with Carbamate as a Removable Directing Group. ACS. Omega. 2018, 3, 4187–4198. [Google Scholar] [CrossRef] [PubMed]
- Ojha, S.; Panda, N. Palladium-Catalyzed Ortho-Benzoylation of Sulfonamides through C−H Activation: Expedient Synthesis of Cyclic N-Sulfonyl Ketimines. Adv. Synth. Catal. 2020, 362, 561–571. [Google Scholar] [CrossRef]
- Das, P.; Biswas, P.; Guin, J. Palladium-Catalyzed Decarboxylative Ortho-C(sp2)−H Aroylation of N-Sulfoximine Benzamides at Room Temperature. Chem. Asian J. 2020, 15, 920–925. [Google Scholar] [CrossRef]
- Li, H.; Li, P.; Wang, L. Direct Access to Acylated Azobenzenes via Pd-Catalyzed C–H Functionalization and Further Transformation into an Indazole Backbone. Org. Lett. 2013, 15, 620–623. [Google Scholar] [CrossRef]
- Xiao, F.; Chen, S.; Huang, H.; Deng, G.J. Palladium-Catalyzed Oxidative Direct Ortho-C–H Acylation of Arenes with Aldehydes under Aqueous Conditions. Eur. J. Org. Chem. 2015, 2015, 7919–7925. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Li, D.-D.; Wang, G.-W. Palladium-Catalyzed Decarboxylative Ortho Acylation of Azobenzenes with α-Oxocarboxylic Acids. J. Org. Chem. 2013, 78, 10414–10420. [Google Scholar] [CrossRef]
- Li, H.; Li, P.; Tan, H.; Wang., L.A. Highly Efficient Palladium-Catalyzed Decarboxylative ortho-Acylation of Azobenzenes with α-Oxocarboxylic Acids: Direct Access to Acylated Azo Compounds. Chem. Eur. J. 2013, 19, 14432–14436. [Google Scholar] [CrossRef]
- Majhi, B.; Ahammed, S.; Kundu, D.; Ranu, B.C. Palladium-Catalyzed Oxidative C−C Bond Cleavage of α-Hydroxyketones: Application to C−H Acylation of Azoarenes and Synthesis of a Liver(X) Receptor Agonist. Asian J. Org. Chem. 2015, 4, 154–163. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, L.; Chen, Y.; Zhou, Q.; Huang, J.W.; Miao, H.; Luo, H.B. Palladium-Catalyzed Decarboxylative Acylation of N-Nitrosoanilines with α-Oxocarboxylic Acids. J. Org. Chem. 2016, 81, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Z.; Guo, P.; Sun, W.; Li, Y.-M.; Sun, M.; Hua, C. Palladium-catalyzed Ortho-acylation of N-Nitrosoanilines with α-Oxocarboxylic Acids: A Convenient Method to Synthesize N-Nitroso Ketones and Indazoles. Tetrahedron Lett. 2016, 57, 2511–2514. [Google Scholar] [CrossRef]
- Yao, J.P.; Wang, G.W. Palladium-catalyzed Decarboxylative Ortho-acylation of N-Nitrosoanilines with α-Oxocarboxylic Acids. Tetrahedron Lett. 2016, 57, 1687–1690. [Google Scholar] [CrossRef]
- De Abreu, M.; Belmont, P.; Brachet, E. Synergistic Photoredox/Transition-Metal Catalysis for Carbon–Carbon Bond Formation Reactions. Eur. J. Org. Chem. 2020, 2020, 1327–1378. [Google Scholar] [CrossRef]
- Zhou, C.; Li, P.; Zhu, X.; Wang, L. Merging Photoredox with Palladium Catalysis: Decarboxylative Ortho-Acylation of Acetanilides with α-Oxocarboxylic Acids under Mild Reaction Conditions. Org. Lett. 2015, 17, 6198–6201. [Google Scholar] [CrossRef]
- Xu, N.; Li, P.; Xie, Z.; Wang, L. Merging Visible-Light Photocatalysis and Palladium Catalysis for C-H Acylation of Azo- and Azoxybenzenes with α-Keto Acids. Chem. Eur. J. 2016, 22, 2236–2242. [Google Scholar] [CrossRef]
- Kianmehr, E.; Kamezi, S.; Foroumadi, A. Palladium-catalyzed Oxidative C−H bond coupling of Indoles and Benzaldehydes: A New Approach to the Synthesis of 3-Benzoylindoles. Tetrahedron 2014, 70, 349–354. [Google Scholar] [CrossRef]
- Pan, C.; Jin, H.; Liu, X.; Cheng, Y.; Zhu, C. Palladium-catalyzed Decarboxylative C2-Acylation of Indoles with α-Oxocarboxylic Acids. Chem. Commun. 2013, 49, 2933–2935. [Google Scholar] [CrossRef]
- Yan, X.-B.; Shen, Y.-W.; Chen, D.-Q.; Gao, P.; Li, Y.-X.; Song, X.-R.; Liu, X.-Y.; Liang, Y.-M. Palladium-catalyzed C2-Acylation of Indoles with Aryl and Alkyl Aldehydes. Tetrahedron 2014, 70, 7490–7495. [Google Scholar] [CrossRef]
- Kumar, G.; Sekar, G. Pd-catalyzed Direct C2-Acylation and C2,C7-Diacylation of Indoles: Pyrimidine as an Easily Removable C–H Directing Group. RSC Adv. 2015, 5, 28292–28298. [Google Scholar] [CrossRef]
- Li, C.; Shu, S.; Wu, X.; Liu, H. Palladium-Catalyzed C2-Acylation of Indoles with α-Diketones Assisted by the Removable N-(2-Pyrimidyl) Group. Eur. J. Org. Chem. 2015, 2015, 3743–3750. [Google Scholar] [CrossRef]
- Zhao, Y.; Sharma, U.K.; Schröder, F.; Sharma, N.; Song, G.; Van der Eycken, E.V. Direct C-2 Acylation of Indoles with Toluene Derivatives via Pd(II)-catalysed C–H Activation. RSC Adv 2017, 7, 32559–32563. [Google Scholar] [CrossRef]
- Zhou, B.; Yang, Y.; Li, Y. Rhodium-catalyzed Oxidative C2-Acylation of Indoles with Aryl and Alkyl Aldehydes. Chem. Commun. 2012, 48, 5163–5165. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.K.; Gemoets, H.P.L.; Schröder, F.; Nöel, T.; Van der Eycken, E.V. Merger of Visible-Light Photoredox Catalysis and C−H Activation for the Room-Temperature C-2 Acylation of Indoles in Batch and Flow. ACS Catal. 2017, 7, 3818–38232. [Google Scholar] [CrossRef]
- Manna, K.M.; Bairy, G.; Jana, R. Dual Visible-light Photoredox and Palladium(II) Catalysis for Dehydrogenative C2-Acylation of Indoles at Room Temperature. Org. Biomol. Chem. 2017, 15, 5899–5903. [Google Scholar] [CrossRef]
- Jiang, G.; Wang, S.; Zhang, J.; Yu, J.; Zhang, Z.; Ji, F. Palladium-Catalyzed Primary Amine-Directed Decarboxylative Annulation of α-Oxocarboxylic Acids: Access to Indolo [1,2-a] Quinazolines. Adv. Synth. Catal. 2019, 361, 1798–1802. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, M.; Fan, J.; Xu, Q.; Xie, M. Selective C–H Acylation of Indoles with α-Oxocarboxylic Acids at the C4 Position by Palladium Catalysis. Chem. Commun. 2019, 55, 8102–8105. [Google Scholar] [CrossRef]
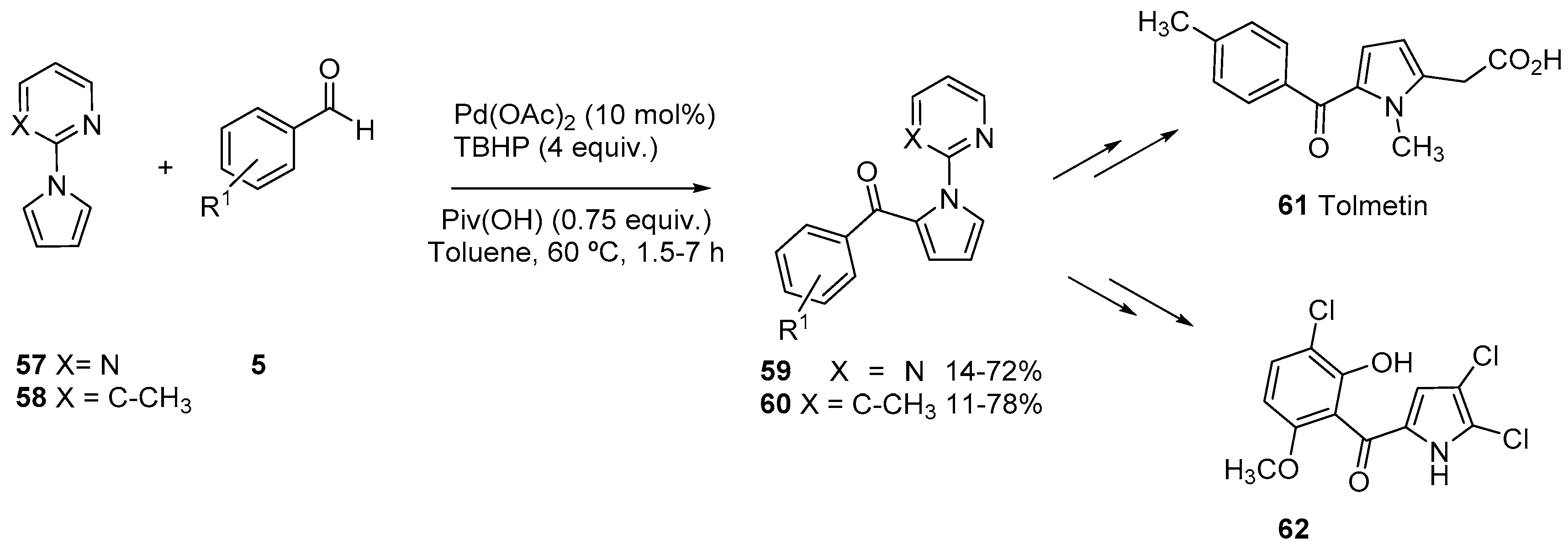
- Santiago, C.; Rubio, I.; Sotomayor, N.; Lete, E. Selective Pd(II)-catalyzed Acylation of Pyrrole with Aldehydes. Application to the Synthesis of Celastramycin analogues and Tolmetin. Eur. J. Org. Chem. 2020, (in press). [CrossRef]
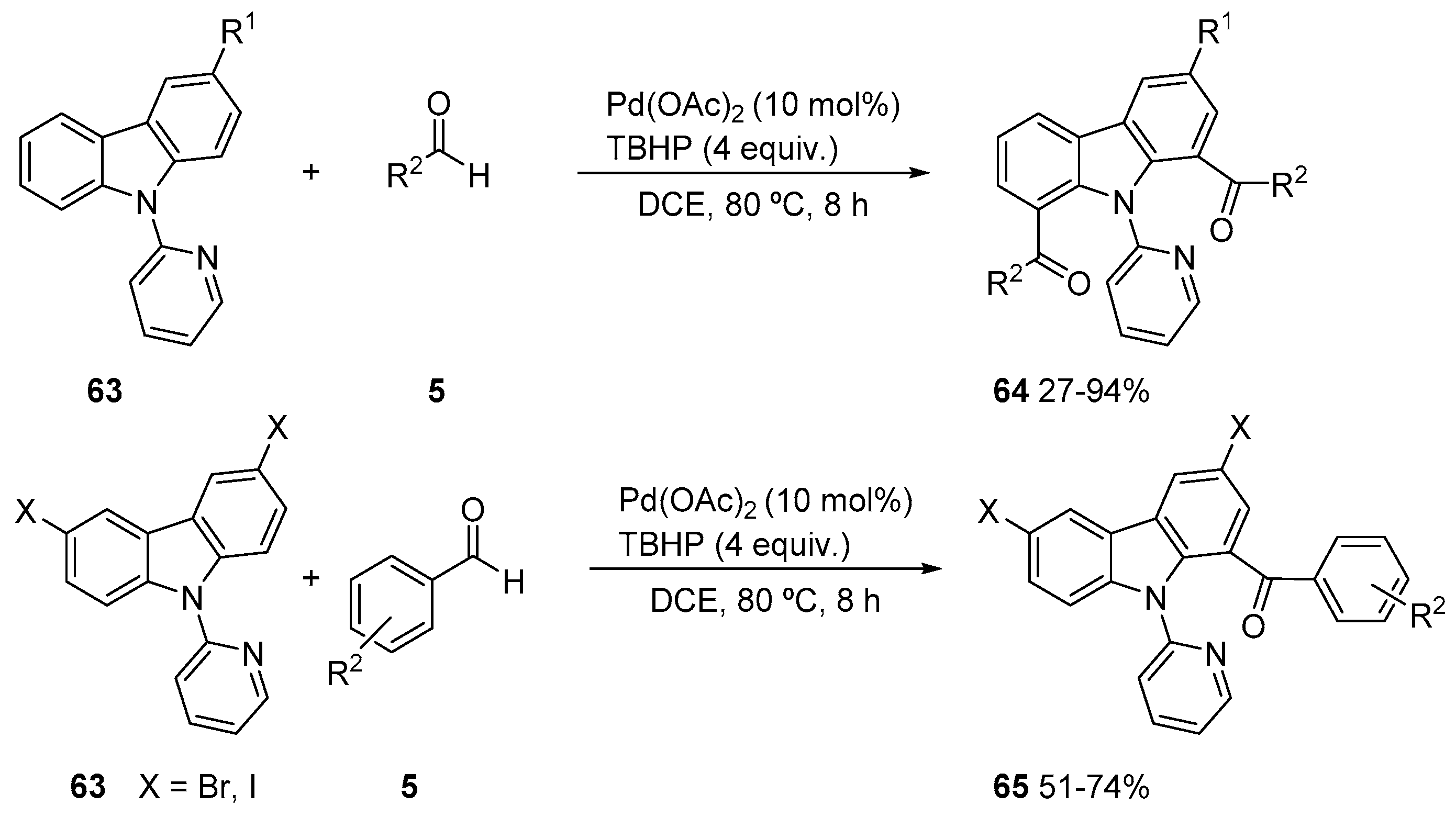
- Maiti, S.; Burgula, L.; Chakraborti, G.; Dash, J. Palladium Catalyzed Pyridine Group Directed Regioselective Oxidative C-H Acylation of Carbazoles using Aldehydes as the Acyl Source. Eur. J. Org. Chem. 2017, 332–340. [Google Scholar] [CrossRef]
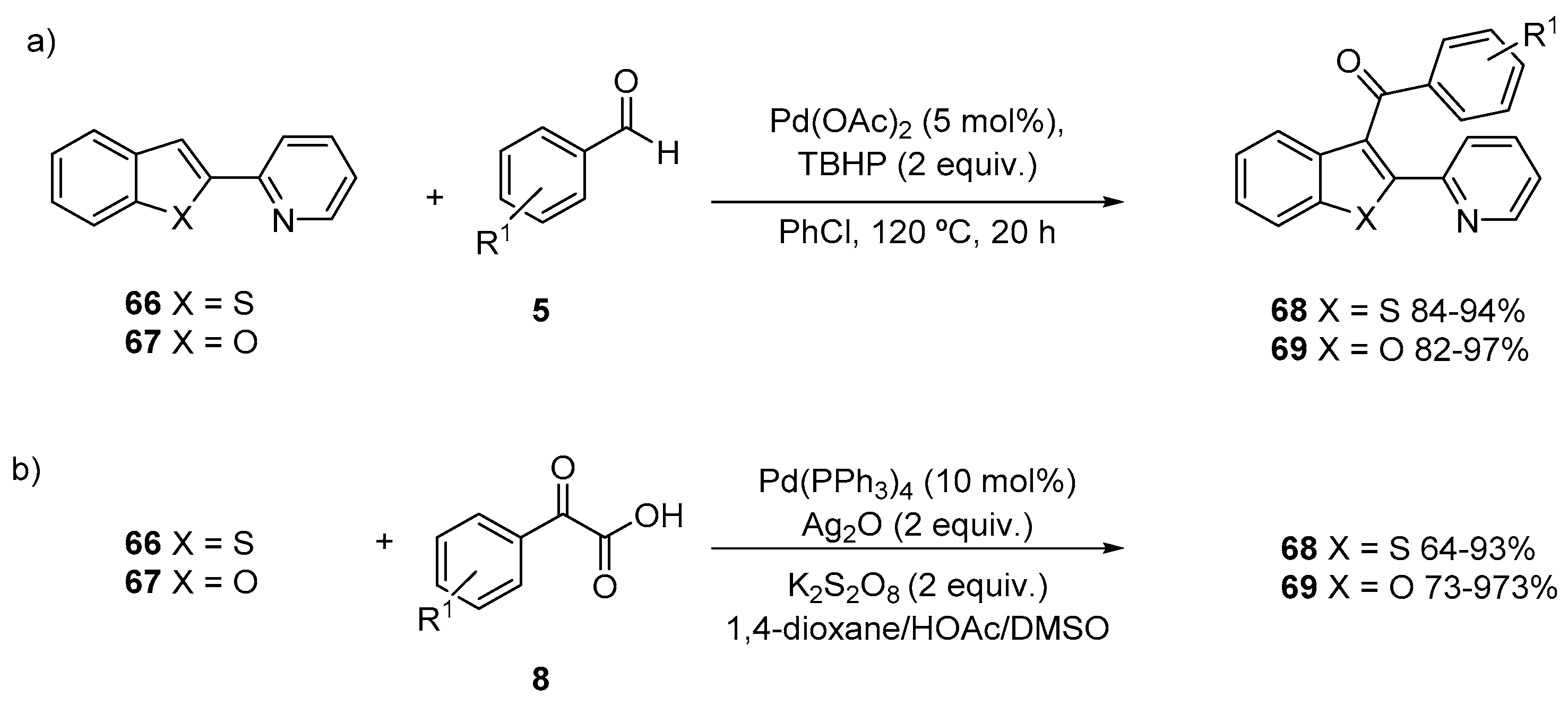
- Zhao, J.; Fang, H.; Xie, C.; Han, J.; Li, G.; Pan, Y. Palladium-Catalyzed C3 Acylation of Benzofurans and Benzothiophenes with Aromatic Aldehydes by Cross-Dehydrogenative Coupling Reactions. Asian J. Org. Chem. 2013, 2, 1044–1047. [Google Scholar] [CrossRef]
- Gong, W.-J.; Liu, D.-X.; Li, F.-L.; Gao, J.; Li, H.-X.; Lang, J.-P. Palladium-catalyzed Decarboxylative C3-Acylation of Benzofurans and Benzothiophenes with α-Oxocarboxylic Acids via Direct sp2 C-H Bond Activation. Tetrahedron 2015, 71, 1269–1275. [Google Scholar] [CrossRef]
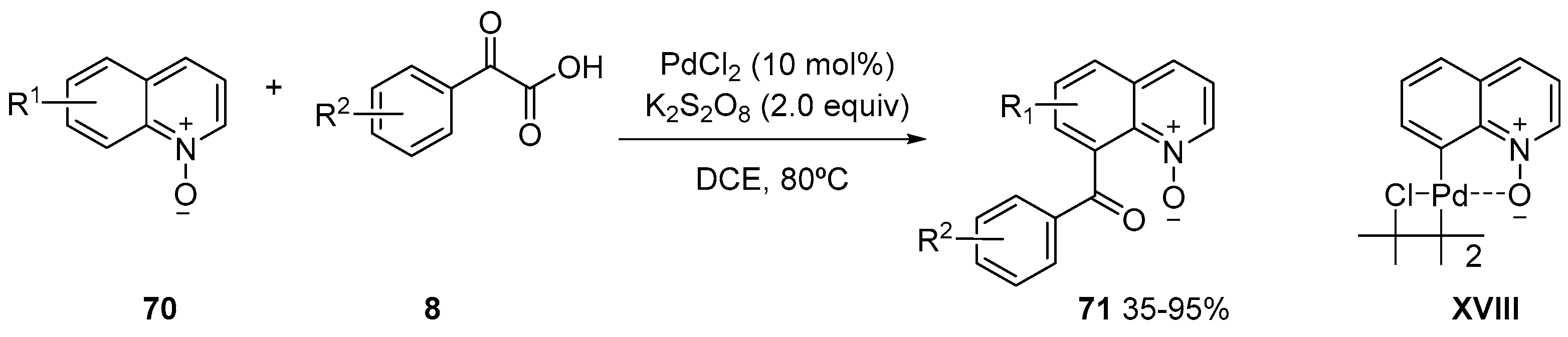
- Chen, X.; Cui, X.; Wu, Y. C8-Selective Acylation of Quinoline N-Oxides with α-Oxocarboxylic Acids via Palladium-Catalyzed Regioselective C−H Bond Activation. Org. Lett. 2016, 18, 3722–3725. [Google Scholar] [CrossRef] [PubMed]
- Cirujano, F.G.; Leo, P.; Vercammen, J.; Smolders, S.; Orcajo, G.; De Vos, D.E. MOFs Extend the Lifetime of Pd(II) Catalyst for Room Temperature Alkenylation of Enamine-Like Arenes. Adv. Synth. Catal. 2018, 360, 3872–3876. [Google Scholar] [CrossRef]
- Gandeepan, P.; Muüller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C−H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef]
- Yang, K.; Zhang, C.; Wang, P.; Zhang, Y.; Ge, H. Nickel-Catalyzed Decarboxylative Acylation of Heteroarenes by sp2 C−H Functionalization. Chem. Eur. J. 2014, 20, 1–5. [Google Scholar] [CrossRef]
- Yang, K.; Chen, X.; Wang, Y.; Li, W.; Kadi, A.A.; Fun, H.-K.; Sun, H.; Zang, Y.; Li, G.; Lu, H. Cobalt-Catalyzed Decarboxylative 2-Benzoylation of Oxazoles and Thiazoles with α-Oxocarboxylic Acids. J. Org. Chem. 2015, 80, 11065–11072. [Google Scholar] [CrossRef]





















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santiago, C.; Sotomayor, N.; Lete, E. Pd(II)-Catalyzed C-H Acylation of (Hetero)arenes—Recent Advances. Molecules 2020, 25, 3247. https://doi.org/10.3390/molecules25143247
Santiago C, Sotomayor N, Lete E. Pd(II)-Catalyzed C-H Acylation of (Hetero)arenes—Recent Advances. Molecules. 2020; 25(14):3247. https://doi.org/10.3390/molecules25143247
Chicago/Turabian StyleSantiago, Carlos, Nuria Sotomayor, and Esther Lete. 2020. "Pd(II)-Catalyzed C-H Acylation of (Hetero)arenes—Recent Advances" Molecules 25, no. 14: 3247. https://doi.org/10.3390/molecules25143247
APA StyleSantiago, C., Sotomayor, N., & Lete, E. (2020). Pd(II)-Catalyzed C-H Acylation of (Hetero)arenes—Recent Advances. Molecules, 25(14), 3247. https://doi.org/10.3390/molecules25143247