Design, Synthesis and Anti-Tumor Activity of Novel Benzimidazole-Chalcone Hybrids as Non-Intercalative Topoisomerase II Catalytic Inhibitors



Abstract
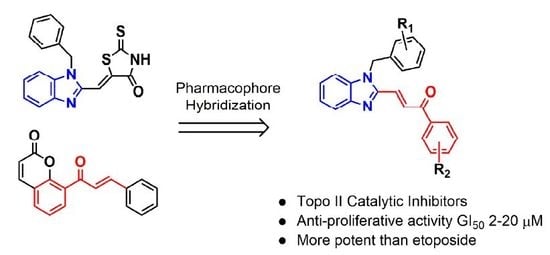
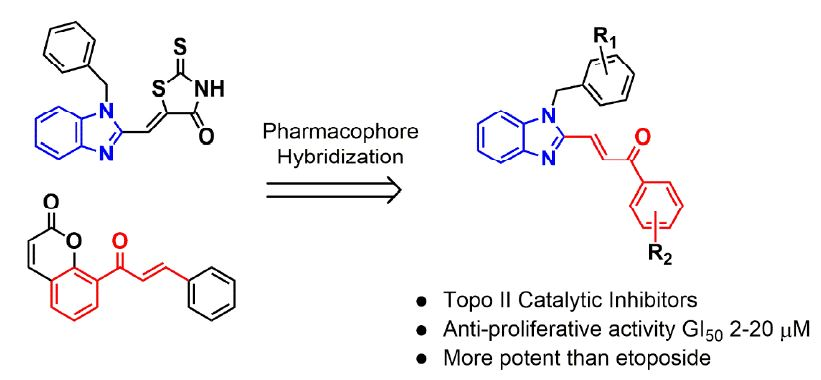
1. Introduction
2. Results and Discussion
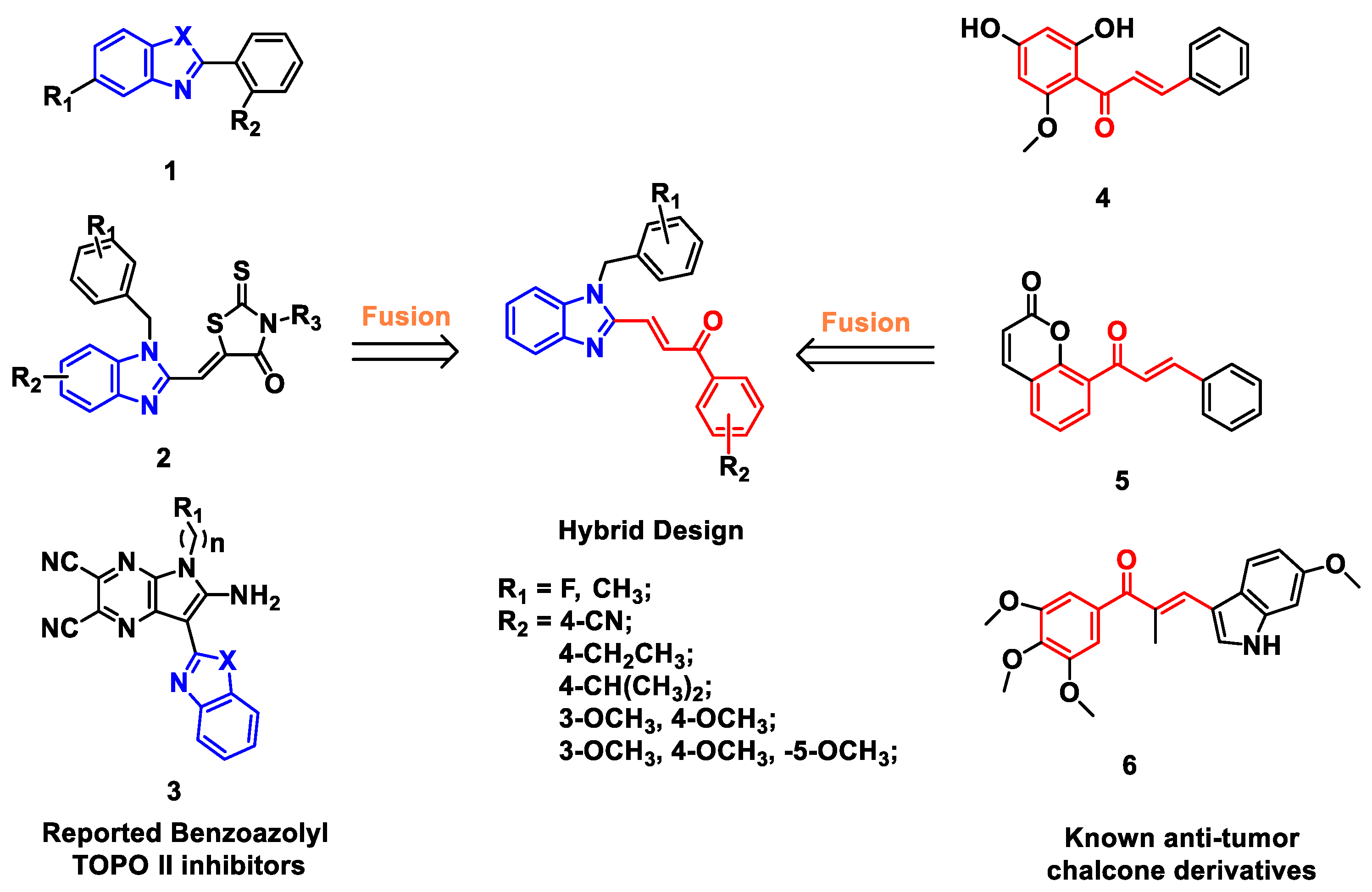
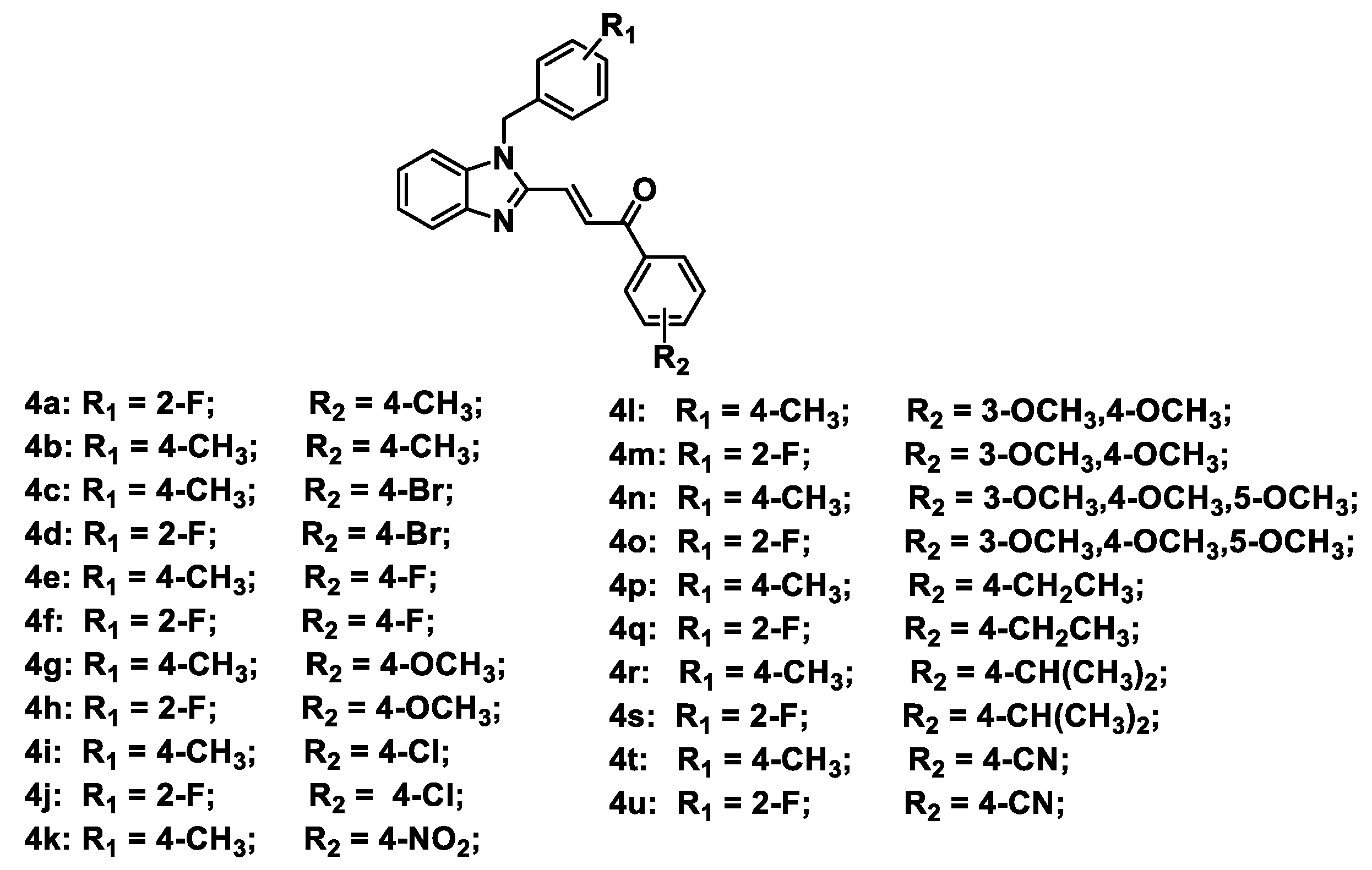
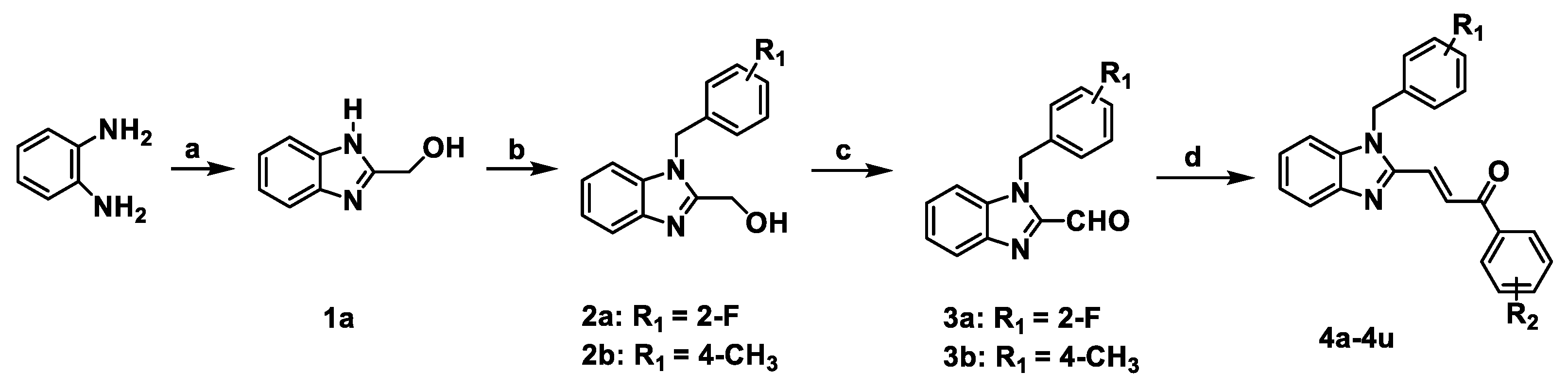
2.1. Chemistry
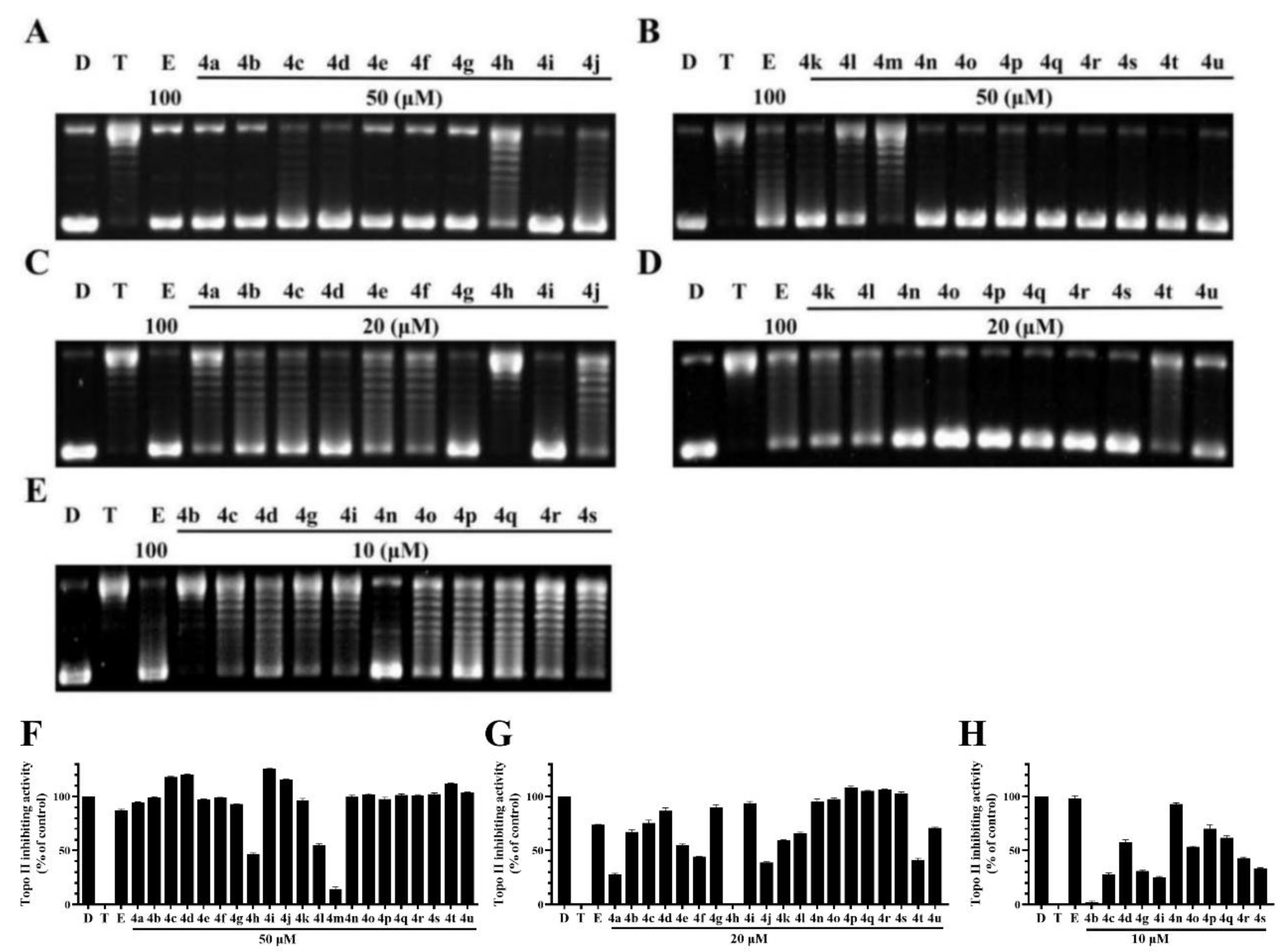
2.2. BCHs as Potent Topo II Inhibitors
2.3. Anti-Proliferative Activity of BCHs on Tumor Cells
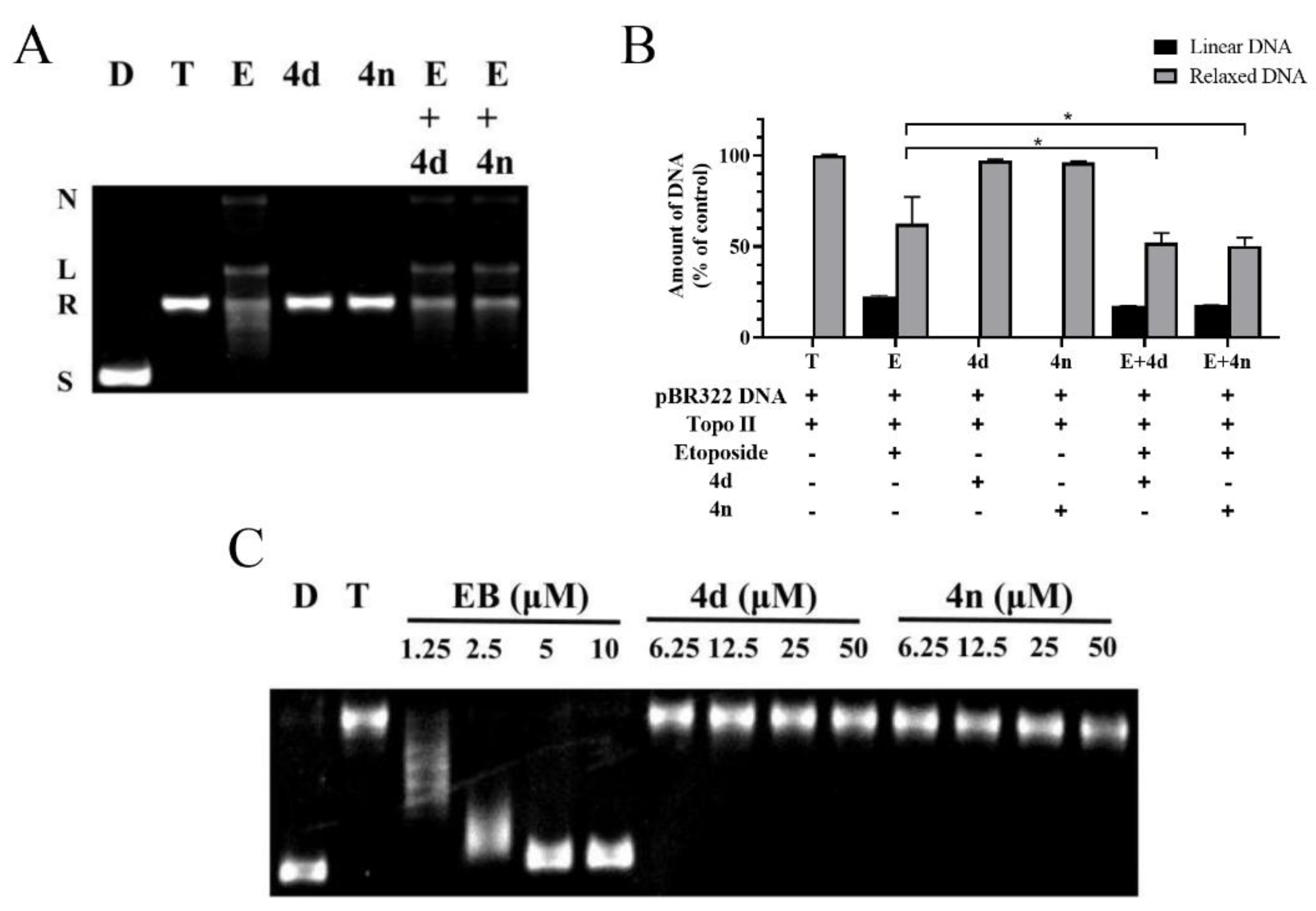
2.4. BCHs as Non-Intercalative Topo II Catalytic Inhibitors
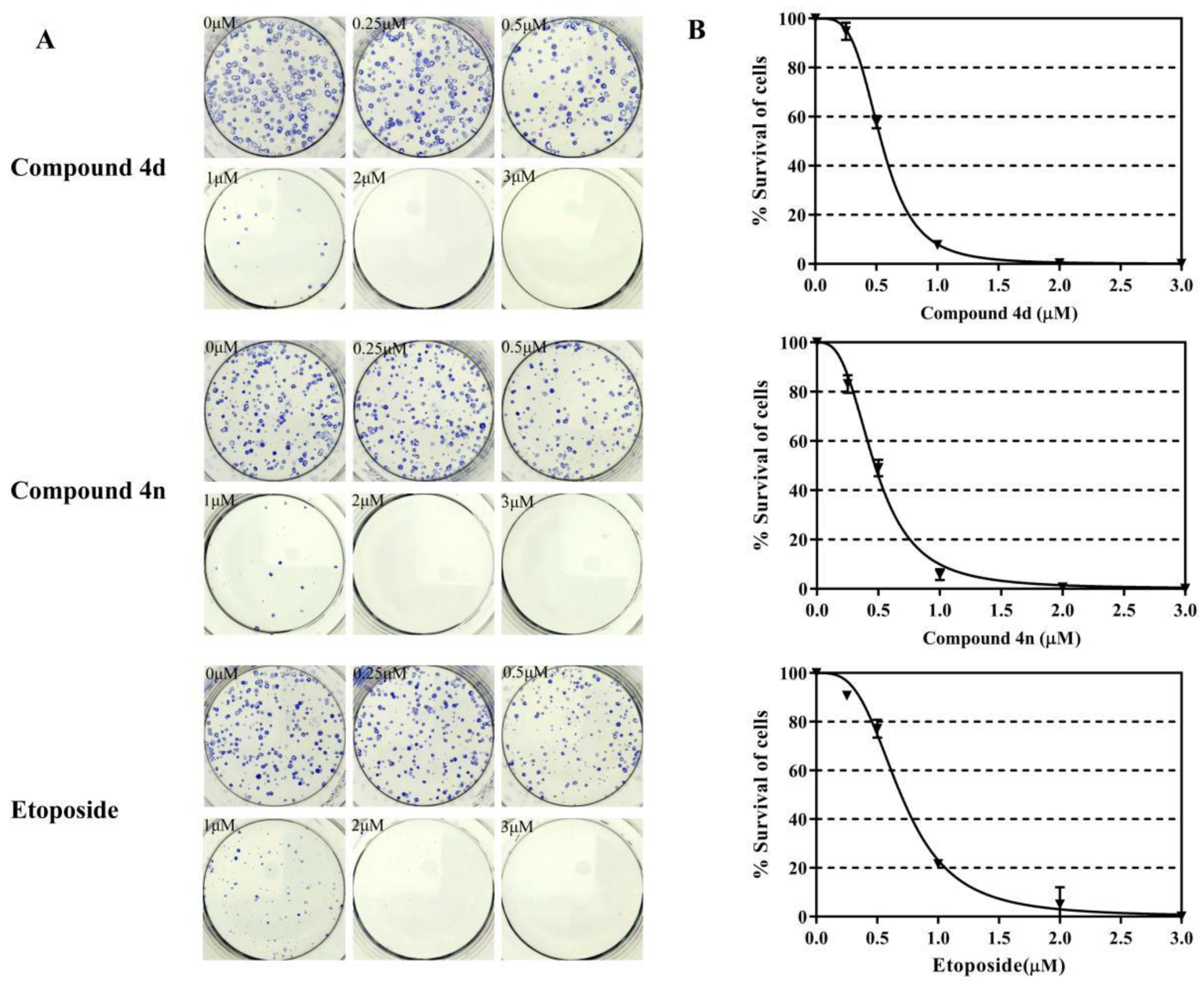
2.5. Inhibition of Clonogenic Survival of A549 Cells by 4d and 4n
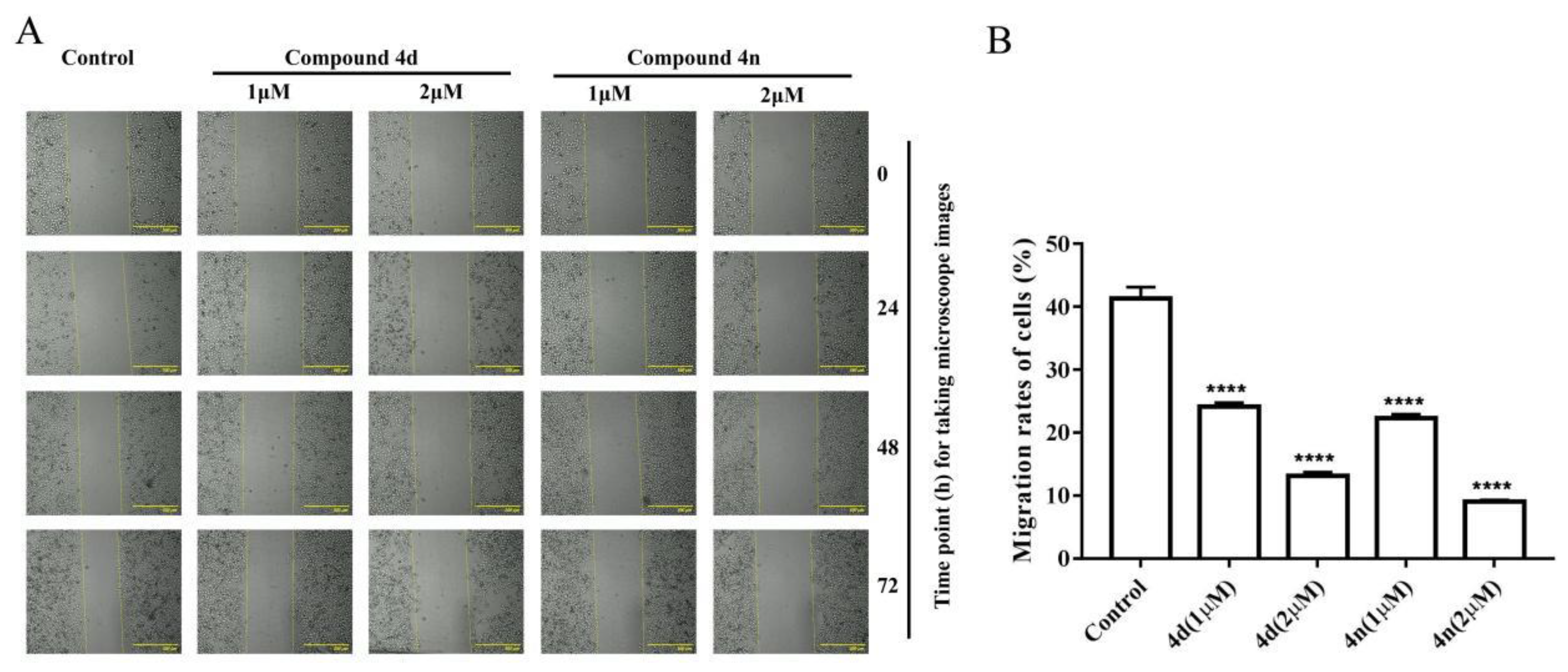
2.6. Inhibition of Wound Healing in A549 Cell by 4d and 4n
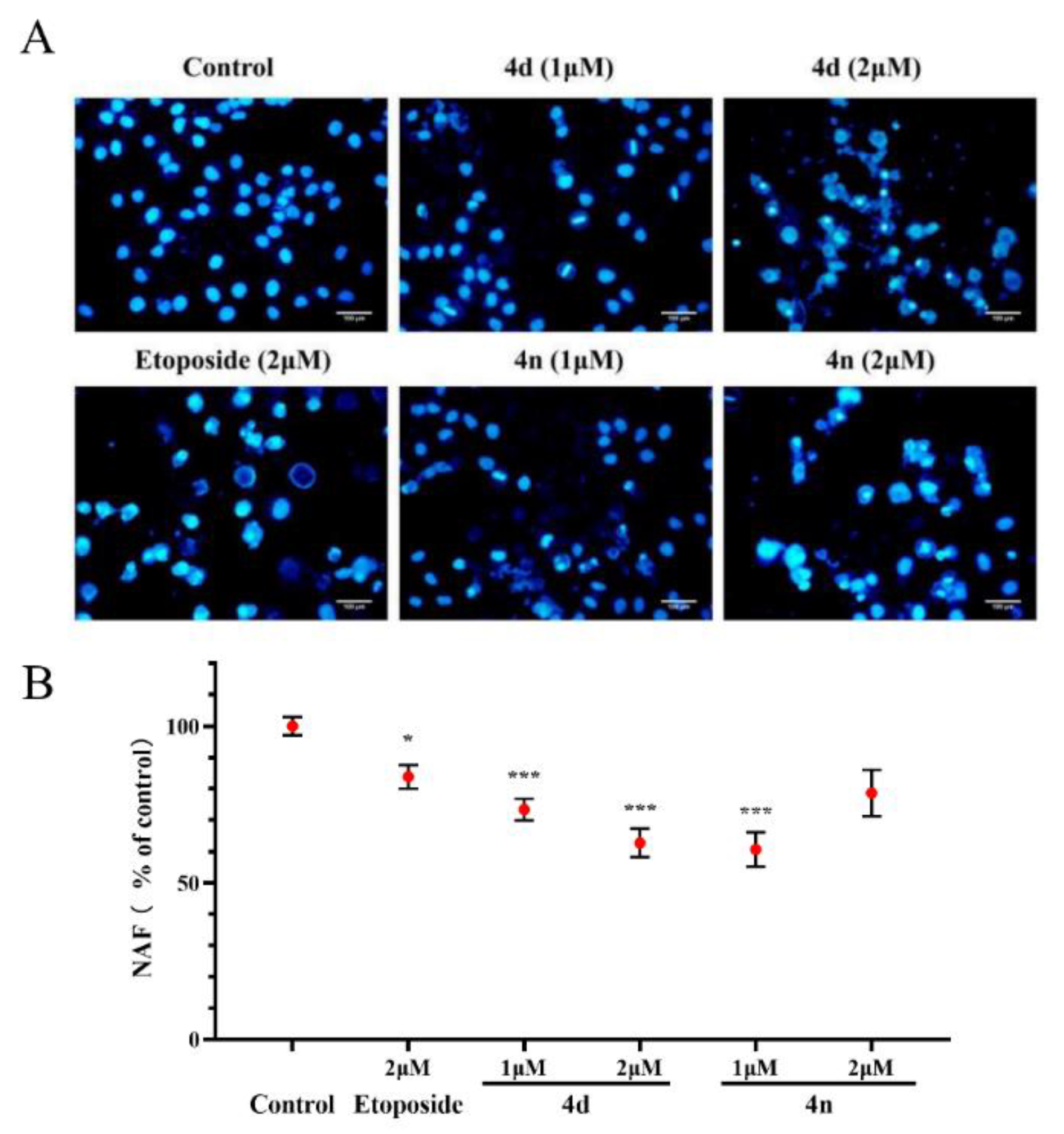
2.7. Cell Apoptosis of A549 Cells by 4d and 4n
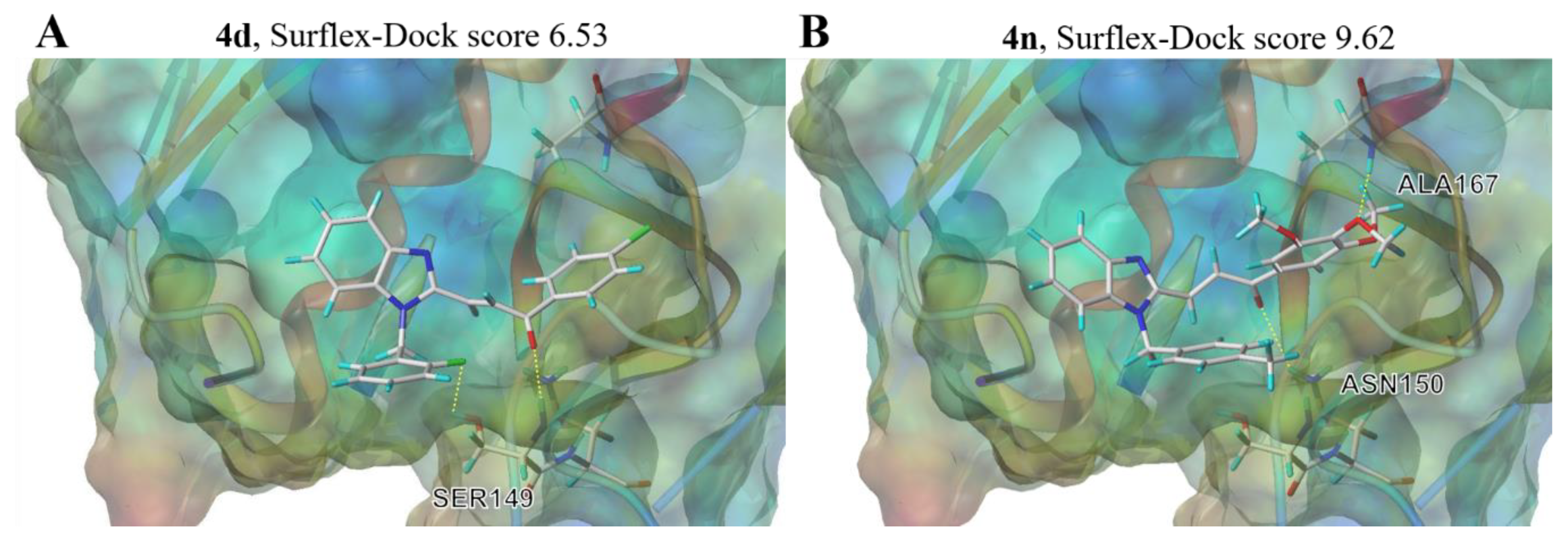
2.8. Molecular Docking
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis and Characterization
3.1.2. Procedure for the Synthesis of Intermediate 1a
3.1.3. General Procedure for the Synthesis of Intermediates 2a and 2b
3.1.4. General Procedure for the Synthesis of Intermediates 3a and 3b
3.1.5. General Procedure for the Synthesis of Compounds 4a–4u
3.2. Biology
3.2.1. Topo II Mediated DNA Relaxation Assay In Vitro
3.2.2. Topo II Mediated DNA Cleavage Assay
3.2.3. DNA Unwinding Assay
3.2.4. In Vitro Anti-Proliferation Assay
3.2.5. Clonogenic Assay
3.2.6. Cell Migration Assay
3.2.7. DAPI Staining
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Topo II | type II topoisomerase |
| BCHs | benzimidazole-chalcone hybrids |
| IC50 | half maximal inhibitory concentration |
| ATP | adenosine triphosphate |
| EB | ethidium bromide |
| DAPI | 4′,6-diamidino-2-phenylindole |
| ADP | adenosine diphosphate |
References
- Delgado, J.L.; Hsieh, C.-M.; Chan, N.-L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Huang, X.-S.; Wu, J.-F.; Yang, L.; Zheng, Y.-T.; Shen, Y.; Li, Z.-Y.; Li, X. Discovery of Novel Topoisomerase II Inhibitors by Medicinal Chemistry Approaches. J. Med. Chem. 2018, 61, 8947–8980. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Kathiravan, M.K.; Khilare, M.M.; Nikoomanesh, K.; Chothe, A.S.; Jain, K.S. Topoisomerase as target for antibacterial and anticancer drug discovery. J. Enzym. Inhib. Med. Chem. 2012, 28, 419–435. [Google Scholar] [CrossRef]
- Kohrt, H.E.; Patel, S.; Ho, M.; Owen, T.; Pollyea, D.A.; Majeti, R.; Gotlib, J.; Coutre, S.; Liedtke, M.; Berube, C.; et al. Second-line mitoxantrone, etoposide, and cytarabine for acute myeloid leukemia: A single-center experience. Am. J. Hematol. 2010, 85, 877–881. [Google Scholar] [CrossRef]
- Wu, C.-C.; Li, T.-K.; Farh, L.; Lin, L.-Y.; Lin, T.-S.; Yu, Y.-J.; Yen, T.-J.; Chiang, C.-W.; Chan, N.-L. Structural Basis of Type II Topoisomerase Inhibition by the Anticancer Drug Etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef]
- Vos, S.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef]
- Darcy, N.; Gabrielli, B. Topoisomerase II inhibitors and poisons, and the influence of cell cycle checkpoints. Curr. Med. Chem. 2017, 24, 1504–1519. [Google Scholar] [CrossRef]
- Huang, H.; Chen, Q.; Ku, X.; Meng, L.; Lin, L.; Wang, X.; Zhu, C.; Wang, Y.; Chen, Z.; Li, M.; et al. A Series of α-Heterocyclic Carboxaldehyde Thiosemicarbazones Inhibit Topoisomerase IIα Catalytic Activity. J. Med. Chem. 2010, 53, 3048–3064. [Google Scholar] [CrossRef]
- Kumar, A.; Ehrenshaft, M.; Tokar, E.J.; Mason, R.P.; Sinha, B.K. Nitric oxide inhibits topoisomerase II activity and induces resistance to topoisomerase II-poisons in human tumor cells. Biochim. Biophys. Acta BBA Gen. Subj. 2016, 1860, 1519–1527. [Google Scholar] [CrossRef]
- Lam, T.; Hilgers, M.T.; Cunningham, M.L.; Kwan, B.P.; Nelson, K.J.; Brown-Driver, V.; Ong, V.; Trzoss, M.; Hough, G.; Shaw, K.; et al. Structure-Based Design of New Dihydrofolate Reductase Antibacterial Agents: 7-(Benzimidazol-1-yl)-2,4-diaminoquinazolines. J. Med. Chem. 2014, 57, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, W.; Khan, M.F.; Verma, G.; Shaquiquzzaman, M.; Rizvi, M.M.A.; Mehdi, S.H.; Akhtar, M.; Alam, M.M. Therapeutic evolution of benzimidazole derivatives in the last quinquennial period. Eur. J. Med. Chem. 2017, 126, 705–753. [Google Scholar] [CrossRef] [PubMed]
- Achar, K.C.; Hosamani, K.M.; Seetharamareddy, H.R. In-vivo analgesic and anti-inflammatory activities of newly synthesized benzimidazole derivatives. Eur. J. Med. Chem. 2010, 45, 2048–2054. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, K.; Kawamine, K.; Ozaki, C.; Ohgiya, T.; Edano, T.; Yoshinaka, Y.; Tsunenari, Y. Discovery of Clinical Candidate 2-(4-(2-((1H-Benzo[d]imidazol-2-yl)thio)ethyl)piperazin-1-yl)-N-(6-methyl-2,4-bis(methylthio)pyridin-3-yl)acetamide Hydrochloride [K-604], an Aqueous-Soluble Acyl-CoA:Cholesterol O-Acyltransferase-1 Inhibitor. J. Med. Chem. 2018, 61, 10635–10650. [Google Scholar] [CrossRef]
- Özkay, Y.; Tunalı, Y.; Karaca, H.; Işıkdağ, I.; Tunali, Y. Antimicrobial activity and a SAR study of some novel benzimidazole derivatives bearing hydrazone moiety. Eur. J. Med. Chem. 2010, 45, 3293–3298. [Google Scholar] [CrossRef]
- Baviskar, A.T.; Madaan, C.; Preet, R.; Mohapatra, P.; Jain, V.; Agarwal, A.; Guchhait, S.K.; Kundu, C.N.; Banerjee, U.C.; Bharatam, P.V. N-Fused Imidazoles As Novel Anticancer Agents That Inhibit Catalytic Activity of Topoisomerase IIα and Induce Apoptosis in G1/S Phase. J. Med. Chem. 2011, 54, 5013–5030. [Google Scholar] [CrossRef]
- Baviskar, A.T.; Amrutkar, S.M.; Trivedi, N.; Chaudhary, V.; Nayak, A.; Guchhait, S.K.; Banerjee, U.C.; Bharatam, P.V.; Kundu, C.N. Switch in Site of Inhibition: A Strategy for Structure-Based Discovery of Human Topoisomerase IIα Catalytic Inhibitors. ACS Med. Chem. Lett. 2015, 6, 481–485. [Google Scholar] [CrossRef]
- Li, P.-H.; Zhang, W.; Jiang, H.; Li, Y.; Dong, C.-Z.; Chen, H.; Zhang, K.; Du, Z. Design, synthesis and biological evaluation of benzimidazole–rhodanine conjugates as potent topoisomerase II inhibitors. MedChemComm 2018, 9, 1194–1205. [Google Scholar] [CrossRef]
- Li, P.-H.; Zeng, P.; Chen, S.-B.; Yao, P.-F.; Mai, Y.-W.; Tan, J.-H.; Ou, T.-M.; Huang, S.-L.; Li, D.; Gu, L.-Q.; et al. Synthesis and Mechanism Studies of 1,3-Benzoazolyl Substituted Pyrrolo[2,3-b]pyrazine Derivatives as Nonintercalative Topoisomerase II Catalytic Inhibitors. J. Med. Chem. 2015, 59, 238–252. [Google Scholar] [CrossRef]
- Sun, L.-Y.; Zhu, L.-W.; Tang, Y.-J. Increasing the distance between two monomers of topoisomerase IIβ under the action of antitumor agent 4 β-sulfur-(benzimidazole) 4′-demethylepipodophyllotoxin. Sci. Rep. 2018, 8, 14949. [Google Scholar] [CrossRef]
- Moriello, A.; Luongo, L.; Guida, F.; Christodoulou, M.S.; Perdicchia, D.; Maione, S.; Passarella, D.; Di Marzo, V.; De Petrocellis, L. Chalcone Derivatives Activate and Desensitize the Transient Receptor Potential Ankyrin 1 Cation Channel, Subfamily A, Member 1 TRPA1 Ion Channel: Structure-Activity Relationships in vitro and Anti-Nociceptive and Anti-inflammatory Activity in vivo. CNS Neurol. Disord. Drug Targets 2016, 15, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Anand, A.; Kumar, V. Recent developments in biological activities of chalcones: A mini review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, L.M.; Valente, I.; Rodrigues, J.A. An Overview on Cardamonin. J. Med. Food 2014, 17, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xu, F.; Shuai, W.; Sun, H.; Yao, H.; Ma, C.; Xu, S.; Yao, H.; Zhu, Z.; Yang, D.-H.; et al. Discovery of Novel Quinoline–Chalcone Derivatives as Potent Antitumor Agents with Microtubule Polymerization Inhibitory Activity. J. Med. Chem. 2018, 62, 993–1013. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Chen, J.; Zhang, S.; Hu, J.; Huang, L.; Li, X. Synthesis, Evaluation, and Mechanism Study of Novel Indole-Chalcone Derivatives Exerting Effective Antitumor Activity Through Microtubule Destabilization in Vitro and in Vivo. J. Med. Chem. 2016, 59, 5264–5283. [Google Scholar] [CrossRef]
- Venturelli, S.; Burkard, M.; Biendl, M.; Lauer, U.M.; Frank, J.; Busch, C. Prenylated chalcones and flavonoids for the prevention and treatment of cancer. Nutrition 2016, 32, 1171–1178. [Google Scholar] [CrossRef]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Ortega, J.A.; Riccardi, L.; Minniti, E.; Borgogno, M.; Arencibia, J.M.; Greco, M.L.; Minarini, A.; Sissi, C.; De Vivo, M. Pharmacophore Hybridization to Discover Novel Topoisomerase II Poisons with Promising Antiproliferative Activity. J. Med. Chem. 2017, 61, 1375–1379. [Google Scholar] [CrossRef]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.; Dhar, K. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef]
- Yao, B.-L.; Mai, Y.-W.; Chen, S.-B.; Xie, H.-T.; Yao, P.-F.; Ou, T.-M.; Tan, J.-H.; Wang, H.-G.; Li, D.; Huang, Z.; et al. Design, synthesis and biological evaluation of novel 7-alkylamino substituted benzo[a]phenazin derivatives as dual topoisomerase I/II inhibitors. Eur. J. Med. Chem. 2015, 92, 540–553. [Google Scholar] [CrossRef]
- Li, P.-H.; Jiang, H.; Zhang, W.-J.; Li, Y.-L.; Zhao, M.-C.; Zhou, W.; Zhang, L.-Y.; Tang, Y.-D.; Dong, C.-Z.; Huang, Z.-S.; et al. Synthesis of carbazole derivatives containing chalcone analogs as non-intercalative topoisomerase II catalytic inhibitors and apoptosis inducers. Eur. J. Med. Chem. 2018, 145, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.; Rodermond, H.M.; Stap, J.; Haveman, J.; Van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.-B.; Park, C.; Jeon, K.-H.; Lee, E.; Park, S.-E.; Jun, K.-Y.; Kadayat, T.M.; Thapa, P.; Karki, R.; Na, Y.; et al. A Series of Novel Terpyridine-Skeleton Molecule Derivants Inhibit Tumor Growth and Metastasis by Targeting Topoisomerases. J. Med. Chem. 2015, 58, 1100–1122. [Google Scholar] [CrossRef] [PubMed]
- Donthiboina, K.; Anchi, P.; Ramya, P.S.; Karri, S.; Srinivasulu, G.; Godugu, C.; Shankaraiah, N.; Kamal, A. Synthesis of substituted biphenyl methylene indolinones as apoptosis inducers and tubulin polymerization inhibitors. Bioorg. Chem. 2019, 86, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, Y.; Wang, X.; Liu, D.; Wu, X.; Xu, C.; Chen, C.; Li, Z. The effects of jolkinolide B on HepG2 cells as revealed by 1H-NMR-based metabolic profiling. Eur. J. Pharmacol. 2019, 842, 10–19. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (Mean ± SD) (μM) a | Topo II Inhibition | ||||
|---|---|---|---|---|---|---|
| A549 | HePG2 | MG63 | LNCaP | Inhibition Rate (%) b | Indicator | |
| 4a | 2.8 ± 0.2 | 4.0 ± 0.6 | 4.0 ± 0.5 | 3.5 ± 0.1 | 28.3 ± 0.7 | + |
| 4b | 3.6 ± 0.6 | 4.6 ± 0.9 | 5.5 ± 0.8 | 4.2 ± 0.2 | 67.1 ± 2.1 | + |
| 4c | 8.3 ± 2.8 | 6.9 ± 1.1 | 6.7 ± 0.6 | 7.7 ± 0.4 | 75.5 ± 2.6 | ++ |
| 4d | 3.6 ± 0.7 | 4.5 ± 1.0 | 4.7 ± 0.8 | 5.4 ± 0.1 | 86.9 ± 2.7 | ++ |
| 4e | 5.5 ± 1.5 | 5.5 ± 1.7 | 6.7 ± 0.6 | 8.6 ± 0.5 | 54.8 ± 1.0 | + |
| 4f | 6.1 ± 4.9 | 10.8 ± 5.7 | 5.5 ± 0.5 | 8.5 ± 1.5 | 44.2 ± 0.3 | + |
| 4g | 3.3 ± 0.6 | 4.4 ± 1.3 | 4.7 ± 0.5 | 3.6 ± 0.2 | 90.2 ± 2.1 | ++ |
| 4h | 2.9 ± 0.2 | 4.6 ± 0.8 | 4.4 ± 0.1 | 3.6 ± 0.1 | −0.9 ± 0.2 | - |
| 4i | 5.6 ± 2.7 | 4.9 ± 1.5 | 5.7 ± 0.6 | 6.1 ± 0.2 | 93.9 ± 1.4 | ++ |
| 4j | 3.1 ± 0.1 | 4.4 ± 0.9 | 5.7 ± 0.8 | 6.1 ± 0.1 | 38.5 ± 1.0 | + |
| 4k | 8.8 ± 1.0 | 7.0 ± 0.5 | 10.7 ± 0.6 | 15.7 ± 2.7 | 59.2 ± 0.9 | + |
| 4l | 3.7 ± 1.1 | 5.1 ± 0.3 | 10.3 ± 0.6 | 3.4 ± 0.2 | 65.8 ± 1.3 | + |
| 4m | 3.1 ± 0.7 | 3.9 ± 0.3 | 5.0 ± 0.4 | 3.5 ± 0.4 | n.d. [c] | n.d. [c] |
| 4n | 3.8 ± 1.8 | 4.6 ± 0.2 | 4.1 ± 0.6 | 3.6 ± 0.3 | 94.9 ± 2.9 | ++ |
| 4o | 3.4 ± 0.4 | 4.5 ± 0.2 | 6.2 ± 0.6 | 3.4 ± 0.5 | 97.4 ± 1.6 | ++ |
| 4p | 5.7 ± 0.8 | 6.5 ± 0.3 | 3.7 ± 0.6 | 5.7 ± 0.5 | 108.6 ± 1.0 | ++ |
| 4q | 4.3 ± 1.0 | 5.5 ± 0.3 | 4.1 ± 0.9 | 4.3 ± 1.4 | 105.2 ± 0.6 | ++ |
| 4r | 5.8 ± 0.7 | 6.5 ± 0.2 | 4.3 ± 1.3 | 5.8 ± 0.3 | 106.4 ± 0.6 | ++ |
| 4s | 5.5 ± 1.0 | 5.6 ± 0.2 | 3.8 ± 0.8 | 4.9 ± 0.3 | 103.1 ± 1.2 | ++ |
| 4t | 12.1 ± 1.1 | 22.4 ± 1.8 | 47.7 ± 1.6 | 20.1 ± 2.4 | 41.2 ± 1.5 | + |
| 4u | 8.2 ± 1.6 | 8.1 ± 0.4 | 18.5 ± 3.0 | 18.0 ± 0.5 | 70.9 ± 0.7 | + |
| 2a | 74.3 ± 2.7 | >100 | >100 | 86.4 ± 2.4 | 23.2 ± 1.6 | - |
| chalcone | 67.3 ± 1.3 | 86.4 ± 0.9 | >100 | 52.4 ± 1.1 | 12.5 ± 2.5 | - |
| etoposide | 11.8 ± 1.2 | 5.2 ± 1.3 | 12.1 ± 1.1 | 13.3 ± 1.6 | 73.6 ± 0.6 | + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, W.; Zhang, W.; Peng, Y.; Jiang, Z.-H.; Zhang, L.; Du, Z. Design, Synthesis and Anti-Tumor Activity of Novel Benzimidazole-Chalcone Hybrids as Non-Intercalative Topoisomerase II Catalytic Inhibitors. Molecules 2020, 25, 3180. https://doi.org/10.3390/molecules25143180
Zhou W, Zhang W, Peng Y, Jiang Z-H, Zhang L, Du Z. Design, Synthesis and Anti-Tumor Activity of Novel Benzimidazole-Chalcone Hybrids as Non-Intercalative Topoisomerase II Catalytic Inhibitors. Molecules. 2020; 25(14):3180. https://doi.org/10.3390/molecules25143180
Chicago/Turabian StyleZhou, Wei, Wenjin Zhang, Yi Peng, Zhi-Hong Jiang, Lanyue Zhang, and Zhiyun Du. 2020. "Design, Synthesis and Anti-Tumor Activity of Novel Benzimidazole-Chalcone Hybrids as Non-Intercalative Topoisomerase II Catalytic Inhibitors" Molecules 25, no. 14: 3180. https://doi.org/10.3390/molecules25143180
APA StyleZhou, W., Zhang, W., Peng, Y., Jiang, Z.-H., Zhang, L., & Du, Z. (2020). Design, Synthesis and Anti-Tumor Activity of Novel Benzimidazole-Chalcone Hybrids as Non-Intercalative Topoisomerase II Catalytic Inhibitors. Molecules, 25(14), 3180. https://doi.org/10.3390/molecules25143180