Cytotoxic Phenanthrene, Dihydrophenanthrene, and Dihydrostilbene Derivatives and Other Aromatic Compounds from Combretum laxum

 , and
, and
Abstract
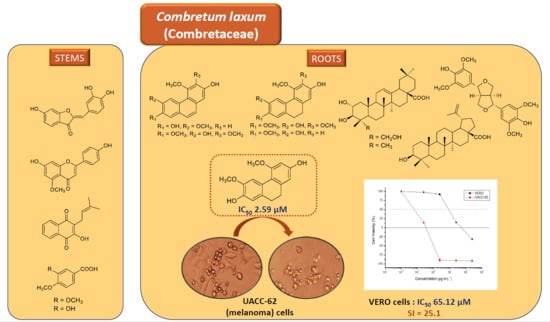
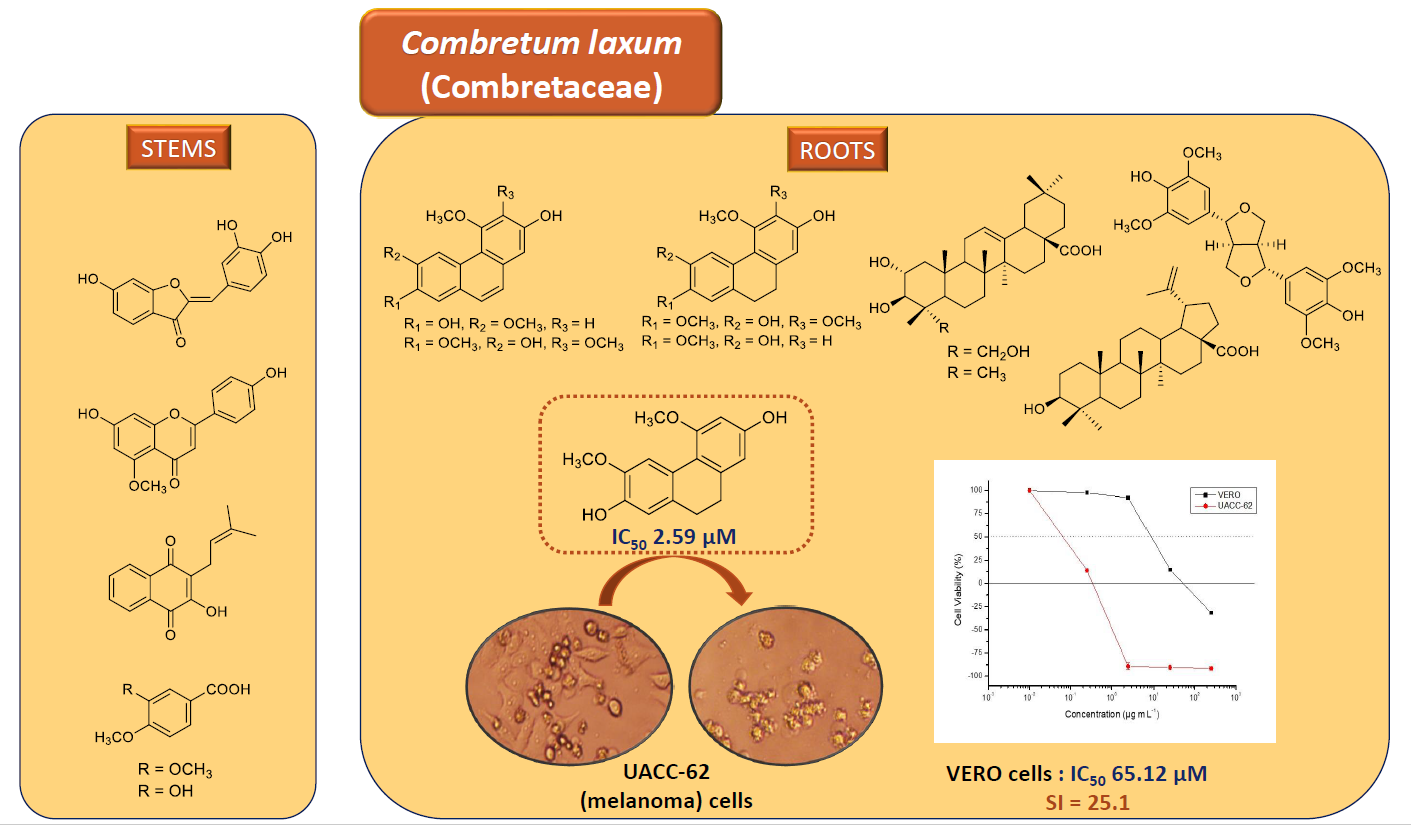
1. Introduction
2. Results and Discussion
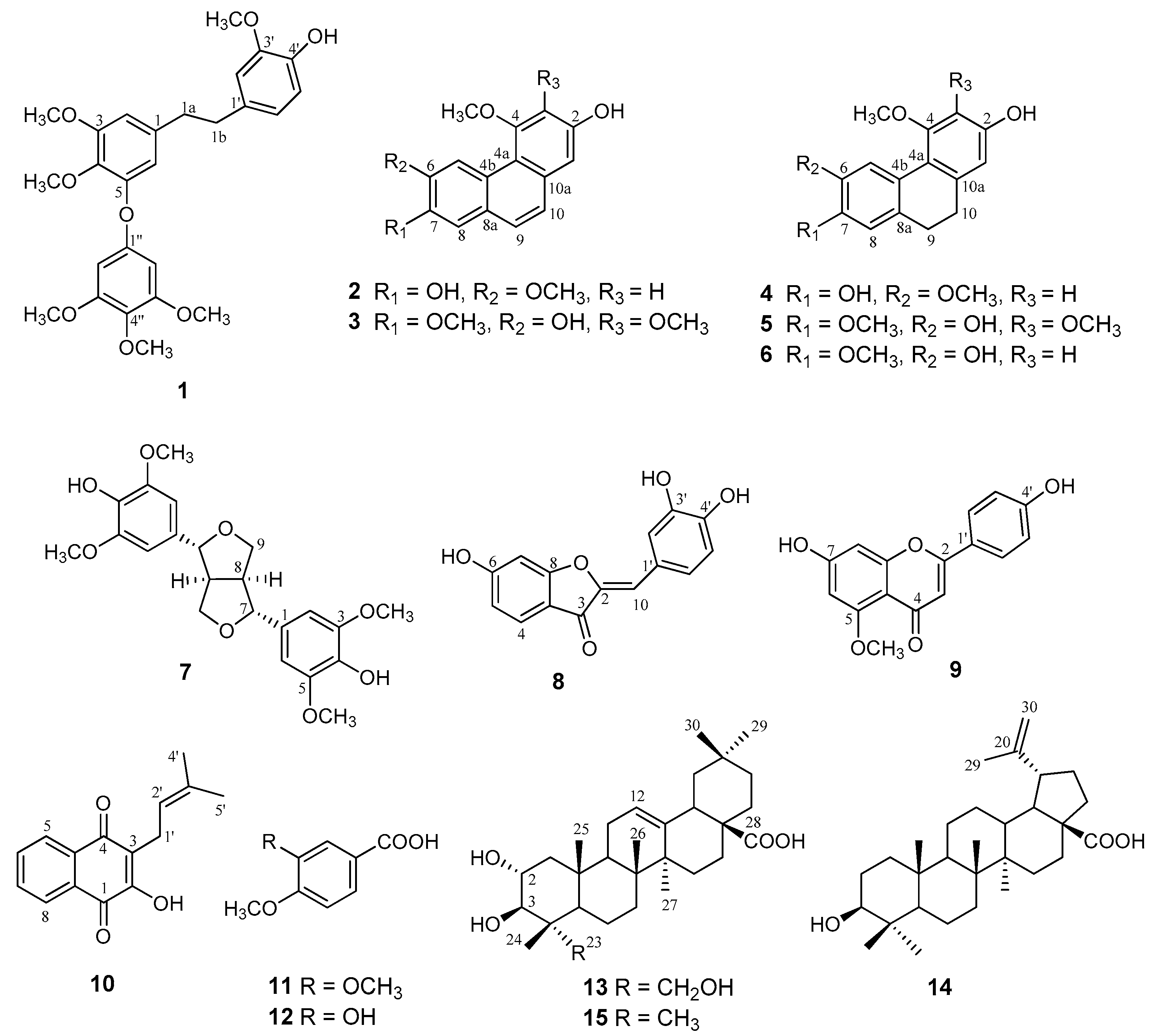
2.1. Extraction, Isolation, and NMR Spectroscopic Data
2.2. In Vitro Cytotoxic Evaluations
2.3. DPPH-Radical-Scavenging Assay
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. In Vitro Cytotoxic Assay
3.5. DPPH-Radical-Scavenging Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pettit, G.R.; Singh, S.B.; Niven, M.L.; Hamel, E.; Schmidt, J.M. Antineoplastic agents.124. Isolation, structure, and synthesis of Combretastatins A-1 and B-1, potent new inhibitors of microtubule assembly, derived from Combretum caffrum. J. Nat. Prod. 1987, 50, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Fyhrquist, P.; Mwasumbi, L.; Haeggstrom, C.A.; Vuorela, H.; Hiltunen, R.; Vuorela, P. Ethnobotanical and antimicrobial investigation on some species of Terminalia and Combretum (Combretaceae) growing in Tanzania. J. Ethnopharmacol. 2002, 79, 169–177. [Google Scholar] [CrossRef]
- de Morais Lima, G.R.; Praxedes de Sales, I.R.; Dutra Caldas Filho, M.R.; Taveira de Jesus, N.Z.; Falcão, H.D.S.; Barbosa-Filho, J.M.; Silveira Cabral, A.G.; Souto, A.L.; Tavares, J.F.; Batista, L.M. Bioactivities of the genus Combretum (Combretaceae): A review. Molecules 2012, 17, 9142–9206. [Google Scholar] [CrossRef]
- Arora, S.; Gonzalez, A.F.; Solanki, K. Combretastatin A-4 and its analogs in cancer therapy. Int. J. Pharm. Sci. Rev. Res. 2013, 22, 168–174. [Google Scholar]
- Fraga, A.G.M. Combretastatins and their analogues: Nature as an alternative source for the therapy of cancer. Rev. Virtual Quim. 2015, 7, 765–790. [Google Scholar] [CrossRef]
- Jaroch, K.; Karolak, M.; Gorski, P.; Jaroch, A.; Krajewski, A.; Ilnicka, A.; Sloderbach, A.; Stefanski, T.; Sobiak, S. Combretastatins: In vitro structure-activity relationship, mode of action and current clinical status. Pharmacol. Rep. 2016, 68, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Faustino, C.; Francisco, A.P.; Isca, V.M.S.; Duarte, N. Cytotoxic stilbenes and derivatives as promising antimitotic leads for cancer therapy. Curr. Pharm. Des. 2018, 24, 4270–4311. [Google Scholar] [CrossRef]
- Bisoli, E.; Garcez, W.S.; Hamerski, L.; Tieppo, C.; Garcez, F.R. Bioactive pentacyclic triterpenes from the stems of Combretum laxum. Molecules 2008, 13, 2717–2728. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J. Spectrometric Identification of Organic Compounds, 7th ed.; Wiley: New York, NY, USA, 2005; pp. 1–502. [Google Scholar]
- Biondi, D.M.; Rocco, C.; Ruberto, G. Dihydrostilbene derivatives from Glycyrrhiza glabra leaves. J. Nat. Prod. 2005, 68, 1099–1102. [Google Scholar] [CrossRef]
- Majumder, P.L.; Guha, S.; Sen, S. Bibenzyl derivatives from the orchid Dendrobium amoenum. Phytochemistry 1999, 52, 1365–1369. [Google Scholar] [CrossRef]
- Majumder, P.L.; Kar, A. Confusarin and confusaridin two phenanthrene derivatives of the orchid Eria confusa. Phytochemistry 1987, 26, 1127–1129. [Google Scholar] [CrossRef]
- Malan, E.; Swinny, E. Substituted bibenzyls, phenanthrenes and 9,10-dihydrophenanthrenes from the heartwood of Combretum apiculatum. Phytochemistry 1993, 34, 1139–1142. [Google Scholar] [CrossRef]
- Leong, Y.W.; Kang, C.C.; Harrison, L.J.; Powell, A.D. Phenanthrenes, dihydrophenanthrenes and bibenzyls from the orchid Bulbophyllum vaginatum. Phytochemistry 1997, 44, 157–165. [Google Scholar] [CrossRef]
- Letcher, R.M.; Nhamo, L.R.M. Chemical constituents of Combretaceae. 1. Substituted phenanthrenes and 9,10-dihydrophenanthrenes from heartwood of Combretum apiculatum. J. Chem. Soc. C 1971, 3070–3076. [Google Scholar] [CrossRef]
- Majumder, P.L.; Sen, S.; Majumder, S. Phenanthrene derivatives from the orchid Coelogyne cristata. Phytochemistry 2001, 58, 581–586. [Google Scholar] [CrossRef]
- Majumder, P.L.; Banerjee, S.; Sen, S. Three stilbenoids from the orchid Agrostophyllum callosum. Phytochemistry 1996, 42, 847–852. [Google Scholar] [CrossRef]
- Juneja, R.K.; Sharma, S.C.; Tandon, J.S. Two substituted bibenzyls and a dihydrophenanthrene from Cymbidium aloifolium. Phytochem 1987, 26, 1123–1125. [Google Scholar] [CrossRef]
- Lee, C.-L.; Chang, F.-R.; Yen, M.-H.; Yu, D.; Liu, Y.-N.; Bastow, K.F.; Morris-Natschke, S.L.; Wu, Y.-C.; Lee, K.-H. Cytotoxic phenanthrenequinones and 9,10-dihydrophenanthrenes from Calanthe arisanensis. J. Nat. Prod. 2009, 72, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Apel, C.; Dumontet, V.; Lozach, O.; Meijer, L.; Gueritte, F.; Litaudon, M. Phenanthrene derivatives from Appendicula reflexa as new CDK1/cyclin B inhibitors. Phytochem. Lett. 2012, 5, 814–818. [Google Scholar] [CrossRef]
- Lu, D.; Liu, J.-P.; Li, H.-J.; Li, P.-Y. Phenanthrene derivatives from the stems and leaves of Dioscorea nipponica Makino. J. Asian Nat. Prod. Res. 2010, 12, 1–6. [Google Scholar] [CrossRef]
- Katerere, D.R.; Gray, A.I.; Nash, R.J.; Waigh, R.D. Phytochemical and antimicrobial investigations of stilbenoids and flavonoids isolated from three species of Combretaceae. Fitoterapia 2012, 83, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Letcher, R.M.; Nhamo, L.R.M.; Gumiro, I.T. Chemical constituents of Combretaceae. 2. Substituted phenanthrenes and 9,10-dihydrophenanthrenes and a substituted bibenzyl from heartwood of Combretum molle. J. Chem. Soc. Perkin 1 1972, 206–210. [Google Scholar] [CrossRef]
- Majumder, P.L.; Banerjee, S.; Maiti, D.C.; Sen, S. Stilbenoids from the orchids Agrostophyllum callosum and Coelogyne flaccida. Phytochemistry 1995, 39, 649–653. [Google Scholar] [CrossRef]
- Agrawal, P.K.; Thakur, R.S. C-13 NMR spectral investigations, part 9. C-13 NMR-spectroscopy of lignan and neolignan derivatives. Magn. Reson. Chem. 1985, 23, 389–418. [Google Scholar] [CrossRef]
- Park, J.A.; Kim, H.J.; Jin, C.B.; Lee, K.T.; Lee, Y.S. A new pterocarpan, (-)-maackiain sulfate, from the roots of Sophora subprostrata. Arch. Pharm. Res. 2003, 26, 1009–1013. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Wu, L.J.; Cai, Y.; Wu, S.Y.; Song, X.P.; Chen, G.Y.; Zheng, C.J.; Han, C.R. One new lignan derivative from the Combretum alfredii Hance. Nat. Prod. Res. 2017, 31, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Moura, A.F.; Lima, K.S.B.; Sousa, T.S.; Marinho, J.D.B.; Pessoa, C.; Silveira, E.R.; Pessoa, O.D.L.; Costa-Lotufo, L.V.; Moraes, M.O.; Araujo, A.J. In vitro antitumor effect of a lignan isolated from Combretum fruticosum, trachelogenin, in HCT-116 human colon cancer cells. Toxicol. In Vitro 2018, 47, 129–136. [Google Scholar] [CrossRef]
- Garcez, F.R.; Garcez, W.S.; Miguel, D.L.S.; Serea, A.A.T.; Prado, F.C. Chemical constituents from Terminalia glabrescens. J. Brazil Chem. Soc. 2003, 14, 461–465. [Google Scholar] [CrossRef]
- Wang, L.Q.; Wu, M.M.; Wang, J.H.; Jiang, J.H.; Chen, Y.G.; Li, T. A new chalcone glycoside from Combretum griffithii. Chem. Nat. Compd. 2014, 50, 258–260. [Google Scholar] [CrossRef]
- Mabry, T.J.; Markham, K.R.; Thomas, M.B. The determination and interpretation of NMR spectra of flavonoids. In The Systematic Identification of Flavonoids; Mabry, T.J., Markham, K.R., Thomas, M.B., Eds.; Springer: Berlin, Germany, 1970; pp. 253–273. [Google Scholar]
- Li, Y.L.; Li, J.; Wang, N.L.; Yao, X.S. Flavonoids and a new polyacetylene from Bidens parviflora Willd. Molecules 2008, 13, 1931–1941. [Google Scholar] [CrossRef]
- Agrawal, P.K.; Thakur, R.S.; Bansal, M.C. Flavonoids. In Carbon-13 NMR of Flavonoids, 1st ed.; Agrawal, P.K., Ed.; Elsevier: Amsterdam, Holland, 1989; pp. 95–182. [Google Scholar]
- Wagner, H.; Chari, V.M.; Sonnenbichler, J. 13C-NMR-spektren natürlich vorkommender flavonoide. Tetrahedron Lett. 1976, 21, 1799–1802. [Google Scholar] [CrossRef]
- Park, B.S.; Lee, H.K.; Lee, S.E.; Piao, X.L.; Takeoka, G.R.; Wong, R.Y.; Ahn, Y.J.; Kim, J.H. Antibacterial activity of Tabebuia impetiginosa Martius ex DC (Taheebo) against Helicobacter pylori. J. Ethnopharmacol. 2006, 105, 255–262. [Google Scholar] [CrossRef]
- Epifano, F.; Genovese, S.; Fiorito, S.; Mathieu, V.; Kiss, R. Lapachol and its congeners as anticancer agents: A review. Phytochem. Rev. 2014, 13, 37–49. [Google Scholar] [CrossRef]
- Rao, K.V.; McBride, T.J.; Oleson, J.J. Recognition and evaluation of lapachol as an antitumor agent. Cancer Res. 1968, 28, 1952–1954. [Google Scholar]
- Pouchert, C.J.; Behnke, J. The Aldrich Library of 13C and 1H NMR Spectra, 1st ed.; Aldrich Chemical Co.: St. Louis, MO, USA, 1993; pp. 1–4300. [Google Scholar]
- Toth, B.; Hohmann, J.; Vasas, A. Phenanthrenes: A promising group of plant secondary metabolites. J. Nat. Prod. 2018, 81, 661–678. [Google Scholar] [CrossRef]
- Kovacs, A.; Vasas, A.; Hohmann, J. Natural phenanthrenes and their biological activity. Phytochemistry 2008, 69, 1084–1110. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-L.; Lin, Y.-T.; Chang, F.-R.; Chen, G.-Y.; Backlund, A.; Yang, J.-C.; Chen, S.-L.; Wu, Y.-C. Synthesis and biological evaluation of phenanthrenes as cytotoxic agents with pharmacophore modeling and ChemGPS-NP prediction as topo II inhibitors. PLoS ONE 2012, 7, e37897. [Google Scholar] [CrossRef] [PubMed]
- Rethy, B.; Kovacs, A.; Zupko, I.; Forgo, P.; Vasas, A.; Falkay, G.; Hohmann, J. Cytotoxic phenanthrenes from the rhizomes of Tamus communis. Planta Med. 2006, 72, 767–770. [Google Scholar] [CrossRef]
- Lee, Y.H.; Park, J.D.; Baek, N.I.; Kim, S.I.; Ahn, B.Z. In vitro and in vivo antitumoral phenanthrenes from the aerial parts of Dendrobium nobile. Planta Med. 1995, 61, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-N.; Zhong, L.-Y.; Bligh, S.W.A.; Guo, Y.-L.; Zhang, C.-F.; Zhang, M.; Wang, Z.-T.; Xu, L.-S. Bi-bicyclic and bi-tricyclic compounds from Dendrobium thyrsiflorum. Phytoochemistry 2005, 66, 1113–1120. [Google Scholar] [CrossRef]
- Ma, W.; Zhang, Y.; Ding, Y.-Y.; Liu, F.; Li, N. Cytotoxic and anti-inflammatory activities of phenanthrenes from the medullae of Juncus effusus L. Arch. Pharm. Res. 2016, 39, 154–160. [Google Scholar] [CrossRef]
- Mena, S.; Ortega, A.; Estrela, J.M. Oxidative stress in environmental-induced carcinogenesis. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2009, 674, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Mumper, R.J. Plant phenolics: Extraction, analysis and their antioxidant and anticancer properties. Molecules 2010, 15, 7313–7352. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, J.-K.; Wang, N.-L.; Kurihara, H.; Yao, X.-S. Antioxidant phenanthrenes and lignans from Dendrobium nobile. J. Chin. Pharm. Sci. 2008, 17, 314–318. [Google Scholar]
- Zhang, X.; Xu, J.-K.; Wang, J.; Wang, N.-L.; Kurihara, H.; Kinataka, S.; Yao, X.-S. Bioactive bibenzyl derivatives and fluorenones from Dendrobium nobile. J. Nat. Prod. 2007, 70, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Araujo-Lima, C.F.; Oliveira, J.P.S.; Coscarella, I.L.; Aiub, C.A.F.; Felzenszwalb, I.; Caprini, E.; Geisa, P.; Macedo, A.F. Metabolomic analysis of Cyrtopodium glutiniferum extract by UHPLC-MS/MS and in vitro antiproliferative and genotoxicity assessment. J. Ethnopharmacol. 2020, 253, 112607. [Google Scholar] [CrossRef]
- Paudel, M.R.; Chand, M.B.; Pant, B.; Pant, B. Antioxidant and cytotoxic activities of Dendrobium moniliforme extracts and the detection of related compounds by GC-MS. BMC Complem. Altern. Med. 2018, 18, 134. [Google Scholar] [CrossRef]
- Yang, M.; Cai, L.; Tai, Z.; Zeng, X.; Ding, Z. Four new phenanthrenes from Monomeria barbata Lindl. Fitoterapia 2010, 81, 992–997. [Google Scholar] [CrossRef]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistika, D. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.; Fang, R.; Techatanawat, I.; Steventon, G.; Hylands, P.J.; Lee, C.C. The sulphorhodamine (SRB) assay and other approaches to testing plant extracts and derived compounds for activities related to reputed anticancer activity. Methods 2007, 42, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Takamura, H.; Matoba, T.; Terao, J. HPLC method for evaluation of the free radical-scavenging activity of foods by using 1,1-diphenyl-2-picrylhydrazyl. Biosci. Biotechnol. Biochem. 1998, 62, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Kapinova, A.; Kubatka, P.; Liskova, A.; Baranenko, D.; Kruzliak, P.; Matta, M.; Büsselberg, D.; Malicherova, B.; Zulli, A.; Kwon, T.K.; et al. Controlling metastatic cancer: The role of phytochemicals in cell signaling. J. Cancer Res. Clin. Oncol. 2019, 145, 1087–1109. [Google Scholar] [CrossRef] [PubMed]
- Samec, M.; Liskova, A.; · Kubatka, P.; Uramova, S.; Zubor, P.; Samuel, S.M.; Zulli, A.; Pec, M.; Bielik, T.; Biringer, K.; et al. The role of dietary phytochemicals in the carcinogenesis via the modulation of miRNA expression. J. Cancer Res. Clin. Oncol. 2019, 145, 1665–1679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, H.; Zhang, J.; Zhao, C.; Lu, S.; Qiao, J.; Han, M. The combinatory effects of natural products and chemotherapy drugs and their mechanisms in breast cancer treatment. Phytochem. Rev. 2019. [Google Scholar] [CrossRef]
- Abotaleb, M.; Kubatka, P.; Caprnda, M.; Varghese, E.; Zolakova, B.; Zubor, P.; Opatrilova, R.; Kruzliak, P.; Stefanicka, P.; Busselberg, D. Chemotherapeutic agents for the treatment of metastatic breast cancer: An update. Biomed. Pharmacother. 2018, 101, 458–477. [Google Scholar] [CrossRef] [PubMed]
- Kapinova, A.; Kubatka, P.; Golubnitschaja, O.; Kello, M.; Zubor, P.; Solar, P.; Pec, M. Dietary phytochemicals in breast cancer research: Anticancer effects and potential utility for effective chemoprevention. Environ. Health Prev. Med. 2018, 23, 1–18. [Google Scholar] [CrossRef]
- Nagaprashantha, L.D.; Adhikari, R.; Singhal, J.; Chikara, S.; Awasthi, S.; Horne, D.; Singha, S.S. Translational opportunities for broad-spectrum natural phytochemicals and targeted agent combinations in breast cancer. Int. J. Cancer 2018, 142, 658–670. [Google Scholar] [CrossRef]
- Wagner, H. Synergy research: Approaching a new generation of phytopharmaceuticals. Fitoterapia 2011, 82, 34–37. [Google Scholar] [CrossRef]
Sample Availability: Samples of compounds 13–15 are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
| Position | δH | δC | HMBC (H → C) | |
|---|---|---|---|---|
| 2J | 3J | |||
| 1 | - | 139.3 | ||
| 2 | 6.25 (d, 3.0) | 105.5 | C-3 | C-4, C-6, C-1a |
| 3 | - | 154.2 | ||
| 4 | - | 135.8 | ||
| 5 | - | 151.2 | ||
| 6 | 6.30 (d, 3.0) | 110.3 | C-5 | C-2, C-4, C-1a |
| 1a | 2.74 (m) | 39.4 | C-1, C-1b | C-2, C-6, C-1′ |
| 1b | 2.76 (m) | 38.6 | C-1a, C-1′ | C-1, C-2′, C-6′ |
| 1′ | - | 134.7 | ||
| 2′ | 6.64 (d, 2.0) | 113.5 | C-1′, C-3′ | C-1b, C-4′, C-6′ |
| 3′ | - | 148.7 | ||
| 4′ | - | 145.6 | ||
| 5′ | 6.68 (d, 9.0) | 116.0 | C-4′ | C-1′, C-3′ |
| 6′ | 6.60 (dd, 9.0, 2.0) | 122.0 | - | C-1b, C-2′, C-4′ |
| OCH3-4 | 3.74 (s) | 61.0 | - | C-4 |
| OCH3-3 | 3.75 (s) | 56.3 | - | C-3 |
| OCH3-3′ | 3.77 (s) | 56.3 | - | C-3′ |
| 1″ | - | 155.4 | - | |
| 2″, 6″ | 6.09 (s) | 94.0 | C-1″, C-3″,5″ | C-4″ |
| 3″, 5″ | - | 155.0 | ||
| 4″ | - | 132.2 | - | |
| OCH3-3″, 5″ | 3.77 (s) | 56.3 | - | C-3″,5″ |
| OCH3-4″ | 3.67 (s) | 61.3 | - | C-4″ |
| Position | 2 | 3 | 4 | 5 | 6 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | δH | δC | δH | δC | |
| 1 | 6.89 (d, 3.0) | 105.4 | 7.04 (s) | 109.8 | 6.30 (d, 3.0) | 108.6 | 6.50 (s) | 112.2 | 6.30 (d, 3.0) | 108.5 |
| 2 | - | 156.1 | - | 150.4 | - | 157.6 | - | 150.2 | - | 157.8 |
| 3 | 6.79 (d, 3.0) | 100.0 | - | 142.9 | 6.40 (d, 3.0) | 99.4 | - | 141.3 | 6.39 (d, 3.0) | 99.3 |
| 4 | - | 160.2 | - | 152.8 | - | 158.9 | - | 152.7 | - | 159.2 |
| 4a | - | 115.5 | - | 118.9 | - | 116.9 | - | 120.9 | - | 116.5 |
| 4b | - | 125.5 | - | 126.1 | - | 125.5 | - | 126.9 | - | 127.3 |
| 5 | 9.11 (s) | 109.6 | 8.90 (s) | 112.4 | 7.83 (s) | 113.7 | 7.78 (s) | 115.6 | 7.75 (s) | 116.5 |
| 6 | - | 148.3 | - | 147.4 | - | 146.6 | - | 145.5 | - | 144.9 |
| 7 | - | 145.8 | - | 148.4 | - | 145.2 | - | 147.2 | - | 146.6 |
| 8 | 7.24 (s) | 112.2 | 7.27 (s) | 109.6 | 6.62 (s) | 115.3 | 6.76 (s) | 112.3 | 6.75 (s) | 112.0 |
| 8a | - | 128.2 | - | 128.1 | - | 132.2 | - | 130.9 | - | 130.8 |
| 9 | 7.56 (d, 9.0) | 127.9 | 7.50 (d, 9.0) | 127.3 | 2.58 (m) | 32.1 | 2.60 (s) | 30.4 | 2.62 (s) | 32.2 |
| 10 | 7.44 (d, 9.0) | 125.4 | 7.33 (d, 9.0) | 124.9 | 2.60 (m) | 30.2 | 2.60 (s) | 31.5 | 2.62 (s) | 30.4 |
| 10a | - | 135.7 | - | 131.5 | - | 141.9 | - | 136.1 | - | 142.2 |
| OCH3-3 | - | - | 4.00 (s) | 61.5 | - | - | 3.85 (s) | 61.3 | - | - |
| OCH3-4 | 4.12 (s) | 56.0 | 3.98 (s) | 60.5 | 3.84 (s) | 56.1 | 3.70 (s) | 60.6 | 3.84 (s) | 56.4 |
| OCH3-6 | 4.02 (s) | 56.2 | - | - | 3.84 (s) | 56.7 | - | - | - | - |
| OCH3-7 | - | - | 3.99 (s) | 56.2 | - | - | 3.84 (s) | 56.4 | 3.83 (s) | 55.9 |
| Compound | 786-0 | MCF-7 | Hep2 | UACC-62 | NCI/ADR-RES |
|---|---|---|---|---|---|
| 1 | 112.86 ± 2.89 | 72.69 ± 4.87 | 218.27 ± 2.52 | NT | 32.09 ± 4.31 |
| 2 | 73.26 ± 7.70 | 118.40 ± 9.29 | > 250 | > 250 | 83.99 ± 5.40 |
| 3 | 64.27 ± 9.62 | 226.10 ± 5.09 | NT | 246.75 ± 10.32 | 116.88 ± 2.66 |
| 4 | 56.98 ± 9.29 | 46.99 ± 5.55 | 207.93 ± 17.09 | 2.59 ± 0.11 | 58.83 ± 2.33 |
| 5 | 199.46 ± 6.75 | 42.01 ± 9.33 | 222.61 ± 2.81 | 221.62 ± 3.04 | 212.03 ± 14.06 |
| 6 | 257.14 ± 6.51 | 160.20 ± 8.21 | 547.58 ± 0.11 | 268.24 ± 13.8 | 303.02 ± 12.58 |
| Cisplatin * | 20.66 ± 2.67 | 22.00 ± 2.93 | 5.00 ± 0.23 | 18.66 ± 3.73 | 25.32 ± 1.43 |
| Compound | IC50 (µM) |
|---|---|
| 1 | 56.5 ± 0.3 |
| 2 | 20.4 ± 0.3 |
| 3 | 45.6 ± 0.3 |
| 4 | 55.6 ± 0.4 |
| 5 | 32.9 ± 0.3 |
| 6 | 17.7 ± 0.3 |
| Caffeic acid (positive control) | 10.9 ± 0.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bisoli, E.; Freire, T.V.; Yoshida, N.C.; Garcez, W.S.; Queiróz, L.M.M.; Matos, M.d.F.C.; Perdomo, R.T.; Garcez, F.R. Cytotoxic Phenanthrene, Dihydrophenanthrene, and Dihydrostilbene Derivatives and Other Aromatic Compounds from Combretum laxum. Molecules 2020, 25, 3154. https://doi.org/10.3390/molecules25143154
Bisoli E, Freire TV, Yoshida NC, Garcez WS, Queiróz LMM, Matos MdFC, Perdomo RT, Garcez FR. Cytotoxic Phenanthrene, Dihydrophenanthrene, and Dihydrostilbene Derivatives and Other Aromatic Compounds from Combretum laxum. Molecules. 2020; 25(14):3154. https://doi.org/10.3390/molecules25143154
Chicago/Turabian StyleBisoli, Eder, Talita Vilalva Freire, Nídia Cristiane Yoshida, Walmir Silva Garcez, Lyara Meira Marinho Queiróz, Maria de Fátima Cepa Matos, Renata Trentin Perdomo, and Fernanda Rodrigues Garcez. 2020. "Cytotoxic Phenanthrene, Dihydrophenanthrene, and Dihydrostilbene Derivatives and Other Aromatic Compounds from Combretum laxum" Molecules 25, no. 14: 3154. https://doi.org/10.3390/molecules25143154
APA StyleBisoli, E., Freire, T. V., Yoshida, N. C., Garcez, W. S., Queiróz, L. M. M., Matos, M. d. F. C., Perdomo, R. T., & Garcez, F. R. (2020). Cytotoxic Phenanthrene, Dihydrophenanthrene, and Dihydrostilbene Derivatives and Other Aromatic Compounds from Combretum laxum. Molecules, 25(14), 3154. https://doi.org/10.3390/molecules25143154