
The Similarities between Human Mitochondria and Bacteria in the Context of Structure, Genome, and Base Excision Repair System



Abstract
1. Introduction
2. Common Features of Mitochondria and Bacteria
2.1. Structure
2.2. Fusion/Fission Systems
3. Human Mitochondrial DNA vs. Bacterial DNA
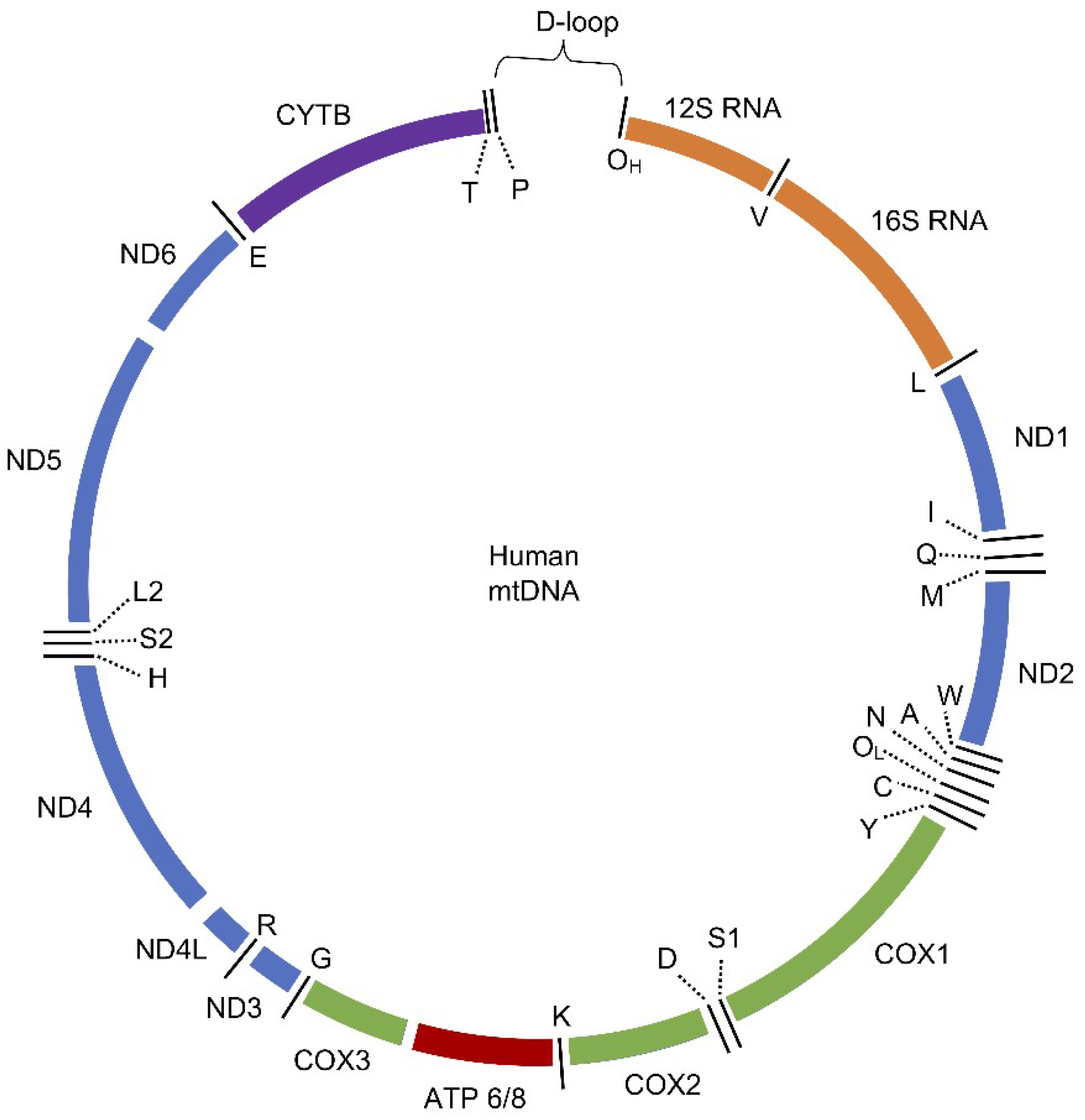
3.1. Human Mitochondrial DNA (mtDNA)
3.2. Comparison of Human mtDNA and Bacterial DNA
4. DNA Damage in Human Mitochondria
4.1. Oxidative DNA Damage
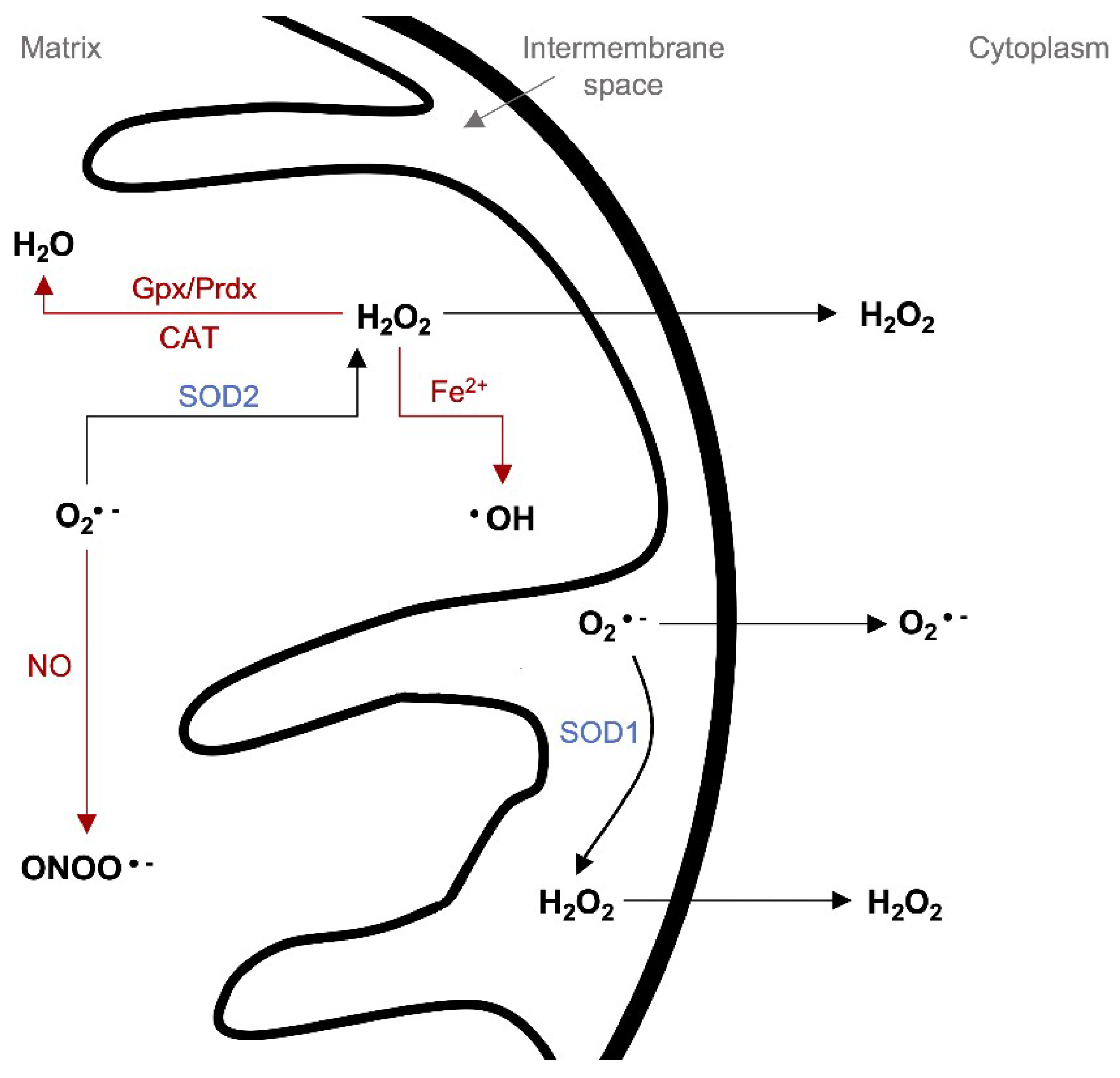
4.2. Mitochondrial ROS Production and Redox Homeostasis
5. Mitochondrial BER System
5.1. Mitochondrial BER Overview
5.2. BER Proteins in Human Mitochondria and Bacteria
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| mtDNA | Mitochondrial DNA |
| nDNA | Nuclear DNA |
| OXPHOS | Oxidative phosphorylation |
| ROS | Reactive oxygen species |
| BER | Base excision repair |
| SP-BER | Short-patch BER |
| LP-BER | Long-patch BER |
| ETC | Electron transport chain |
| IMM | Inner mitochondrial membrane |
| OMM | Outer mitochondrial membrane |
| CL | Cardiolipin |
| PG | Peptidoglycan |
| D-loop | Displacement loop |
| Polγ | DNA polymerase γ |
| PolI | DNA polymerase I |
| POLRMT | DNA-dependent RNA polymerase |
| TFAM | Mitochondrial transcription factor A |
| LigIII | DNA ligase III |
| 8-oxo-G | 8-oxoguanine |
| SSB | Single-strand break |
| DSB | Double strand break |
| AP site | Apurinic/apyrimidinic site |
| SOD | Superoxide dismutase |
| CAT | Catalase |
| Gpx | Glutathione peroxidase |
| Trx | Thioredoxin |
| Prdx | Peroxiredoxin |
| GSH | Glutathione |
| OGG1 | 8-oxoguanine glycosylase |
| NEIL | Nei-like endonuclease |
| NTH1 | Endonuclease III homolog 1 |
| MUTYH | Human adenine DNA glycosylase |
| UDG | Uracil DNA glycosylase |
| UNG | Uracil N-glycosylase |
| APE1 | AP endonuclease |
| PNKP | Polynucleotide kinase 3′-phosphatase |
| FEN1 | Flap endonuclease |
| MTS | Mitochondrial targeting sequence |
| Fpg | Formamidopyrimidine-DNA glycosylase |
| Nei | Endonuclease VIII |
| AAG | N-methylpurine DNA glycosylase |
| AlkA | Alkyladenine DNA glycosylase |
| MutY | Adenine DNA glycosylase |
| MutT | 8-oxo-dGTP diphosphatase |
References
- Martin, W.F.; Garg, S.; Zimorski, V. Endosymbiotic theories for eukaryote origin. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140330. [Google Scholar] [CrossRef] [PubMed]
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, 1177–1192. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, M. An integrated phylogenomic approach toward pinpointing the origin of mitochondria. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Eme, L.; Ettema, T.J.G. The eukaryotic ancestor shapes up. Nature 2018, 562, 352–353. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.H.; Conway, T. Evolution of carbohydrate metabolic pathways. Res. Microbiol. 1996, 147, 448–455. [Google Scholar] [CrossRef]
- Lange, B.M.; Rujan, T.; Martin, W.; Croteau, R. Isoprenoid biosynthesis: The evolution of two ancient and distinct pathways across genomes. Proc. Natl. Acad. Sci. USA 2000, 97, 13172–13177. [Google Scholar] [CrossRef]
- Gray, M.W. Mitochondrial evolution. Science 1999, 283. [Google Scholar] [CrossRef]
- Booth, A.; Doolittle, W.F. Eukaryogenesis, how special really? Proc. Natl. Acad. Sci. USA 2015, 112, 10278–10285. [Google Scholar] [CrossRef]
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxid. Med. Cell. Longev. 2017, 2017, 1–15. [Google Scholar] [CrossRef]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Qian, L.; Sung, J.-S.; De Souza-Pinto, N.C.; Zheng, L.; Bogenhagen, D.F.; Bohr, V.A.; Wilson, D.M.; Shen, B.; Demple, B. Removal of Oxidative DNA Damage via FEN1-Dependent Long-Patch Base Excision Repair in Human Cell Mitochondria. Mol. Cell. Biol. 2008, 28, 4975–4987. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Visnes, T.; Krokan, H.E.; Otterlei, M. Mitochondrial base excision repair of uracil and AP sites takes place by single-nucleotide insertion and long-patch DNA synthesis. DNA Repair 2008, 7, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Holzerová, E.; Prokisch, H. Mitochondria: Much ado about nothing? How dangerous is reactive oxygen species production? Int. J. Biochem. Cell Biol. 2015, 63, 16–20. [Google Scholar] [CrossRef]
- Burke, P.J. Mitochondria, Bioenergetics & Apoptosis in Cancer. Trends Cancer 2017, 3, 857–870. [Google Scholar] [CrossRef]
- Meyer, A.; Laverny, G.; Bernardi, L.; Charles, A.L.; Alsaleh, G.; Pottecher, J.; Sibilia, J.; Geny, B. Mitochondria: An organelle of bacterial origin controlling inflammation. Front. Immunol. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A 40 kDa protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Lill, R.; Hoffmann, B.; Molik, S.; Pierik, A.J.; Rietzschel, N.; Stehling, O.; Uzarska, M.A.; Webert, H.; Wilbrecht, C.; Mühlenhoff, U. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta 2012, 1823, 1491–1508. [Google Scholar] [CrossRef]
- Veatch, J.R.; McMurray, M.A.; Nelson, Z.W.; Gottschling, D.E. Mitochondrial dysfunction leads to nuclear genome instability: A link through iron-sulfur clusters. Cell 2009, 137, 1247–1258. [Google Scholar] [CrossRef]
- Lackner, L.L. Determining the shape and cellular distribution of mitochondria: The integration of multiple activities. Curr. Opin. Cell Biol. 2013, 25, 471–476. [Google Scholar] [CrossRef]
- D’Erchia, A.M.; Atlante, A.; Gadaleta, G.; Pavesi, G.; Chiara, M.; De Virgilio, C.; Manzari, C.; Mastropasqua, F.; Prazzoli, G.M.; Picardi, E.; et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 2015, 20, 13–21. [Google Scholar] [CrossRef]
- Taylor, S.W.; Fahy, E.; Zhang, B.; Glenn, G.M.; Warnock, D.E.; Wiley, S.; Murphy, A.N.; Gaucher, S.P.; Capaldi, R.A.; Gibson, B.W.; et al. Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 2003, 21, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Pagliuso, A.; Cossart, P.; Stavru, F. The ever-growing complexity of the mitochondrial fission machinery. Cell. Mol. Life Sci. 2018, 75, 355–374. [Google Scholar] [CrossRef]
- Bartolák-Suki, E.; Imsirovic, J.; Nishibori, Y.; Krishnan, R.; Suki, B. Regulation of Mitochondrial Structure and Dynamics by the Cytoskeleton and Mechanical Factors. Int. J. Mol. Sci. 2017, 18, 1812. [Google Scholar] [CrossRef] [PubMed]
- Boesch, P.; Weber-Lotfi, F.; Ibrahim, N.; Tarasenko, V.; Cosset, A.; Paulus, F.; Lightowlers, R.N.; Dietrich, A. DNA repair in organelles: Pathways, organization, regulation, relevance in disease and aging. Biochim. Biophys. Acta 2011, 1813, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, D.P.; Salter, M.W. Strangers in strange lands: Mitochondrial proteins found at extra-mitochondrial locations. Biochem. J. 2019, 476, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Mootha, V.K. The Mitochondrial Proteome and Human Disease. Annu. Rev. Genomics Hum. Genet. 2010, 11, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.; Song, J.; Pfanner, N. Versatility of Preprotein Transfer from the Cytosol to Mitochondria. Trends Cell Biol. 2019, 29, 534–548. [Google Scholar] [CrossRef]
- Ryan, M.T. Mitochondria—The energy powerhouses. Semin. Cell Dev. Biol. 2018, 76, 130–131. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef] [PubMed]
- Frohman, M.A. Role of mitochondrial lipids in guiding fission and fusion. J. Mol. Med. 2015, 93, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Mileykovskaya, E.; Dowhan, W. Cardiolipin Membrane Domains in Prokaryotes and Eukaryotes. Biochim. Biophys. Acta 2009, 1788, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Kojima, R.; Kakimoto, Y.; Furuta, S.; Itoh, K.; Sesaki, H.; Endo, T.; Tamura, Y. Maintenance of Cardiolipin and Crista Structure Requires Cooperative Functions of Mitochondrial Dynamics and Phospholipid Transport. Cell Rep. 2019, 26, 518–528. [Google Scholar] [CrossRef]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.-G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef]
- Schulz, H.N.; Jørgensen, B.B. Big acteria. Annu. Rev. Microbiol. 2001, 55, 105–137. [Google Scholar] [CrossRef]
- Taheri-Araghi, S.; Bradde, S.; Sauls, J.T.; Hill, N.S.; Levin, P.A.; Paulsson, J.; Vergassola, M.; Jun, S. Cell-size control and homeostasis in bacteria. Curr. Biol. 2015, 25, 385–391. [Google Scholar] [CrossRef]
- Prashar, A.; Bhatia, S.; Gigliozzi, D.; Martin, T.; Duncan, C.; Guyard, C.; Terebiznik, M.R. Filamentous morphology of bacteria delays the timing of phagosome morphogenesis in macrophages. J. Cell Biol. 2013, 203, 1081–1097. [Google Scholar] [CrossRef]
- Williams, C. Who are you calling simple? New Sci. 2011, 211, 38–41. [Google Scholar] [CrossRef]
- Ng, W.; Bassler, B.L. Bacterial Quorum-Sensing Network Architectures. Annu. Rev. Genet. 2009, 43, 197–222. [Google Scholar] [CrossRef]
- Sochacki, K.A.; Shkel, I.A.; Record, M.T.; Weisshaar, J.C. Protein diffusion in the periplasm of E. coli under osmotic stress. Biophys. J. 2011, 100, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Cabeen, M.T.; Jacobs-Wagner, C. Bacterial cell shape. Nat. Rev. Microbiol. 2005, 3, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; London, E. Cholesterol lipids and cholesterol-containing lipid rafts in bacteria. Chem. Phys. Lipids 2016, 199, 11–16. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bramkamp, M.; Lopez, D. Exploring the Existence of Lipid Rafts in Bacteria. Microbiol. Mol. Biol. Rev. 2015, 79, 81–100. [Google Scholar] [CrossRef]
- Michel, V.; Bakovic, M. Lipid rafts in health and disease. Biol. Cell 2007, 99, 129–140. [Google Scholar] [CrossRef]
- Cossins, B.P.; Jacobson, M.P.; Guallar, V. A new view of the bacterial cytosol environment. PLoS Comput. Biol. 2011, 7. [Google Scholar] [CrossRef]
- Hoppert, M.; Mayer, F. Principles of macromolecular organization and cell function in bacteria and archaea. Cell Biochem. Biophys. 1999, 31, 247–284. [Google Scholar] [CrossRef]
- Lutkenhaus, J. Bacterial cytokinesis: Let the light shine in. Curr. Biol. 1997, 7, 573–575. [Google Scholar] [CrossRef]
- Baranova, N.; Radler, P.; Hernández-Rocamora, V.M.; Alfonso, C.; López-Pelegrín, M.; Rivas, G.; Vollmer, W.; Loose, M. Diffusion and capture permits dynamic coupling between treadmilling FtsZ filaments and cell division proteins. Nat. Microbiol. 2020, 5, 407–417. [Google Scholar] [CrossRef]
- Strahl, H.; Bürmann, F.; Hamoen, L.W. The actin homologue MreB organizes the bacterial cell membrane. Nat. Commun. 2014, 5, 3442. [Google Scholar] [CrossRef]
- Wernegreen, J.J. Endosymbiont evolution: Predictions from theory and surprises From Genomes. Ann. N. Y. Acad. Sci. 2016, 1360, 71. [Google Scholar] [CrossRef] [PubMed]
- Harish, A.; Kurland, C.G. Akaryotes and Eukaryotes are independent descendants of a universal common ancestor. Biochimie 2017, 138, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Archibald, J.M. Endosymbiosis and eukaryotic cell evolution. Curr. Biol. 2015, 25, R911–R921. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Benador, I.; Miller, N.; Shirihai, O.S. Mitochondrial Function, Dynamics, and Quality Control. In The Liver: Biology and Pathology; Arias, I.M., Alter, H.J., Boyer, J.L., Cohen, D.E., Shafritz, D.A., Thorgeirsson, S.S., Wolkoff, A.W., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2020; pp. 86–93. [Google Scholar]
- Carré, M.; André, N.; Carles, G.; Borghi, H.; Brichese, L.; Briand, C.; Braguer, D. Tubulin Is an Inherent Component of Mitochondrial Membranes That Interacts with the Voltage-dependent Anion Channel. J. Biol. Chem. 2002, 277, 33664–33669. [Google Scholar] [CrossRef]
- Torralba, D.; Baixauli, F.; Sánchez-Madrid, F. Mitochondria know no boundaries: Mechanisms and functions of intercellular mitochondrial transfer. Front. Cell Dev. Biol. 2016, 4, 1–11. [Google Scholar] [CrossRef]
- Hahn, A.; Zuryn, S. The Cellular Mitochondrial Genome Landscape in Disease. Trends Cell Biol. 2019, 29, 227–240. [Google Scholar] [CrossRef]
- Berridge, M.V.; McConnell, M.J.; Grasso, C.; Bajzikova, M.; Kovarova, J.; Neuzil, J. Horizontal transfer of mitochondria between mammalian cells: Beyond co-culture approaches. Curr. Opin. Genet. Dev. 2016, 38, 75–82. [Google Scholar] [CrossRef]
- Xia, M.F.; Zhang, Y.Z.; Jin, K.; Lu, Z.T.; Zeng, Z.; Xiong, W. Communication between mitochondria and other organelles: A brand-new perspective on mitochondria in cancer. Cell Biosci. 2019, 9, 1–19. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef]
- Yutin, N.; Wolf, M.Y.; Wolf, Y.I.; Koonin, E.V. The origins of phagocytosis and eukaryogenesis. Biol. Direct 2009, 4. [Google Scholar] [CrossRef]
- Muñoz-Gómez, S.A.; Wideman, J.G.; Roger, A.J.; Slamovits, C.H.; Agashe, D. The origin of mitochondrial cristae from alphaproteobacteria. Mol. Biol. Evol. 2017, 34, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.E. The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 2013, 41, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Suzuki, T.; Rubinstein, J.L. Structure of a bacterial atp synthase. Elife 2019, 8, 1–17. [Google Scholar] [CrossRef]
- Pallen, M.J. Time to recognise that mitochondria are bacteria? Trends Microbiol. 2011, 19, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.; Pich, S.; Soriano, F.X.; Vega, N.; Baumgartner, B.; Oriola, J.; Daugaard, J.R.; Lloberas, J.; Camps, M.; Zierath, J.R.; et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism: A novel regulatory mechanism altered in obesity. J. Biol. Chem. 2003, 278, 17190–17197. [Google Scholar] [CrossRef]
- Farmer, T.; Naslavsky, N.; Caplan, S. Tying trafficking to fusion and fission at the mighty mitochondria. Traffic 2018, 19, 569–577. [Google Scholar] [CrossRef]
- Ciarlo, L.; Vona, R.; Manganelli, V.; Gambardella, L.; Raggi, C.; Marconi, M.; Malorni, W.; Sorice, M.; Garofalo, T.; Matarrese, P. Recruitment of mitofusin 2 into “lipid rafts” drives mitochondria fusion induced by Mdivi-1. Oncotarget 2018, 9, 18869–18884. [Google Scholar] [CrossRef]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtdna stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Tábara, L.C.; Tilokani, L.; Paupe, V.; Anand, H.; Pogson, J.H.; Zunino, R.; McBride, H.M.; Prudent, J. Golgi-derived PI(4)P-containing vesicles drive late steps of mitochondrial division. Science 2020, 367, 1366–1371. [Google Scholar] [CrossRef] [PubMed]
- Schnupf, P.; Gaboriau-Routhiau, V.; Gros, M.; Friedman, R.; Moya-Nilges, M.; Nigro, G.; Cerf-Bensussan, N.; Sansonetti, P.J. Growth and host interaction of mouse segmented filamentous bacteria in vitro. Nature 2015, 520, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Vedyaykin, A.D.; Ponomareva, E.V.; Khodorkovskii, M.A.; Borchsenius, S.N.; Vishnyakov, I.E. Mechanisms of Bacterial Cell Division. Microbiology 2019, 88, 245–260. [Google Scholar] [CrossRef]
- Harry, E.; Monahan, L.; Thompson, L. Bacterial Cell Division: The Mechanism and Its Precison. Int. Rev. Cytol. 2006, 253, 27–94. [Google Scholar] [CrossRef]
- Spier, A.; Sachse, M.; Tham, N.T.; Matondo, M.; Cossart, P.; Stavru, F. Bacterial FtsZ Induces Mitochondrial Fission in Human Cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Med. 2013, 3, 1–47. [Google Scholar] [CrossRef]
- Wang, Y.; Bogenhagen, D.F. Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J. Biol. Chem. 2006, 281, 25791–25802. [Google Scholar] [CrossRef]
- Harner, M.; Körner, C.; Walther, D.; Mokranjac, D.; Kaesmacher, J.; Welsch, U.; Griffith, J.; Mann, M.; Reggiori, F.; Neupert, W. The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J. 2011, 30, 4356–4370. [Google Scholar] [CrossRef]
- Bonekamp, N.A.; Larsson, N.G. SnapShot: Mitochondrial Nucleoid. Cell 2018, 172, 388–388.e1. [Google Scholar] [CrossRef] [PubMed]
- Kukat, C.; Wurm, C.A.; Spåhr, H.; Falkenberg, M.; Larsson, N.G.; Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H.S.; Temiakov, D.; Cramer, P. Structural basis of mitochondrial transcription. Nat. Struct. Mol. Biol. 2018, 25, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Robberson, D.L.; Kasamatsu, H.; Vinograd, J. Replication of mitochondrial DNA. Circular replicative intermediates in mouse L cells. Proc. Natl. Acad. Sci. USA 1972, 69, 737–741. [Google Scholar] [CrossRef]
- Yasukawa, T.; Kang, D. An overview of mammalian mitochondrial DNA replication mechanisms. J. Biochem. 2018, 164, 183–193. [Google Scholar] [CrossRef]
- Zinovkina, L.A. DNA Replication in Human Mitochondria. Biochemistry 2019, 84, 884–895. [Google Scholar] [CrossRef]
- Herbers, E.; Kekäläinen, N.J.; Hangas, A.; Pohjoismäki, J.L.; Goffart, S. Tissue specific differences in mitochondrial DNA maintenance and expression. Mitochondrion 2019, 44, 85–92. [Google Scholar] [CrossRef]
- Zheng, W.; Khrapko, K.; Coller, H.A.; Thilly, W.G.; Copeland, W.C. Origins of human mitochondrial point mutations as DNA polymerase γ-mediated errors. Mutat. Res. Fundam. Mol. Mech. Mutagenes. 2006, 599, 11–20. [Google Scholar] [CrossRef]
- Kotrys, A.V.; Szczesny, R.J. Mitochondrial Gene Expression and Beyond—Novel Aspects of Cellular Physiology. Cells 2020, 9, 17. [Google Scholar] [CrossRef]
- Litonin, D.; Sologub, M.; Shi, Y.; Savkina, M.; Anikin, M.; Falkenberg, M.; Gustafsson, C.M.; Temiakov, D. Human mitochondrial transcription revisited: Only TFAM and TFB2M are required for transcription of the mitochondrial genes in vitro. J. Biol. Chem. 2010, 285, 18129–18133. [Google Scholar] [CrossRef]
- Terzioglu, M.; Ruzzenente, B.; Harmel, J.; Mourier, A.; Jemt, E.; López, M.D.; Kukat, C.; Stewart, J.B.; Wibom, R.; Meharg, C.; et al. MTERF1 Binds mtDNA to prevent transcriptional interference at the light-strand promoter but is dispensable for rRNA gene transcription regulation. Cell Metab. 2013, 17, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Hällberg, B.M.; Larsson, N.G. Making proteins in the powerhouse. Cell Metab. 2014, 20, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.K.; Sharma, M.R. Structural aspects of mitochondrial translational apparatus. Curr. Opin. Struct. Biol. 2012, 22, 797–803. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.R.; Minczuk, M. Mitochondrial transcription and translation: Overview. Essays Biochem. 2018, 62, 309–320. [Google Scholar] [CrossRef]
- Yarham, J.W.; Al-Dosary, M.; Blakely, E.L.; Alston, C.L.; Taylor, R.W.; Elson, J.L.; Mcfarland, R. A comparative analysis approach to determining the pathogenicity of mitochondrial tRNA mutations. Hum. Mutat. 2011, 32, 1319–1325. [Google Scholar] [CrossRef]
- Blakely, E.L.; Yarham, J.W.; Alston, C.L.; Craig, K.; Poulton, J.; Brierley, C.; Park, S.M.; Dean, A.; Xuereb, J.H.; Anderson, K.N.; et al. Pathogenic mitochondrial tRNA point mutations: Nine novel mutations affirm their importance as a cause of mitochondrial disease. Hum. Mutat. 2013, 34, 1260–1268. [Google Scholar] [CrossRef]
- Garcia, I.; Jones, E.; Ramos, M.; Innis-Whitehouse, W.; Gilkerson, R. The little big genome: The organization of mitochondrial DNA. Front. Biosci. 2017, 22, 710–721. [Google Scholar]
- Van Haute, L.; Powell, C.A.; Minczuk, M. Dealing with an unconventional genetic code in mitochondria: The biogenesis and pathogenic defects of the 5-formylcytosine modification in mitochondrial tRNAMet. Biomolecules 2017, 7, 24. [Google Scholar] [CrossRef]
- Chemale, G.; Paneto, G.G.; Menezes, M.A.M.; De Freitas, J.M.; Jacques, G.S.; Cicarelli, R.M.B.; Fagundes, P.R. Development and validation of a D-loop mtDNA SNP assay for the screening of specimens in forensic casework. Forensic Sci. Int. Genet. 2013, 7, 353–358. [Google Scholar] [CrossRef]
- Stroud, D.A.; Surgenor, E.E.; Formosa, L.E.; Reljic, B.; Frazier, A.E.; Dibley, M.G.; Osellame, L.D.; Stait, T.; Beilharz, T.H.; Thorburn, D.R.; et al. Accessory subunits are integral for assembly and function of human mitochondrial complex i. Nature 2016, 538, 123–126. [Google Scholar] [CrossRef]
- Sousa, J.S.; D’Imprima, E.; Vonck, J. Mitochondrial Respiratory Chain Complexes. In Subcellular Biochemistry; Springer Nature Switzerland AG.: Cham, Switzerland, 2018; ISBN 9789811077579. [Google Scholar]
- Allen, J.F. Why chloroplasts and mitochondria retain their own genomes and genetic systems: Colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, 10231–10238. [Google Scholar] [CrossRef] [PubMed]
- Björkholm, P.; Ernst, A.M.; Hagström, E.; Andersson, S.G.E. Why mitochondria need a genome revisited. FEBS Lett. 2017, 591, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Bobay, L.M.; Ochman, H. The evolution of bacterial genome architecture. Front. Genet. 2017, 8, 1–6. [Google Scholar] [CrossRef]
- Trevors, J.T. Genome size in bacteria. Antonie Leeuwenhoek Int. J. Gen. Mol. Microbiol. 1996, 69, 293–303. [Google Scholar] [CrossRef]
- Nilsson, A.I.; Koskiniemi, S.; Eriksson, S.; Kugelberg, E.; Hinton, J.C.D.; Andersson, D.I. Bacterial genome size reduction by experimental evolution. Proc. Natl. Acad. Sci. USA 2005, 102, 12112–12116. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.C.; Dorman, C.J. Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 2010, 8, 185–195. [Google Scholar] [CrossRef]
- Wang, W.; Li, G.W.; Chen, C.; Xie, X.S.; Zhuang, X. Chromosome organization by a nucleoid-associated protein in live bacteria. Science 2011, 333, 1445–1449. [Google Scholar] [CrossRef] [PubMed]
- Ranea, J.A.G.; Buchan, D.W.A.; Thornton, J.M.; Orengo, C.A. Evolution of Protein Superfamilies and Bacterial Genome Size. J. Mol. Biol. 2004, 336, 871–887. [Google Scholar] [CrossRef]
- Almpanis, A.; Swain, M.; Gatherer, D.; McEwan, N. Correlation between bacterial G+C content, genome size and the G+C content of associated plasmids and bacteriophages. Microb. Genom. 2018, 4, 168. [Google Scholar] [CrossRef]
- Woldringh, C.L.; Jensen, P.R.; Westerhoff, H.V. Structure and partitioning of bacterial DNA: Determined by a balance of compaction and expansion forces? FEMS Microbiol. Lett. 1995, 131, 235–242. [Google Scholar] [CrossRef]
- Robinson, A.; Van Oijen, A.M. Bacterial replication, transcription and translation: Mechanistic insights from single-molecule biochemical studies. Nat. Rev. Microbiol. 2013, 11, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Rodnina, M.V. Translation in prokaryotes. Cold Spring Harb. Perspect. Biol. 2018, 10, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Dorman, C.J.; Dorman, M.J. DNA supercoiling is a fundamental regulatory principle in the control of bacterial gene expression. Biophys. Rev. 2016, 8, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.E. β-Lactamase-producing enterococci. Antimicrob. Agents Chemother. 1992, 36, 2355–2359. [Google Scholar] [CrossRef] [PubMed]
- Barshad, G.; Marom, S.; Cohen, T.; Mishmar, D. Mitochondrial DNA Transcription and Its Regulation: An Evolutionary Perspective. Trends Genet. 2018, 34, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Akhmedov, A.T.; Marín-García, J. Mitochondrial DNA maintenance: An appraisal. Mol. Cell. Biochem. 2015, 409, 283–305. [Google Scholar] [CrossRef]
- Whitaker, A.M.; Schaich, M.A.; Smith, M.S.; Flynn, T.S.; Freudenthal, B.D. Base excision repair of oxidative DNA damage from mechanism to disease. Front. Biosci. 2017, 22, 1493–1522. [Google Scholar] [CrossRef]
- Ba, X.; Boldogh, L. 8-Oxoguanine DNA glycosylase 1: Beyond repair of the oxidatively modified base lesions. Redox Biol. 2018, 14, 669–678. [Google Scholar] [CrossRef]
- Greenberg, M.M. The Formamidopyrimidines: Purine Lesions Formed in Competition With 8-Oxopurines From Oxidative Stress. ACC Chem. Res. 2012, 45, 588–597. [Google Scholar] [CrossRef]
- Kusumoto, R.; Masutani, C.; Iwai, S.; Hanaoka, F. Translesion synthesis by human DNA polymerase η across thymine glycol lesions. Biochemistry 2002, 41, 6090–6099. [Google Scholar] [CrossRef]
- Dobbs, T.A.; Palmer, P.; Maniou, Z.; Lomax, M.E.; O’Neill, P. Interplay of two major repair pathways in the processing of complex double-strand DNA breaks. DNA Repair (Amst) 2008, 7, 1372–1383. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lepe, U.O.; Bermudez-Cruz, R. Mitochondrial Genome Maintenance: Damage and Repair Pathways. In DNA Repair—An Update; IntechOpen: London, UK, 2019; p. 21. ISBN 978-1-83880-784-9. [Google Scholar]
- Posse, V.; Shahzad, S.; Falkenberg, M.; Hällberg, B.M.; Gustafsson, C.M. TEFM is a potent stimulator of mitochondrial transcription elongation in vitro. Nucleic Acids Res. 2015, 43, 2615–2624. [Google Scholar] [CrossRef] [PubMed]
- Das, K.C.; White, C.W. Redox systems of the cell: Possible links and implications. Proc. Natl. Acad. Sci. USA 2002, 99, 9617–9618. [Google Scholar] [CrossRef] [PubMed]
- Bazinet, L.; Doyen, A. Antioxidants, mechanisms, and recovery by membrane processes. Crit. Rev. Food Sci. Nutr. 2017, 57, 677–700. [Google Scholar] [CrossRef] [PubMed]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Laude, K.; Cai, H. Mitochondrial Pathophysiology, Reactive Oxygen Species, and Cardiovascular Diseases. Vet. Clin. N. Am. Small Anim. Pract. 2008, 38, 137–154. [Google Scholar] [CrossRef]
- Inoue, M.; Sato, E.F.; Nishikawa, M.; Park, A.-M.; Kira, Y.; Imada, I.; Utsumi, K. Mitochondrial Generation of Reactive Oxygen Species and its Role in Aerobic Life. Curr. Med. Chem. 2005, 10, 2495–2505. [Google Scholar] [CrossRef]
- Venditti, P.; Di Stefano, L.; Di Meo, S. Mitochondrial metabolism of reactive oxygen species. Mitochondrion 2013, 13, 71–82. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Antonarakis, S.E.; Lyle, R.; Dermitzakis, E.T.; Reymond, A.; Deutsch, S. Chromosome 21 and Down syndrome: From genomics to pathophysiology. Nat. Rev. Genet. 2004, 5, 725–738. [Google Scholar] [CrossRef]
- Coskun, P.E.; Busciglio, J. Oxidative stress and mitochondrial dysfunction in Down’s syndrome: Relevance to aging and dementia. Curr. Gerontol. Geriatr. Res. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Gulesserian, T.; Seidl, R.; Hardmeier, R.; Cairns, N.; Lubec, G. Superoxide dismutase SOD1, encoded on chromosome 21, but not SOD2 is overexpressed in brains of patients with Down Syndrome. J. Investig. Med. 2001, 49, 41–46. [Google Scholar] [CrossRef]
- Patel, Y.; Moraes, Y.C.; Latchman, D.; Coffin, R.; De Belleroche, J. Neuroprotective effects of copper/zinc-dependent superoxide dismutase against a wide variety of death-inducing stimuli and proapoptotic effect of familial amyotrophic lateral sclerosis mutations. Mol. Brain Res. 2002, 109, 189–197. [Google Scholar] [CrossRef]
- Cristiano, F.; De Haan, J.B.; Iannello, R.C.; Kola, I. Changes in the levels of enzymes which modulate the antioxidant balance occur during aging and correlate with cellular damage. Mech. Ageing Dev. 1995, 80, 93–105. [Google Scholar] [CrossRef]
- Lei, X.G.; Zhu, J.H.; Cheng, W.H.; Bao, Y.; Ho, Y.S.; Reddi, A.R.; Holmgren, A.; Arnér, E.S.J. Paradoxical roles of antioxidant enzymes: Basic mechanisms and health implications. Physiol. Rev. 2015, 96, 307–364. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R. DNA Base Damage by Reactive Oxygen Species, Oxidizing Agents, and UV Radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Semenza, G.L. Maintenance of redox homeostasis by hypoxia-inducible factors. Redox Biol. 2017, 13, 331–335. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef]
- Ji, Y.; Dai, Z.; Wu, G.; Wu, Z. 4-Hydroxy-2-nonenal induces apoptosis by activating ERK1/2 signaling and depleting intracellular glutathione in intestinal epithelial cells. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed]
- McBee, M.E.; Chionh, Y.H.; Sharaf, M.L.; Ho, P.; Cai, M.W.L.; Dedon, P.C. Production of superoxide in bacteria is stress- and cell state-dependent: A gating-optimized flow cytometry method that minimizes ROS measurement artifacts with fluorescent dyes. Front. Microbiol. 2017, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A. The molecular mechanisms and physiological consequences of oxidative stress: Lessons from a model bacterium. Nat. Rev. Microbiol. 2013, 11, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Janowska, B.; Komisarski, M.; Prorok, P.; Sokołowska, B.; Kuśmierek, J.; Janion, C.; Tudek, B. Nucleotide excision repair and recombination are engaged in repair of trans-4-hydroxy-2-nonenal adducts to DNA bases in Escherichia coli. Int. J. Biol. Sci. 2009, 5, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Alnajjar, K.S.; Sweasy, J.B. A new perspective on oxidation of DNA repair proteins and cancer. DNA Repair (Amst) 2019, 76, 60–69. [Google Scholar] [CrossRef]
- Muftuoglu, M.; Mori, M.P.; Souza-Pinto, N.C.D. Formation and repair of oxidative damage in the mitochondrial DNA. Mitochondrion 2014, 17, 164–181. [Google Scholar] [CrossRef]
- Copeland, W.C. The Mitochondrial DNA Polymerase in Health and Disease. Subcell. Biochem. 2010, 50, 17–42. [Google Scholar] [CrossRef]
- Zharkov, D.O. Base excision DNA repair. Cell. Mol. Life Sci. 2008, 65, 1544–1565. [Google Scholar] [CrossRef]
- Krwawicz, J.; Arczewska, K.D.; Speina, E.; Maciejewska, A.; Grzesiuk, E. Bacterial DNA repair genes and their eukaryotic homologues: 1. Mutations in genes involved in base excision repair (BER) and DNA-end processors and their implication in mutagenesis and human disease. Acta Biochim. Pol. 2007, 54, 413–434. [Google Scholar] [CrossRef]
- Wallace, S. Base excision repair: A critical player in many games. DNA Repair (Amst) 2014, 19, 14–26. [Google Scholar] [CrossRef]
- Krasich, R.; Copeland, W.C. DNA polymerases in the mitochondria: A critical review of the evidence. Front. Biosci. 2017, 22, 692–709. [Google Scholar] [CrossRef]
- Zinovkina, L.A. Mechanisms of Mitochondrial DNA Repair in Mammals. Biochemistry 2018, 83, 233–249. [Google Scholar] [CrossRef] [PubMed]
- De Souza-Pinto, N.C.; Aamann, M.D.; Kulikowicz, T.; Stevnsner, T.V.; Bohr, V.A. Mitochondrial helicases and mitochondrial genome maintenance. Mech. Ageing Dev. 2010, 131, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Ravanat, J.-L.; TavernaPorro, M.; Menoni, H.; Angelov, D. Oxidatively generated complex DNA damage: Tandem and clustered lesions. Cancer Lett. 2012, 327, 5–15. [Google Scholar] [CrossRef]
- Lomax, M.E.; Cunniffe, S.; O’Neill, P. 8-OxoG retards the activity of the ligase III/XRCC1 complex during the repair of a single-strand break, when present within a clustered DNA damage site. DNA Repair (Amst) 2004, 3, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Cunniffe, S.; O’Neill, P. Efficiency of repair of an abasic site within DNA clustered damage sites by mammalian cell nuclear extracts. Biochemistry 2004, 43, 11017–11026. [Google Scholar] [CrossRef]
- Canugovi, C.; Maynard, S.; Bayne, A.C.V.; Sykora, P.; Tian, J.; De Souza-Pinto, N.C.; Croteau, D.L.; Bohr, V.A. The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair (Amst) 2010, 9, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Doublie, S. Base Excision Repair in the Mitochondria. J. Cell Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base excision repair. DNA Repair Genet. Instab. Cancer 2013, 5, 1–22. [Google Scholar] [CrossRef]
- Van Der Veen, S.; Tang, C.M. The BER necessities: The repair of DNA damage in human-adapted bacterial pathogens. Nat. Rev. Microbiol. 2015, 13, 83–94. [Google Scholar] [CrossRef]
- Lindahl, T. An N-Glycosidase from Escherichia coli That Releases Free Uracil from DNA Containing Deaminated Cytosine Residues Biochemistry: Lindahl. Proc. Natl. Acad. Sci. USA 1974, 71, 3649–3653. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Nilsen, H.; Skorpen, F.; Skjelbred, C.; Akbari, M.; Arne, P.; As, A.; Slupphaug, G. Properties and Functions of Human UraciI-DNA Glycosylase from the UNG Gene. Nucleic Acids Res. 2001, 68, 365–386. [Google Scholar]
- De Souza-Pinto, N.C.; Eide, L.; Hogue, B.A.; Thybo, T.; Stevnsner, T.; Seeberg, E.; Klungland, A.; Bohr, V.A. Repair of 8-oxodeoxyguanosine lesions in mitochondrial DNA depends on the oxoguanine DNA glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial DNA of OGG1-defective mice. Cancer Res. 2001, 61, 5378–5381. [Google Scholar]
- Hwang, B.J.; Shi, G.; Lu, A.L. Mammalian MutY homolog (MYH or MUTYH) protects cells from oxidative DNA damage. DNA Repair (Amst) 2014, 13, 10–21. [Google Scholar] [CrossRef]
- Takahashi, M.; Yamagata, Y.; Iwai, S.; Nakabeppu, Y. Structure of Human MTH1, a Nudix Family Hydrolase That Selectively Degrades Oxidized Purine Nucleoside Triphosphates. J. Biol. Chem. 2004, 279, 33806–33815. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Kajitani, K.; Sakamoto, K. Mini review MTH1, an oxidized purine nucleoside triphosphatase, prevents the cytotoxicity and neurotoxicity of oxidized purine nucleotides. DNA Repair (Amst) 2006, 5, 761–772. [Google Scholar] [CrossRef]
- Mullins, E.A.; Shi, R.; Eichman, B.F. Toxicity and repair of DNA adducts produced by the natural product yatakemycin. Nat. Chem. Biol. 2017, 13, 1002–1008. [Google Scholar] [CrossRef]
- Shi, R.; Mullins, E.A.; Shen, X.; Lay, K.T.; Yuen, P.K.; David, S.S.; Rokas, A.; Eichman, B.F. Selective base excision repair of DNA damage by the non-base-flipping DNA glycosylase AlkC. EMBO J. 2018, 37, 63–74. [Google Scholar] [CrossRef]
- Wang, L.K.; Das, U.; Smith, P.; Shuman, S. Structure and mechanism of the polynucleotide kinase component of the bacterial Pnkp-Hen1 RNA repair system. RNA 2012, 18, 2277–2286. [Google Scholar] [CrossRef]
- Tsuchimoto, D. Human APE2 protein is mostly localized in the nuclei and to some extent in the mitochondria, while nuclear APE2 is partly associated with proliferating cell nuclear antigen. Nucleic Acids Res. 2001, 29, 2349–2360. [Google Scholar] [CrossRef]
- Frossi, B.; Tell, G.; Spessotto, P.; Colombatti, A.; Vitale, G.; Pucillo, C. H2O2 induces translocation of APE/Ref-1 to mitochondria in the Raji B-cell line. J. Cell. Physiol. 2002, 193, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Hadi, M.Z.; Wilson, D.M. Second human protein with homology to the escherichia coli abasic endonuclease exonuclease III. Environ. Mol. Mutagen. 2000, 36, 312–324. [Google Scholar] [CrossRef]
- De Souza-Pinto, N.C.; Wilson, D.M.; Stevnsner, T.V.; Bohr, V.A. Mitochondrial DNA, base excision repair and neurodegeneration. DNA Repair (Amst) 2008, 7, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Prased, R.; Caglayan, M.; Dai, D.-P.; Nadalutti, C.A.; Zhao, M.-L.; Gressman, N.; Janoshazi, A.K.; Stefanick, D.; Horton, J.K.; Krasich, R.; et al. DNA polymerase β: A missing link of the base excision repair machinery in mammalian mitochondria. DNA Repair (Amst) 2017, 60, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Rudd, S.G.; Bianchi, J.; Doherty, A.J. PrimPol—A new polymerase on the block. Mol. Cell. Oncol. 2014, 1, 1–10. [Google Scholar] [CrossRef]
- Singh, B.; Li, X.; Owens, K.M.; Vanniarajan, A.; Liang, P.; Singh, K.K. Human REV3 DNA polymerase zeta localizes to mitochondria and protects the mitochondrial genome. PLoS ONE 2015, 10, e0140409. [Google Scholar] [CrossRef]
- Wisnovsky, S.; Sack, T.; Pagliarini, D.J.; Laposa, R.R.; Kelley, S.O. DNA Polymerase θ Increases Mutational Rates in Mitochondrial DNA. ACS Chem. Biol. 2018, 13, 900–908. [Google Scholar] [CrossRef]
- Bailey, L.J.; Bianchi, J.; Doherty, A.J. PrimPol is required for the maintenance of efficient nuclear and mitochondrial DNA replication in human cells. Nucleic Acids Res. 2019, 47, 4026–4038. [Google Scholar] [CrossRef]
- Torregrosa-Muñumer, R.; Forslund, J.M.E.; Goffart, S.; Pfeiffer, A.; Stojkovič, G.; Carvalho, G.; Al-Furoukh, N.; Blanco, L.; Wanrooij, S.; Pohjoismäki, J.L.O. PrimPol is required for replication reinitiation after mtDNA damage. Proc. Natl. Acad. Sci. USA 2017, 114, 11398–11403. [Google Scholar] [CrossRef]
- Sun, H.; Wang, X. Mitochondrial DNA and Diseases; Springer: New York, NY, USA, 2017; ISBN 9789811066733. [Google Scholar]
- Akbari, M.; Keijzers, G.; Maynard, S.; Scheibye-Knudsen, M.; Desler, C.; Hickson, I.D.; Bohr, V.A. Overexpression of DNA ligase III in mitochondria protects cells against oxidative stress and improves mitochondrial DNA base excision repair. DNA Repair (Amst) 2014, 16, 44–53. [Google Scholar] [CrossRef]
- Balakrishnan, L.; Bambara, R.A. Flap Endonuclease 1. Annual 2013, 82, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Perina, D.; Mikoč, A.; Ahel, J.; Ćetković, H.; Žaja, R.; Ahel, I. Distribution of protein poly(ADP-ribosyl)ation systems across all domains of life. DNA Repair (Amst) 2014, 23, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Olsen, L.C.; Aasland, R.; Wittwer, C.U.; Krokan, H.E.; Helland, D.E. Molecular cloning of human uracil-DNA glycosylase, a highly conserved DNA repair enzyme. EMBO J. 1989, 8, 3121–3125. [Google Scholar] [CrossRef] [PubMed]
- Grasso, S.; Tell, G. Base excision repair in Archaea: Back to the future in DNA repair. DNA Repair (Amst) 2014, 21, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Steele, H.E.; Horvath, R.; Lyon, J.J.; Chinnery, P.F. Monitoring clinical progression with mitochondrial disease biomarkers. Brain 2017, 140, 2530–2540. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent Advances in Mitochondrial Disease. Annu. Rev. Genomics Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef] [PubMed]
- McKusick, V.A. # 515000 CHLORAMPHENICOL TOXICITY Dostępne na. Available online: https://omim.org/entry/515000 (accessed on 16 June 2019).
- Akbari, M.; Morevati, M.; Croteau, D.; Bohr, V.A. The role of DNA base excision repair in brain homeostasis and disease. DNA Repair (Amst) 2015, 32, 172–179. [Google Scholar] [CrossRef]
- Leandro, G.S.; Sykora, P.; Bohr, V.A. The impact of base excision DNA repair in age-related neurodegenerative diseases. Mutat. Res. Fundam. Mol. Mech. Mutagenes. 2015, 776, 31–39. [Google Scholar] [CrossRef]
- Sykora, P.; Croteau, D.L.; Bohr, V.A.; Wilson, D.M. Aprataxin localizes to mitochondria and preserves mitochondrial function. Proc. Natl. Acad. Sci. USA 2011, 108, 7437–7442. [Google Scholar] [CrossRef]
- Croteau, D.L.; Fang, E.F.; Nilsen, H.; Bohr, V.A. NAD+ in DNA repair and mitochondrial maintenance. Cell Cycle 2017, 16, 491–492. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2010, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Fakouri, N.B.; Hou, Y.; Demarest, T.G.; Christiansen, L.S.; Okur, M.N.; Mohanty, J.G.; Croteau, D.L.; Bohr, V.A. Toward understanding genomic instability, mitochondrial dysfunction and aging. FEBS J. 2019, 286, 1058–1073. [Google Scholar] [CrossRef] [PubMed]
- Son, J.M.; Lee, C. Mitochondria: Multifaceted regulators of aging. BMB Rep. 2019, 52, 13–23. [Google Scholar] [CrossRef]
- Reznik, E.; Miller, M.L.; Şenbabaoğlu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. Elife 2016, 5, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, L.; Yu, X.; Zhou, H.; Luo, Y.; Wang, W.; Wang, L. Clinical application of plasma mitochondrial DNA content in patients with lung cancer. Oncol. Lett. 2018, 16, 7074–7081. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Li, S.H.; Lin, J.C.; Wu, C.C.; Yeh, D.C.; Wei, Y.H. Somatic mutations in the D-loop and decrease in the copy number of mitochondrial DNA in human hepatocellular carcinoma. Mutat. Res. Fundam. Mol. Mech. Mutagenes. 2004, 547, 71–78. [Google Scholar] [CrossRef]
- Shoshan, M. On mitochondrial metabolism in tumor biology. Curr. Opin. Oncol. 2017, 29, 48–54. [Google Scholar] [CrossRef]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef]
- Greenberg, E.F.; Vatolin, S. Symbiotic Origin of Aging. Rejuvenation Res. 2018, 21, 225–231. [Google Scholar] [CrossRef]
- Picard, M.; McEwen, B.S. Psychological Stress and Mitochondria: A Systematic Review. Psychosom. Med. 2018, 80, 141–153. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| H. sapiens | E. coli | |
|---|---|---|
| DNA glycosylases | ||
| UDG family | UNG1 | Ung |
| Mug | ||
| HtH family | NTH1 | Nth |
| OGG1 | ||
| MUTYH | MutY | |
| MTH1 | MutT | |
| MpgII | ||
| AAG family | AAG-A, AAG-B | AlkA |
| Fpg/Nei family | NEIL1, NEIL2 | Fpg (mutM), Nei |
| AP endonucleases | ||
| Xth family | APE1, APE2 | ExoIII (XthA) |
| Nfo family | Endo IV (Nfo) | |
| DNA polymerases | ||
| Family A | Polγ | PolI |
| Family X | Polβ | |
| Family AEP | PrimPol | |
| DNA ligases | ||
| ATP-dependent | LigIIIα | |
| NAD+-dependent | LigA | |
| Other | ||
| Flap endonuclease | FEN1 |
| Disease | Clinical Symptoms of Disease | Impaired Protein | Protein Function in Mitochondria |
|---|---|---|---|
| Huntington’s disease (HD) OMIM #143100 | decrease in cognitive and motor functions | FEN1 | Endonuclease, takes part in LP-BER |
| Microcephaly, seizures, and developmental delay (MCSZ) OMIM #613402 | early infantile epileptic encephalopathy, cerebellar atrophy, peripheral neuropathy | PNKP | Occurs with Polγ and NEIL2, major 3′-phosphatase |
| Ataxia-oculomotor apraxia 1 (AOA1) OMIM #208920 | cerebellar ataxia with peripheral axonal neuropathy, oculomotor apraxia, hypoalbuminemia | APTX | Removes 5′-AMP and 5′-AMP-dRP from DNA |
| Spinocerebellar ataxia with axonal neuropathy-1 (SCAN1) OMIM #607250 | cerebellar atrophy, peripheral neuropathy, gait disturbance, sensory impairment | TDP1 | Takes part in the repair of 3′-abasic sites and topoisomerase I-linked DNA adducts |
| Ataxia-telangiectasia (A-T) OMIM #208900 | cerebellar ataxia, immune defects, cells are highly sensitive to ionizing radiation | ATM | Regulates mtDNA copy number, LigIII, and mitophagy |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boguszewska, K.; Szewczuk, M.; Kaźmierczak-Barańska, J.; Karwowski, B.T. The Similarities between Human Mitochondria and Bacteria in the Context of Structure, Genome, and Base Excision Repair System. Molecules 2020, 25, 2857. https://doi.org/10.3390/molecules25122857
Boguszewska K, Szewczuk M, Kaźmierczak-Barańska J, Karwowski BT. The Similarities between Human Mitochondria and Bacteria in the Context of Structure, Genome, and Base Excision Repair System. Molecules. 2020; 25(12):2857. https://doi.org/10.3390/molecules25122857
Chicago/Turabian StyleBoguszewska, Karolina, Michał Szewczuk, Julia Kaźmierczak-Barańska, and Bolesław T. Karwowski. 2020. "The Similarities between Human Mitochondria and Bacteria in the Context of Structure, Genome, and Base Excision Repair System" Molecules 25, no. 12: 2857. https://doi.org/10.3390/molecules25122857
APA StyleBoguszewska, K., Szewczuk, M., Kaźmierczak-Barańska, J., & Karwowski, B. T. (2020). The Similarities between Human Mitochondria and Bacteria in the Context of Structure, Genome, and Base Excision Repair System. Molecules, 25(12), 2857. https://doi.org/10.3390/molecules25122857