
Site-Selective Modification of a Porpholactone—Selective Synthesis of 12,13- and 17,18-Dihydroporpholactones

 ,
,  ,
,  and
and 
Abstract
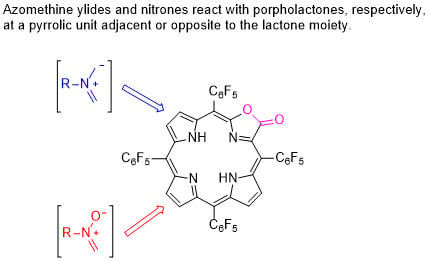
1. Introduction
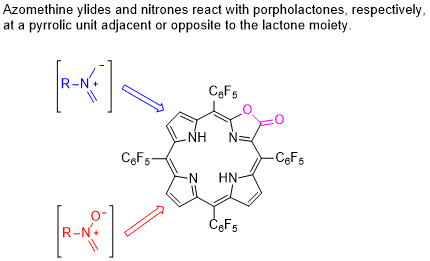
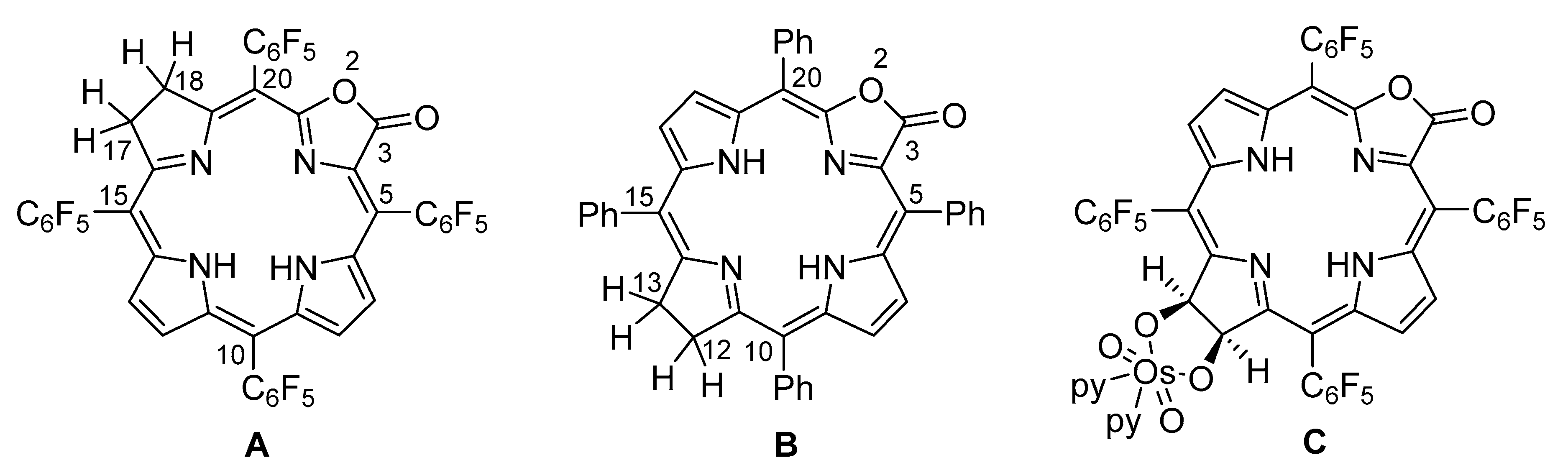
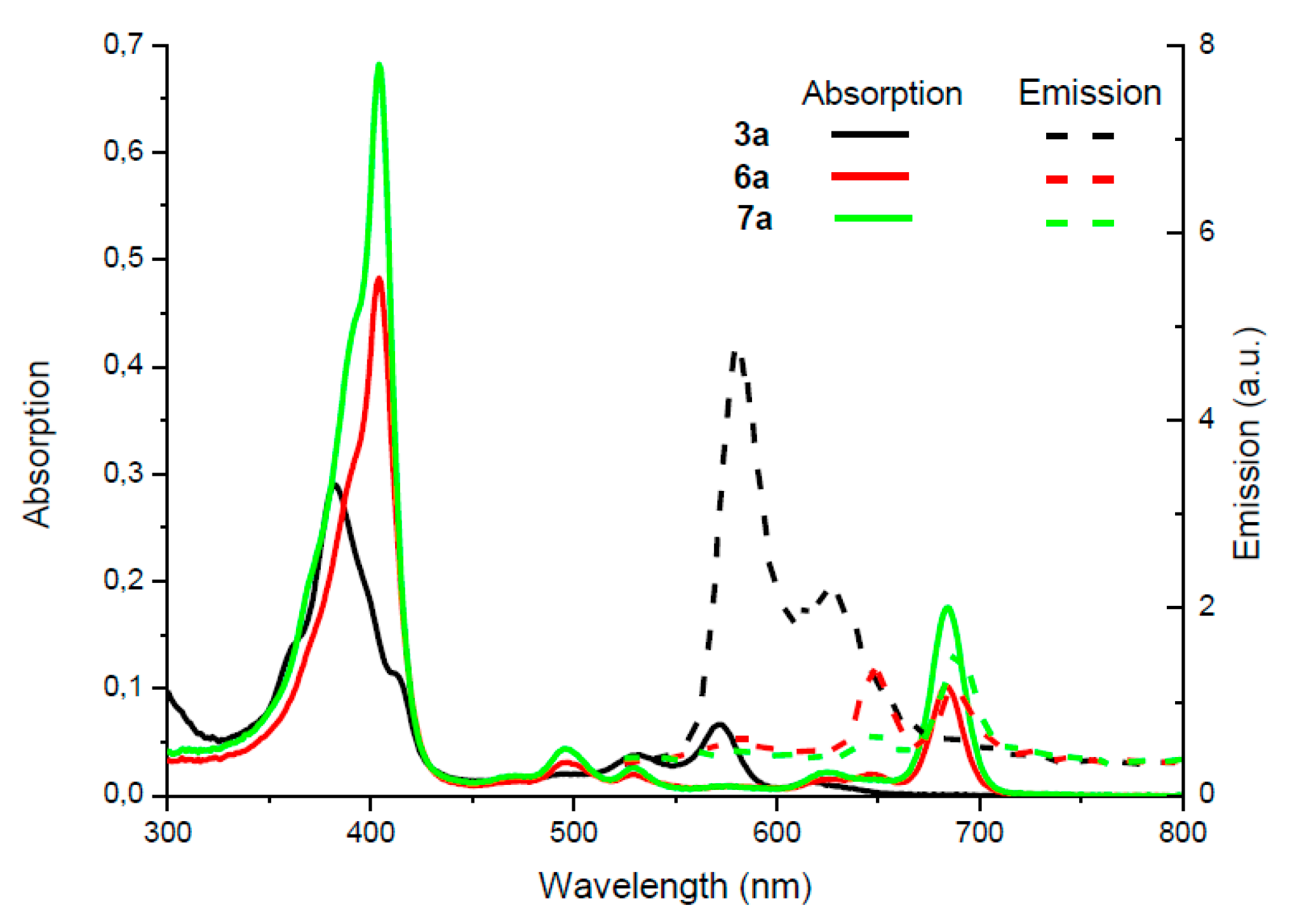
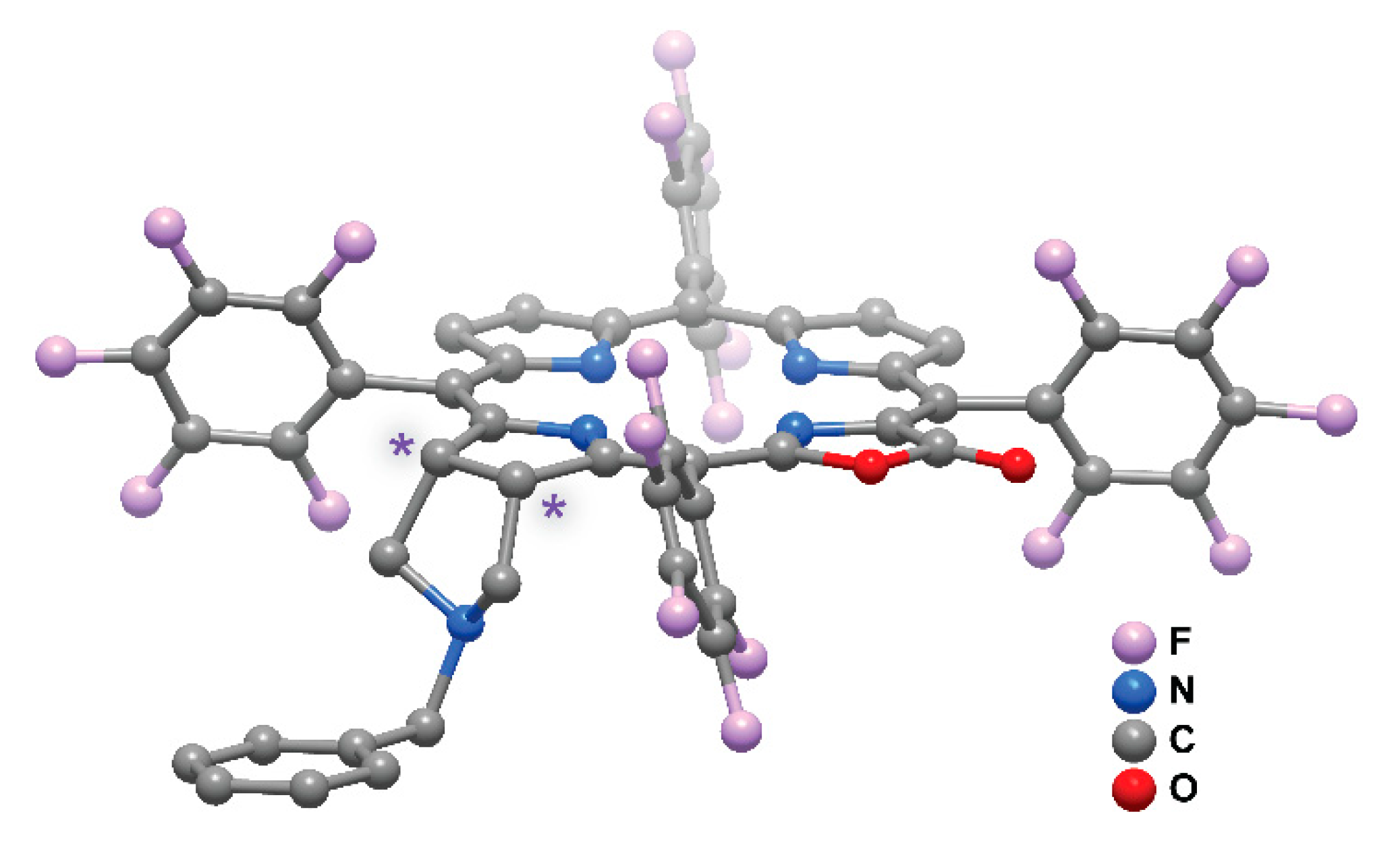
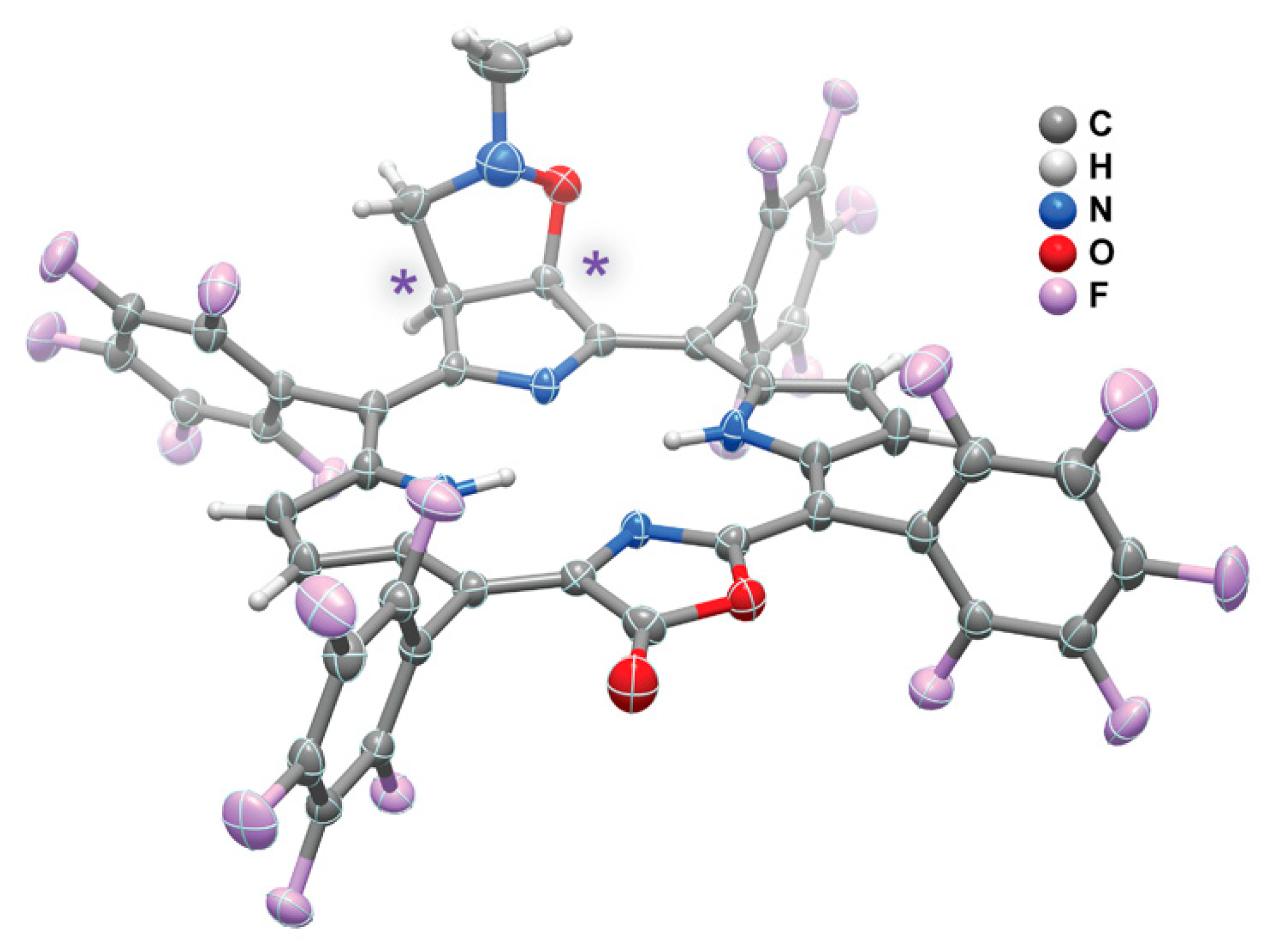
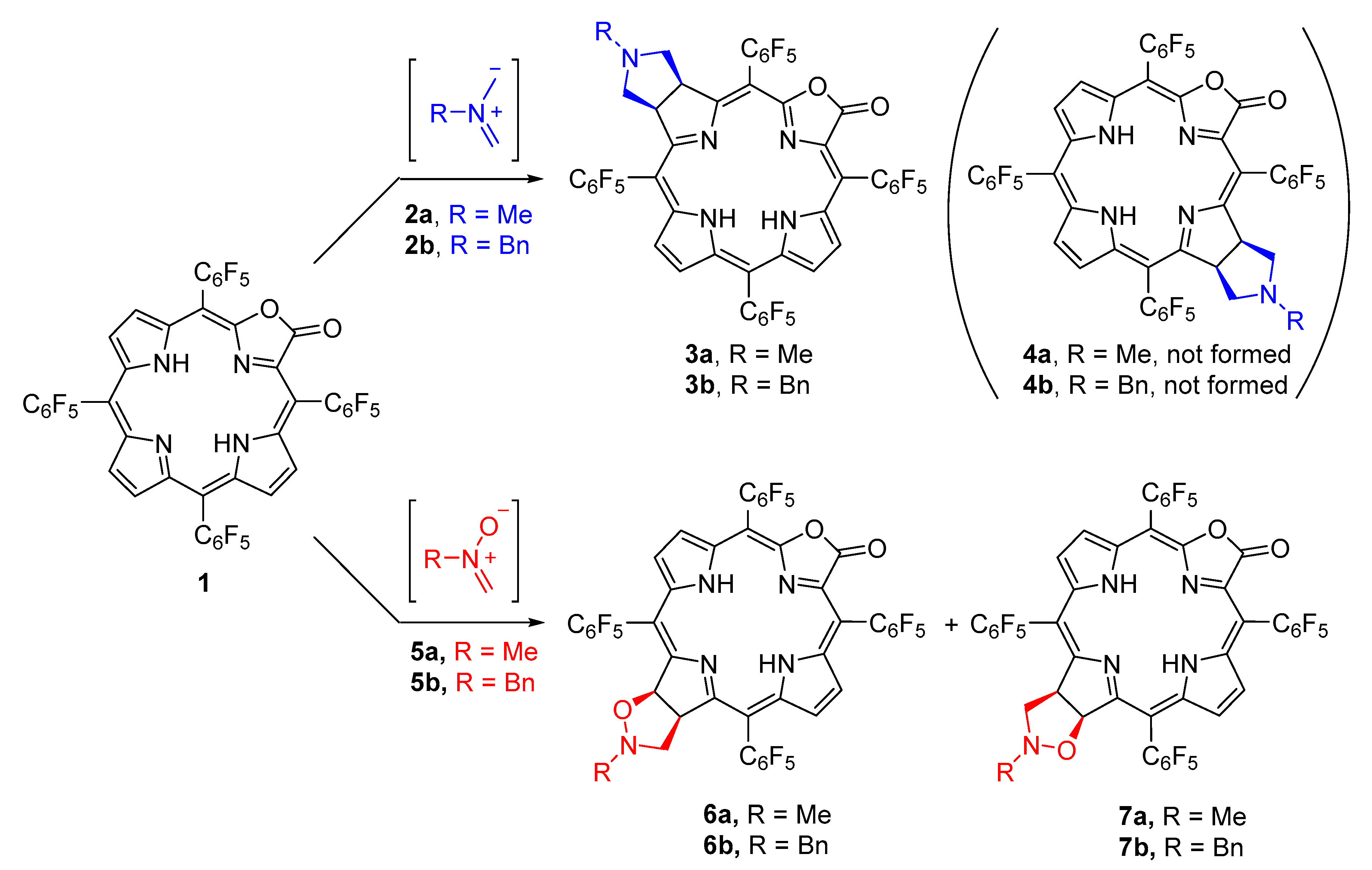
2. Results and Discussion
3. Materials and Methods
3.1. Chemicals and Instrumentation
3.2. Synthesis
3.2.1. General Procedure for the Synthesis of Cycloadducts 3.
3.2.2. General Procedure for the Synthesis of Cycloadducts 6 and 7.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brückner, C.; Akhigbe, J.; Samankumara, L.P. Porphyrin analogs containing non-pyrrolic heterocycles. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: River Edge, NY, USA, 2014; Volume 31, pp. 1–276. [Google Scholar]
- Brückner, C. The breaking and mending of meso-tetraarylporphyrins: Transmuting the pyrrolic building blocks. Acc. Chem. Res. 2016, 49, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.D.; Costa, J.I.T.; Tomé, A.C. Porphyrin macrocycle modification: Pyrrole ring-contracted or -expanded porphyrinoids. Molecules 2016, 21, 320. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Lindsey, J.S. Synthetic chlorins, possible surrogates for chlorophylls, prepared by derivatization of porphyrins. Chem. Rev. 2017, 117, 344–535. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, T.; Shetti, V.S.; Sharma, R.; Ravikanth, M. Heteroatom-containing porphyrin analogues. Chem. Rev. 2017, 117, 3254–3328. [Google Scholar] [CrossRef]
- Ning, Y.; Jin, G.-Q.; Zhang, J.-L. Porpholactone chemistry: An emerging approach to bioinspired photosensitizers with tunable near-infrared photophysical properties. Acc. Chem. Res. 2019, 52, 2620–2633. [Google Scholar] [CrossRef]
- Akhigbe, J.; Samankumara, L.P.; Brückner, C. The breaking and mending of porphyrins: Reductive coupling of secochlorin bisaldehydes. Tetrahedron Lett. 2012, 53, 3524–3526. [Google Scholar] [CrossRef]
- Ke, X.-S.; Ning, Y.; Tang, J.; Hu, J.-Y.; Yin, H.-Y.; Wang, G.-X.; Yang, Z.-S.; Jie, J.; Liu, K.; Meng, Z.-S.; et al. Gadolinium(III) porpholactones as efficient and robust singlet oxygen photosensitizers. Chem. Eur. J. 2016, 22, 9676–9686. [Google Scholar] [CrossRef]
- Ning, Y.; Tang, J.; Liu, Y.-W.; Jing, J.; Sun, Y.; Zhang, J.-L. Highly luminescent, biocompatible ytterbium (III) complexes as near-infrared fluorophores for living cell imaging. Chem. Sci. 2018, 9, 3742–3753. [Google Scholar] [CrossRef]
- Yang, Z.-S.; Ning, Y.; Yin, H.-Y.; Zhang, J.-L. Lutetium (III) porphyrinoids as effective triplet photosensitizers for photon upconversion based on triplet-triplet annihilation (TTA). Inorg. Chem. Front. 2018, 5, 2291–2299. [Google Scholar] [CrossRef]
- Hewage, N.; Daddario, P.; Lau, K.S.F.; Guberman-Pfeffer, M.J.; Gascón, J.A.; Zeller, M.; Lee, C.O.; Khalil, G.E.; Gouterman, M.; Brückner, C. Bacterio- and isobacteriodilactones by stepwise or direct oxidations of meso-tetrakis(pentafluorophenyl)porphyrin. J. Org. Chem. 2019, 84, 239–256. [Google Scholar] [CrossRef]
- Jin, G.-Q.; Ning, Y.; Geng, J.-X.; Jiang, Z.-F.; Wang, Y.; Zhang, J.-L. Joining the journey to near infrared (NIR) imaging: The emerging role of lanthanides in the designing of molecular probes. Inorg. Chem. Front. 2020, 7, 289–299. [Google Scholar] [CrossRef]
- McCarthy, J.R.; Jenkins, H.A.; Brückner, C. Free base meso-tetraaryl-morpholinochlorins and porpholactone from meso-tetraaryl-2,3-dihydroxy-chlorin. Org. Lett. 2003, 5, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lv, H.; Ke, X.; Yang, B.; Zhang, J.-L. Ruthenium-catalyzed oxidation of the porphyrin β,β’-pyrrolic ring: A general and efficient approach to porpholactones. Adv. Synth. Catal. 2012, 354, 3509–3516. [Google Scholar] [CrossRef]
- Tomé, A.C.; Lacerda, P.S.S.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. meso-Arylporphyrins as dienophiles in Diels–Alder reactions: A novel approach to the synthesis of chlorins, bacteriochlorins and naphthoporphyrins. Chem. Commun. 1997, 1199–1200. [Google Scholar] [CrossRef]
- Silva, A.M.G.; Tomé, A.C.; Neves, M.G.P.M.S.; Silva, A.M.S.; Cavaleiro, J.A.S. meso-Tetraarylporphyrins as dipolarophiles in 1,3-dipolar cycloaddition reactions. Chem. Commun. 1999, 1767–1768. [Google Scholar] [CrossRef]
- Tomé, A.C.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Porphyrins and other pyrrolic macrocycles in cycloaddition reactions. J. Porphyrins Phthalocyanines 2009, 13, 408–414. [Google Scholar] [CrossRef]
- Silva, A.M.G.; Tomé, A.C.; Neves, M.G.P.M.S.; Silva, A.M.S.; Cavaleiro, J.A.S. 1,3-Dipolar cycloaddition reactions of porphyrins with azomethine ylides. J. Org. Chem. 2005, 70, 2306–2314. [Google Scholar] [CrossRef]
- Silva, A.M.G.; Tomé, A.C.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.; Perrone, D.; Dondoni, A. Porphyrins in 1,3-dipolar cycloadditions with sugar azomethine ylides. Synthesis of pyrrolidinoporphyrin glycoconjugates. Synlett 2005, 857–859. [Google Scholar] [CrossRef]
- Aguiar, A.; Leite, A.; Silva, A.M.N.; Tomé, A.C.; Cunha-Silva, L.; Castro, B.; Rangel, M.; Silva, A.M.G. Isoxazolidine-fused meso-tetraarylchlorins as key tools for the synthesis of mono- and bis-annulated chlorins. Org. Biomol. Chem. 2015, 13, 7131–7135. [Google Scholar] [CrossRef]
- Almeida, J.; Aguiar, A.; Leite, A.; Silva, A.M.N.; Cunha-Silva, L.; Castro, B.; Rangel, M.; Barone, G.; Tomé, A.C.; Silva, A.M.G. 1,3-Dipolar cycloadditions with meso-tetraarylchlorins – site selectivity and mixed bisadducts. Org. Chem. Front. 2017, 4, 534–544. [Google Scholar] [CrossRef]
- Silva, A.M.G.; Lacerda, P.S.S.; Tomé, A.C.; Neves, M.G.P.M.S.; Silva, A.M.S.; Cavaleiro, J.A.S.; Makarova, E.A.; Lukyanets, E.A. Porphyrins in 1,3-dipolar cycloaddition reactions. Synthesis of new porphyrin-chlorin and porphyrin-tetraazachlorin dyads. J. Org. Chem. 2006, 71, 8352–8356. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, B.; Yu, X.; Yi, P.; Yi, R.; Chmielewski, P.J. 1,3-Dipolar cycloaddition of 2,6-dichlorobenzonitrile oxide to 2-methyl-N-confused porphyrin. Regio- and stereoselective synthesis and structural characterization of 2-aza-21-carbabacteriochlorin and resolution of 2-aza-21-carbachlorin enantiomers. J. Org. Chem. 2012, 77, 2431–2440. [Google Scholar] [CrossRef] [PubMed]
- Hata, H.; Kamimura, Y.; Shinokubo, H.; Osuka, A. Synthesis of pyrrolidine-fused [34]- and [36]octaphyrins via 1,3-dipolar cycloaddition. Org. Lett. 2006, 8, 1169−1172. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nakamura, Y.; Aratani, N.; Osuka, A. Regioselective [3+4] cycloaddition of an azomethine ylide to meso−meso, β−β, β’−β’ triply linked diporphyrins. Tetrahedron Lett. 2008, 49, 3308–3311. [Google Scholar] [CrossRef]
- Yao, Y.; Rao, Y.; Liu, Y.; Jiang, L.; Xiong, J.; Fan, Y.-J.; Shen, Z.; Sessler, J.L.; Zhang, J.-L. Aromaticity versus regioisomeric effect of β-substituents in porphyrinoids. Phys. Chem. Chem. Phys. 2019, 21, 10152–10162. [Google Scholar] [CrossRef] [PubMed]
- Brückner, C.; Ogikubo, J.; McCarthy, J.R.; Akhigbe, J.; Hyland, M.A.; Daddario, P.; Worlinsky, J.L.; Zeller, M.; Engle, J.T.; Ziegler, C.J.; et al. meso-Arylporpholactones and their reduction products. J. Org. Chem. 2012, 77, 6480–6494. [Google Scholar] [CrossRef]
- Yu, Y.; Furuyama, T.; Tang, J.; Wu, Z.-Y.; Chen, J.-Z.; Kobayashi, N.; Zhang, J.-L. Stable iso-bacteriochlorin mimics from porpholactone: Effect of a β-oxazolone moiety on the frontier π-molecular orbitals. Inorg. Chem. Front. 2015, 2, 671–677. [Google Scholar] [CrossRef]
- Wu, Z.-Y.; Wang, T.; Meng, Y.-S.; Rao, Y.; Wang, B.-W.; Zheng, J.; Gao, S.; Zhang, J.-L. Enhancing the reactivity of nickel(II) in hydrogen evolution reactions (HERs) by β-hydrogenation of porphyrinoid ligands. Chem. Sci. 2017, 8, 5953–5961. [Google Scholar] [CrossRef]
- Akhigbe, J.; Haskoor, J.; Zeller, M.; Brückner, C. Porpholactams and chlorolactams: Replacement of a β, β’-double bond in meso-tetraphenyl-porphyrin and -chlorin by a lactam moiety. Chem. Commun. 2011, 47, 8599–8601. [Google Scholar] [CrossRef]
- Akhigbe, J.; Haskoor, J.; Krause, J.A.; Zeller, M.; Brückner, C. Formation, structure, and reactivity of meso-tetraarylchlorolactones, -porpholactams, and -chlorolactams, porphyrin and chlorin analogues incorporating oxazolone or imidazolone moieties. Org. Biomol. Chem. 2013, 11, 3616–3628. [Google Scholar] [CrossRef]
- Ke, X.-S.; Chang, Y.; Chen, J.-Z.; Tian, J.; Mack, J.; Cheng, X.; Shen, Z.; Zhang, J.-L. Porphodilactones as synthetic chlorophylls: Relative orientation of β-substituents on a pyrrolic ring tunes NIR absorption. J. Am. Chem. Soc. 2014, 136, 9598–9607. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.K. Synthesis and characterization of alkylated isobacteriochlorins, models of siroheme and sirohydrochlorin. Biochemistry 1980, 19, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Stolzenberg, A.M.; Spreer, L.O.; Holm, R.H. Octaethylisobacteriochlorin, a model of the sirohydrochlorin ring-system of siroheme enzymes - preparation and spectroscopic and electrochemical properties of the free base and its Zn(II) complex. J. Am. Chem. Soc. 1980, 102, 364–370. [Google Scholar] [CrossRef]
- A mixture of isomers 6a/7a was analyzed by HPLC: The two compounds show retention times at 32.15 min (7a, minor isomer) and 33.62 min (6a, major isomer) (see Supp. mat.)
- Jimenez-Oses, G.; Garcia, J.I.; Silva, A.M.G.; Santos, A.R.N.; Tomé, A.C.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Mechanistic insights on the site selectivity in successive 1,3-dipolar cycloadditions to meso-tetraarylporphyrins. Tetrahedron 2008, 64, 7937–7943. [Google Scholar] [CrossRef]
- Costa, J.I.T.; Tomé, A.C.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin: A versatile platform to novel porphyrinic materials. J. Porphyrins Phthalocyanines 2011, 15, 1116–1133. [Google Scholar] [CrossRef]
- Worlinsky, J.L.; Halepas, S.; Brückner, C. PEGylated meso-arylporpholactone metal complexes as optical cyanide sensors in water. Org. Biomol. Chem. 2014, 12, 3991–4001. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerqueira, A.F.R.; Snarskis, G.; Zurauskas, J.; Guieu, S.; Paz, F.A.A.; Tomé, A.C. Site-Selective Modification of a Porpholactone—Selective Synthesis of 12,13- and 17,18-Dihydroporpholactones. Molecules 2020, 25, 2642. https://doi.org/10.3390/molecules25112642
Cerqueira AFR, Snarskis G, Zurauskas J, Guieu S, Paz FAA, Tomé AC. Site-Selective Modification of a Porpholactone—Selective Synthesis of 12,13- and 17,18-Dihydroporpholactones. Molecules. 2020; 25(11):2642. https://doi.org/10.3390/molecules25112642
Chicago/Turabian StyleCerqueira, Ana F. R., Gustautas Snarskis, Jonas Zurauskas, Samuel Guieu, Filipe A. Almeida Paz, and Augusto C. Tomé. 2020. "Site-Selective Modification of a Porpholactone—Selective Synthesis of 12,13- and 17,18-Dihydroporpholactones" Molecules 25, no. 11: 2642. https://doi.org/10.3390/molecules25112642
APA StyleCerqueira, A. F. R., Snarskis, G., Zurauskas, J., Guieu, S., Paz, F. A. A., & Tomé, A. C. (2020). Site-Selective Modification of a Porpholactone—Selective Synthesis of 12,13- and 17,18-Dihydroporpholactones. Molecules, 25(11), 2642. https://doi.org/10.3390/molecules25112642