Evaluation of Bronopol and Disulfiram as Potential Candidatus Liberibacter asiaticus Inosine 5′-Monophosphate Dehydrogenase Inhibitors by Using Molecular Docking and Enzyme Kinetic



Abstract
1. Introduction
2. Results
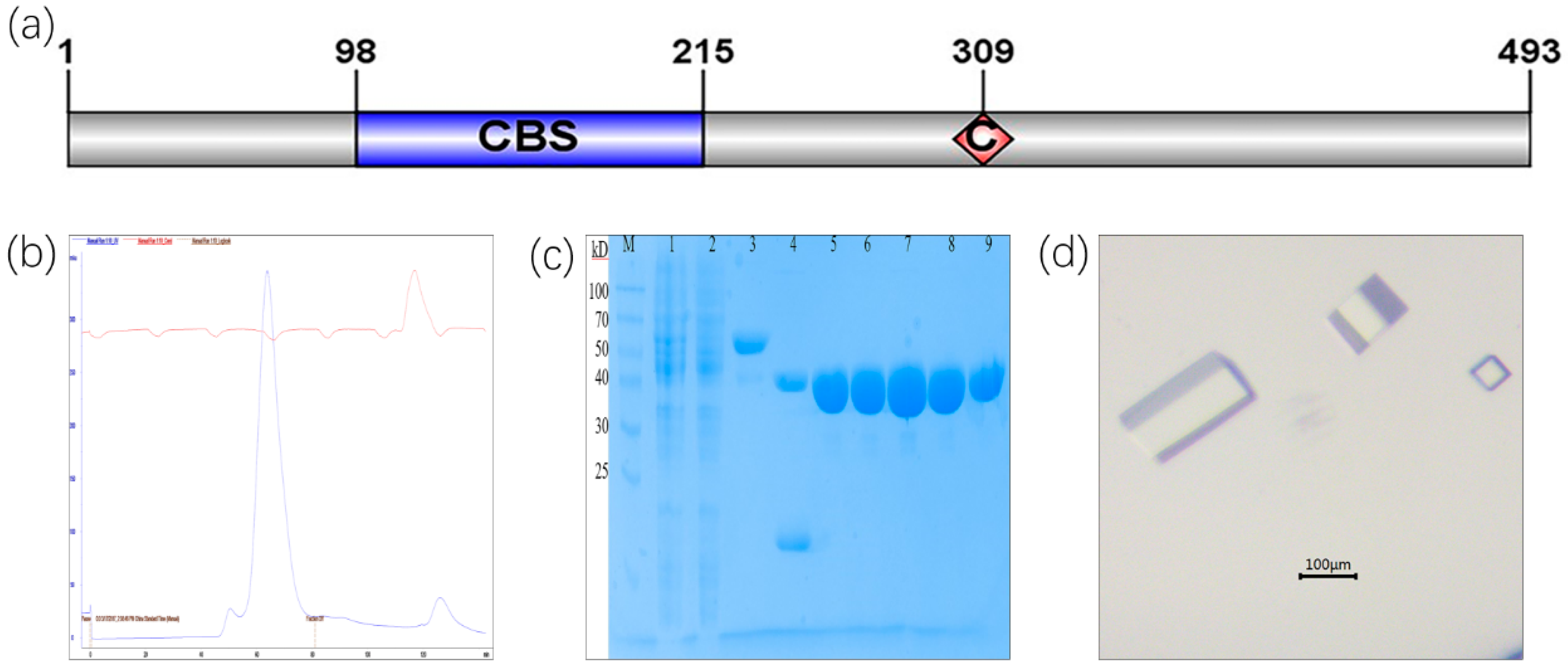
2.1. Protein Purification of CLas IMPDHΔ98-201 and Crystallization Screening
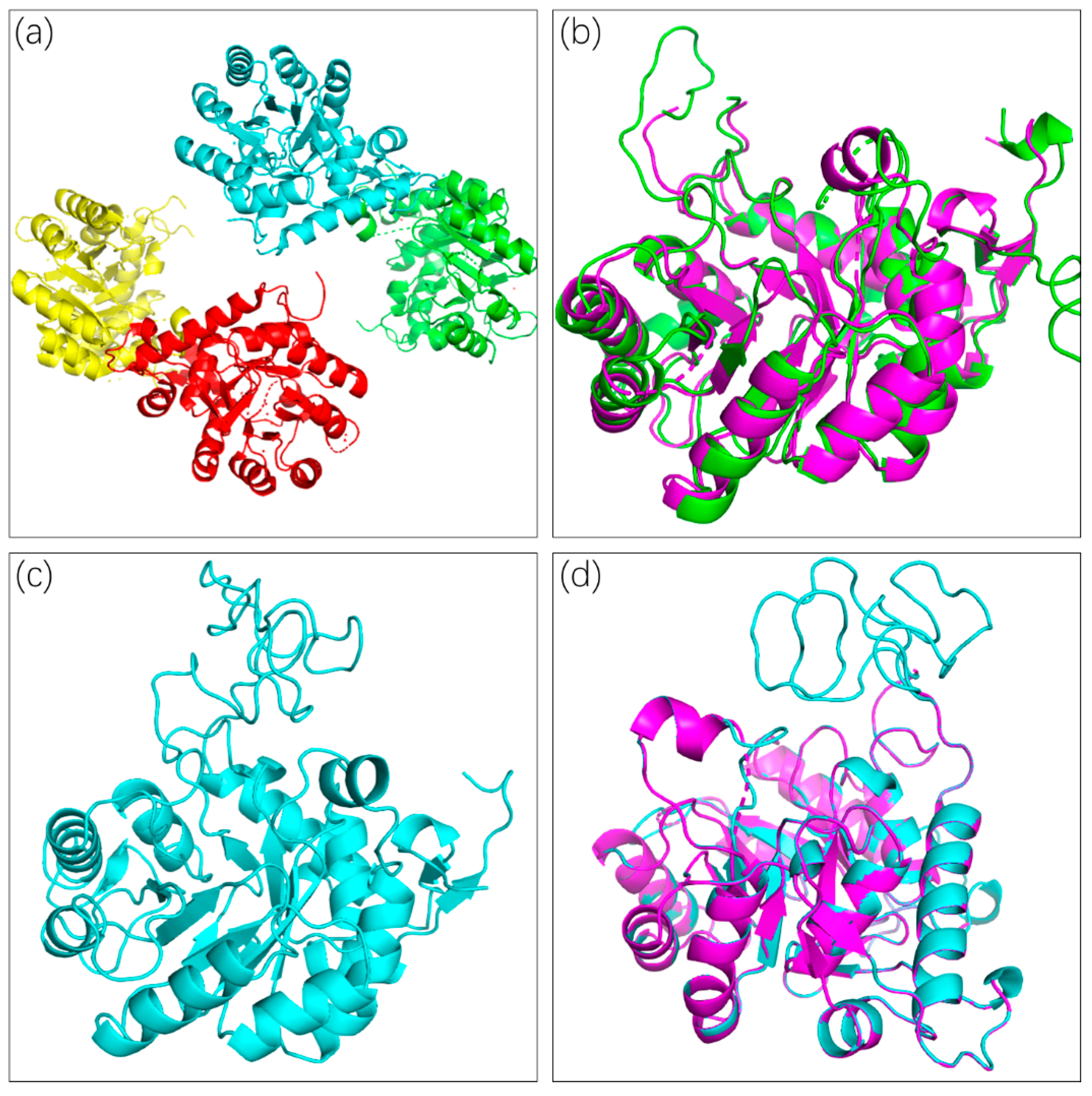
2.2. Crystal Structure and Loop Refinement of CLas IMPDHΔ98-201
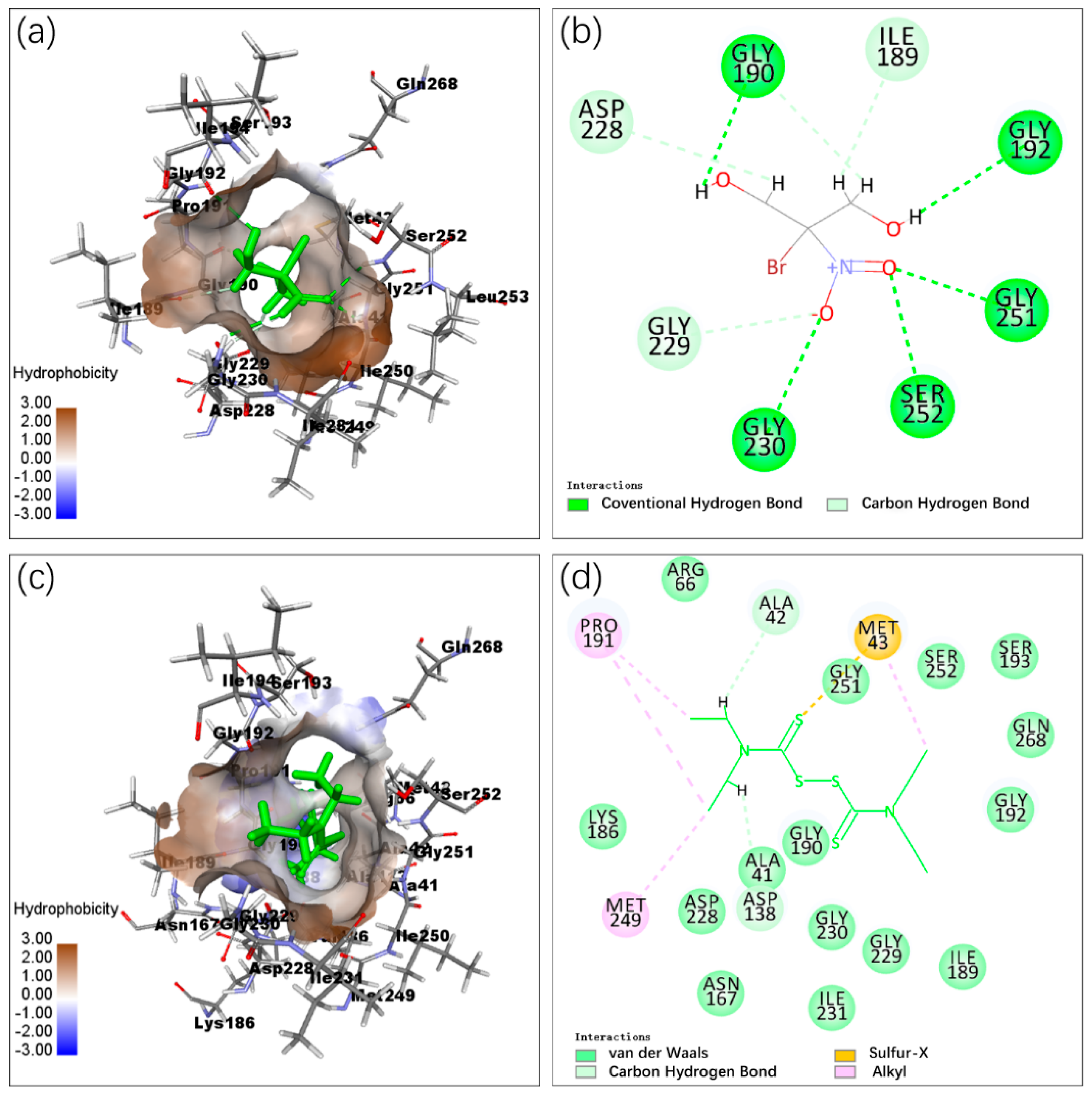
2.3. Molecular Docking
2.4. Kinetic Characterization of CLas IMPDHΔ98-201
2.5. Inhibitory Assay against CLas IMPDHΔ98-201 Enzyme Activity
3. Discussion
3.1. Purification and Crystallization of CLas IMPDHΔ98-201
3.2. Docking Interaction Analysis of CLas IMPDHΔ98-201 with Molecules
3.3. Inhibitory Assay against CLas IMPDHΔ98-201 Activity
4. Materials and Methods
4.1. Cloning and Mutant Construction of CLas IMPDH Gene
4.2. Protein Purification and Crystallization of CLas IMPDHΔ98-201
4.3. Data Collection and Processing
4.4. Loop Refinement and Molecular Docking
4.5. Steady-State Kinetics
4.6. Inhibition Assay against IMPDHΔ98-201 of CLas
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bove, J.M. Huanglongbing: A destructive, newly-emerging, century-old disease of citrus. J. Plant Pathol. 2006, 88, 7–37. [Google Scholar]
- Wang, N.; Pierson, E.A.; Setubal, J.C.; Xu, J.; Levy, J.G.; Zhang, Y.; Li, J.; Rangel, L.T.; Martins, J., Jr. The Candidatus Liberibacter-Host Interface: Insights into Pathogenesis Mechanisms and Disease Control. Annu. Rev. Phytopathol. 2017, 55, 451–482. [Google Scholar] [CrossRef]
- Selvaraj, V.; Maheshwari, Y.; Hajeri, S.; Chen, J.; McCollum, T.G.; Yokomi, R. Development of a duplex droplet digital PCR assay for absolute quantitative detection of “Candidatus Liberibacter asiaticus”. PLoS ONE 2018, 13, e0197184. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Munoz-Bodnar, A.; Zhang, S.; Gabriel, D.W. A Secreted ‘Candidatus Liberibacter asiaticus’ Peroxiredoxin Simultaneously Suppresses Both Localized and Systemic Innate Immune Responses In Planta. Mol. Plant Microbe Interact. MPMI 2018, 31, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Franco, J.Y.; Schwizer, S.; Pang, Z.; Hawara, E.; Liebrand, T.W.H.; Pagliaccia, D.; Zeng, L.; Gurung, F.B.; Wang, P.; et al. An effector from the Huanglongbing-associated pathogen targets citrus proteases. Nat. Commun. 2018, 9, 1718. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Munoz-Bodnar, A.; Gabriel, D.W. ‘Candidatus Liberibacter asiaticus’ peroxiredoxin (LasBCP) suppresses oxylipin-mediated defense signaling in citrus. J. Plant Physiol. 2019, 236, 61–65. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, X.; Liu, X.; Fan, Y.; Zhang, Y.; Zhou, X.; Li, W. A Novel ‘Candidatus Liberibacter asiaticus‘-Encoded Sec-Dependent Secretory Protein Suppresses Programmed Cell Death in Nicotiana benthamiana. Int. J. Mol. Sci. 2019, 20, 5802. [Google Scholar] [CrossRef]
- Shi, Q.; Pitino, M.; Zhang, S.; Krystel, J.; Cano, L.M.; Shatters, R.G., Jr.; Hall, D.G.; Stover, E. Temporal and spatial detection of Candidatus Liberibacter asiaticus putative effector transcripts during interaction with Huanglongbing-susceptible, -tolerant, and -resistant citrus hosts. BMC Plant Biol. 2019, 19, 122. [Google Scholar] [CrossRef]
- Ramadugu, C.; Keremane, M.L.; Halbert, S.E.; Duan, Y.P.; Roose, M.L.; Stover, E.; Lee, R.F. Long-Term Field Evaluation Reveals Huanglongbing Resistance in Citrus Relatives. Plant Dis. 2016, 100, 1858–1869. [Google Scholar] [CrossRef]
- Phuc, T.H.; He, R.F.; Nabil, K.; Judith, K.B.; Anders, O.; David, R.G.; Haluk, B. Host-free biofilm culture of “Candidatus Liberibacter asiaticus”, the bacterium associated with Huanglongbing. Biofilm 2019, 1, 100005. [Google Scholar]
- Wang, N. The Citrus Huanglongbing Crisis and Potential Solutions. Mol. Plant 2019, 12, 607–609. [Google Scholar] [CrossRef]
- Blaustein, R.A.; Lorca, G.L.; Teplitski, M. Challenges for Managing Candidatus Liberibacter spp. (Huanglongbing Disease Pathogen): Current Control Measures and Future Directions. Phytopathology 2018, 108, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Powell, C.A.; Zhou, L.; He, Z.; Stover, E.; Duan, Y. Chemical compounds effective against the citrus Huanglongbing bacterium ‘Candidatus Liberibacter asiaticus’ in planta. Phytopathology 2011, 101, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Ascunce, M.S.; Narouei-Khandan, H.A.; Sun, X.A.; Jones, D.; Kolawole, O.O.; Goss, E.M.; van Bruggen, A.H.C. Effects and side effects of penicillin injection in huanglongbing affected grapefruit trees. Crop Prot. 2016, 90, 106–116. [Google Scholar] [CrossRef]
- Hu, J.; Wang, N. Evaluation of the Spatiotemporal Dynamics of Oxytetracycline and Its Control Effect against Citrus Huanglongbing via Trunk Injection. Phytopathology 2016, 106, 1495–1503. [Google Scholar] [CrossRef]
- Akula, N.; Trivedi, P.; Han, F.Q.; Wang, N. Identification of small molecule inhibitors against SecA of Candidatus Liberibacter asiaticus by structure based design. Eur. J. Med. Chem. 2012, 54, 919–924. [Google Scholar] [CrossRef]
- Gardner, C.L.; Pagliai, F.A.; Pan, L.; Bojilova, L.; Torino, M.I.; Lorca, G.L.; Gonzalez, C.F. Drug Repurposing: Tolfenamic Acid Inactivates PrbP, a Transcriptional Accessory Protein in Liberibacter asiaticus. Front. Microbiol. 2016, 7, 1630. [Google Scholar] [CrossRef]
- Barnett, M.J.; Solow-Cordero, D.E.; Long, S.R. A high-throughput system to identify inhibitors of Candidatus Liberibacter asiaticus transcription regulators. Proc. Natl. Acad. Sci. USA 2019, 116, 18009–18014. [Google Scholar] [CrossRef]
- Saini, G.; Dalal, V.; Savita, B.K.; Sharma, N.; Kumar, P.; Sharma, A.K. Molecular docking and dynamic approach to virtual screen inhibitors against Esbp of Candidatus Liberibacter asiaticus. J. Mol. Graph. Model. 2019, 92, 329–340. [Google Scholar] [CrossRef]
- Jewett, M.W.; Lawrence, K.A.; Bestor, A.; Byram, R.; Gherardini, F.; Rosa, P.A. GuaA and GuaB are essential for Borrelia burgdorferi survival in the tick-mouse infection cycle. J. Bacteriol. 2009, 191, 6231–6241. [Google Scholar] [CrossRef]
- Yoshioka, S.; Newell, P.D. Disruption of de novo purine biosynthesis in Pseudomonas fluorescens Pf0-1 leads to reduced biofilm formation and a reduction in cell size of surface-attached but not planktonic cells. PeerJ 2016, 4, e1543. [Google Scholar] [CrossRef] [PubMed]
- Boitz, J.M.; Jardim, A.; Ullman, B. GMP reductase and genetic uncoupling of adenylate and guanylate metabolism in Leishmania donovani parasites. Mol. Biochem. Parasitol. 2016, 208, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Kofoed, E.M.; Yan, D.; Katakam, A.K.; Reichelt, M.; Lin, B.; Kim, J.; Park, S.; Date, S.V.; Monk, I.R.; Xu, M.; et al. De Novo Guanine Biosynthesis but Not the Riboswitch-Regulated Purine Salvage Pathway Is Required for Staphylococcus aureus Infection in vivo. J. Bacteriol. 2016, 198, 2001–2015. [Google Scholar] [CrossRef] [PubMed]
- Morrison, H.G.; McArthur, A.G.; Gillin, F.D.; Aley, S.B.; Adam, R.D.; Olsen, G.J.; Best, A.A.; Cande, W.Z.; Chen, F.; Cipriano, M.J.; et al. Genomic minimalism in the early diverging intestinal parasite Giardia lamblia. Science 2007, 317, 1921–1926. [Google Scholar] [CrossRef]
- Carlton, J.M.; Hirt, R.P.; Silva, J.C.; Delcher, A.L.; Schatz, M.; Zhao, Q.; Wortman, J.R.; Bidwell, S.L.; Alsmark, U.C.; Besteiro, S.; et al. Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science 2007, 315, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C.; Almquist, S.J.; Muller, C.D.; Eugui, E.M. In vitro immunosuppressive effects of mycophenolic acid and an ester pro-drug, RS-61443. Transplant. Proc. 1991, 23, 10–14. [Google Scholar]
- Robins, R.K.; Revankar, G.R.; McKernan, P.A.; Murray, B.K.; Kirsi, J.J.; North, J.A. The importance of IMP dehydrogenase inhibition in the broad spectrum antiviral activity of ribavirin and selenazofurin. Adv. Enzyme Regul. 1985, 24, 29–43. [Google Scholar] [CrossRef]
- Weber, G. Biochemical strategy of cancer cells and the design of chemotherapy: G. H. A. Clowes Memorial Lecture. Cancer Res. 1983, 43, 3466–3492. [Google Scholar]
- Hedstrom, L.; Liechti, G.; Goldberg, J.B.; Gollapalli, D.R. The antibiotic potential of prokaryotic IMP dehydrogenase inhibitors. Curr. Med. Chem. 2011, 18, 1909–1918. [Google Scholar] [CrossRef]
- Usha, V.; Hobrath, J.V.; Gurcha, S.S.; Reynolds, R.C.; Besra, G.S. Identification of novel Mt-Guab2 inhibitor series active against M. tuberculosis. PLoS ONE 2012, 7, e33886. [Google Scholar] [CrossRef]
- Rao, V.A.; Shepherd, S.M.; Owen, R.; Hunter, W.N. Structure of Pseudomonas aeruginosa inosine 5′-monophosphate dehydrogenase. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 2013, 69, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Mandapati, K.; Gorla, S.K.; House, A.L.; McKenney, E.S.; Zhang, M.; Rao, S.N.; Gollapalli, D.R.; Mann, B.J.; Goldberg, J.B.; Cuny, G.D.; et al. Repurposing cryptosporidium inosine 5′-monophosphate dehydrogenase inhibitors as potential antibacterial agents. ACS Med. Chem. Lett. 2014, 5, 846–850. [Google Scholar] [CrossRef]
- Prosise, G.L.; Luecke, H. Crystal structures of Tritrichomonasfoetus inosine monophosphate dehydrogenase in complex with substrate, cofactor and analogs: A structural basis for the random-in ordered-out kinetic mechanism. J. Mol. Biol. 2003, 326, 517–527. [Google Scholar] [CrossRef]
- Makowska-Grzyska, M.; Kim, Y.; Wu, R.; Wilton, R.; Gollapalli, D.R.; Wang, X.K.; Zhang, R.; Jedrzejczak, R.; Mack, J.C.; Maltseva, N.; et al. Bacillus anthracis inosine 5′-monophosphate dehydrogenase in action: The first bacterial series of structures of phosphate ion-, substrate-, and product-bound complexes. Biochemistry 2012, 51, 6148–6163. [Google Scholar] [CrossRef] [PubMed]
- Labesse, G.; Alexandre, T.; Gelin, M.; Haouz, A.; Munier-Lehmann, H. Crystallographic studies of two variants of Pseudomonas aeruginosa IMPDH with impaired allosteric regulation. Acta Crystallograph. Sect. D Biol. Crystallograph. 2015, 71, 1890–1899. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, L. IMP dehydrogenase: Structure, mechanism, and inhibition. Chem. Rev. 2009, 109, 2903–2928. [Google Scholar] [CrossRef]
- Nimmesgern, E.; Black, J.; Futer, O.; Fulghum, J.R.; Chambers, S.P.; Brummel, C.L.; Raybuck, S.A.; Sintchak, M.D. Biochemical analysis of the modular enzyme inosine 5′-monophosphate dehydrogenase. Protein Expr. Purif. 1999, 17, 282–289. [Google Scholar] [CrossRef]
- Pimkin, M.; Markham, G.D. The CBS subdomain of inosine 5′-monophosphate dehydrogenase regulates purine nucleotide turnover. Mol. Microbiol. 2008, 68, 342–359. [Google Scholar] [CrossRef]
- Makowska-Grzyska, M.; Kim, Y.; Maltseva, N.; Osipiuk, J.; Gu, M.; Zhang, M.; Mandapati, K.; Gollapalli, D.R.; Gorla, S.K.; Hedstrom, L.; et al. A novel cofactor-binding mode in bacterial IMP dehydrogenases explains inhibitor selectivity. J. Biol. Chem. 2015, 290, 5893–5911. [Google Scholar] [CrossRef]
- Gollapalli, D.R.; Macpherson, I.S.; Liechti, G.; Gorla, S.K.; Goldberg, J.B.; Hedstrom, L. Structural determinants of inhibitor selectivity in prokaryotic IMP dehydrogenases. Chem. Biol. 2010, 17, 1084–1091. [Google Scholar] [CrossRef]
- Chen, L.; Wilson, D.J.; Xu, Y.; Aldrich, C.C.; Felczak, K.; Sham, Y.Y.; Pankiewicz, K.W. Triazole-linked inhibitors of inosine monophosphate dehydrogenase from human and Mycobacterium tuberculosis. J. Med. Chem. 2010, 53, 4768–4778. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Umejiego, N.N.; Gollapalli, D.; Sharling, L.; Volftsun, A.; Lu, J.; Benjamin, N.N.; Stroupe, A.H.; Riera, T.V.; Striepen, B.; Hedstrom, L. Targeting a prokaryotic protein in a eukaryotic pathogen: Identification of lead compounds against cryptosporidiosis. Chem. Biol. 2008, 15, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Kirubakaran, S.; Gorla, S.K.; Sharling, L.; Zhang, M.; Liu, X.; Ray, S.S.; Macpherson, I.S.; Striepen, B.; Hedstrom, L.; Cuny, G.D. Structure-activity relationship study of selective benzimidazole-based inhibitors of Cryptosporidium parvum IMPDH. Bioorg. Med. Chem. Lett. 2012, 22, 1985–1988. [Google Scholar] [CrossRef] [PubMed]
- Gorla, S.K.; Kavitha, M.; Zhang, M.; Chin, J.E.W.; Liu, X.; Striepen, B.; Makowska-Grzyska, M.; Kim, Y.; Joachimiak, A.; Hedstrom, L.; et al. Optimization of benzoxazole-based inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J. Med. Chem. 2013, 56, 4028–4043. [Google Scholar] [CrossRef] [PubMed]
- Gorla, S.K.; Zhang, Y.; Rabideau, M.M.; Qin, A.; Chacko, S.; House, A.L.; Johnson, C.R.; Mandapati, K.; Bernstein, H.M.; McKenney, E.S.; et al. Benzoxazoles, Phthalazinones, and Arylurea-Based Compounds with IMP Dehydrogenase-Independent Antibacterial Activity against Francisella tularensis. Antimicrob. Agents Chemother. 2017, 61, e00939-17. [Google Scholar] [CrossRef]
- Chacko, S.; Boshoff, H.I.M.; Singh, V.; Ferraris, D.M.; Gollapalli, D.R.; Zhang, M.; Lawson, A.P.; Pepi, M.J.; Joachimiak, A.; Rizzi, M.; et al. Expanding Benzoxazole-Based Inosine 5′-Monophosphate Dehydrogenase (IMPDH) Inhibitor Structure-Activity As Potential Antituberculosis Agents. J. Med. Chem. 2018, 61, 4739–4756. [Google Scholar] [CrossRef]
- Park, Y.; Pacitto, A.; Bayliss, T.; Cleghorn, L.A.T.; Wang, Z.; Hartman, T.; Arora, K.; Ioerger, T.R.; Sacchettini, J.; Rizzi, M.; et al. Essential but Not Vulnerable: Indazole Sulfonamides Targeting Inosine Monophosphate Dehydrogenase as Potential Leads against Mycobacterium tuberculosis. ACS Infect. Dis. 2017, 3, 18–33. [Google Scholar] [CrossRef]
- Maurya, S.K.; Gollapalli, D.R.; Kirubakaran, S.; Zhang, M.; Johnson, C.R.; Benjamin, N.N.; Hedstrom, L.; Cuny, G.D. Triazole inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J. Med. Chem. 2009, 52, 4623–4630. [Google Scholar] [CrossRef]
- Sahu, N.U.; Singh, V.; Ferraris, D.M.; Rizzi, M.; Kharkar, P.S. Hit discovery of Mycobacterium tuberculosis inosine 5′-monophosphate dehydrogenase, GuaB2, inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 1714–1718. [Google Scholar] [CrossRef]
- Sahu, N.U.; Purushothaman, G.; Thiruvenkatam, V.; Kharkar, P.S. Design, synthesis, and biological evaluation of Helicobacter pylori inosine 5′-monophosphate dehydrogenase (HpIMPDH) inhibitors. Drug Dev. Res. 2019, 80, 125–132. [Google Scholar] [CrossRef]
- Makowska-Grzyska, M.; Kim, Y.; Gorla, S.K.; Wei, Y.; Mandapati, K.; Zhang, M.; Maltseva, N.; Modi, G.; Boshoff, H.I.; Gu, M.; et al. Mycobacterium tuberculosis IMPDH in Complexes with Substrates, Products and Antitubercular Compounds. PLoS ONE 2015, 10, e0138976. [Google Scholar] [CrossRef] [PubMed]
- Usha, V.; Gurcha, S.S.; Lovering, A.L.; Lloyd, A.J.; Papaemmanouil, A.; Reynolds, R.C.; Besra, G.S. Identification of novel diphenyl urea inhibitors of Mt-GuaB2 active against Mycobacterium tuberculosis. Microbiology 2011, 157, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Gorla, S.K.; Kavitha, M.; Zhang, M.; Liu, X.; Sharling, L.; Gollapalli, D.R.; Striepen, B.; Hedstrom, L.; Cuny, G.D. Selective and potent urea inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J. Med. Chem. 2012, 55, 7759–7771. [Google Scholar] [CrossRef] [PubMed]
- Beevers, R.E.; Buckley, G.M.; Davies, N.; Fraser, J.L.; Galvin, F.C.; Hannah, D.R.; Haughan, A.F.; Jenkins, K.; Mack, S.R.; Pitt, W.R.; et al. Low molecular weight indole fragments as IMPDH inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 2535–2538. [Google Scholar] [CrossRef]
- Beevers, R.E.; Buckley, G.M.; Davies, N.; Fraser, J.L.; Galvin, F.C.; Hannah, D.R.; Haughan, A.F.; Jenkins, K.; Mack, S.R.; Pitt, W.R.; et al. Novel indole inhibitors of IMPDH from fragments: Synthesis and initial structure-activity relationships. Bioorg. Med. Chem. Lett. 2006, 16, 2539–2542. [Google Scholar] [CrossRef]
- Watterson, S.H.; Carlsen, M.; Dhar, T.G.M.; Shen, Z.; Pitts, W.J.; Guo, J.; Gu, H.H.; Norris, D.; Chorba, J.; Chen, P.; et al. Novel inhibitors of IMPDH: A highly potent and selective quinolone-based series. Bioorg. Med. Chem. Lett. 2003, 13, 543–546. [Google Scholar] [CrossRef]
- Jangra, S.; Purushothaman, G.; Juvale, K.; Ravi, S.; Menon, A.; Thiruvenkatam, V.; Kirubakaran, S. Synthesis and In Vitro Enzymatic Studies of New 3-Aryldiazenyl Indoles as Promising Helicobacter pylori IMPDH Inhibitors. Curr. Top. Med. Chem. 2019, 19, 376–382. [Google Scholar] [CrossRef]
- Nystroem, K.; Waldenstroem, J.; Tang, K.-W.; Lagging, M. Ribavirin: Pharmacology, multiple modes of action and possible future perspectives. Future Virol. 2019, 14, 153–160. [Google Scholar] [CrossRef]
- Ishikawa, H. Mizoribine and mycophenolate mofetil. Curr. Med. Chem. 1999, 6, 575–597. [Google Scholar] [PubMed]
- Cao, S.; Aboge, G.O.; Terkawi, M.A.; Zhou, M.; Kamyingkird, K.; Moumouni, P.F.A.; Masatani, T.; Igarashi, I.; Nishikawa, Y.; Suzuki, H.; et al. Mycophenolic acid, mycophenolate mofetil, mizoribine, ribavirin, and 7-nitroindole inhibit propagation of Babesia parasites by targeting inosine 5′-monophosphate dehydrogenase. J. Parasitol. 2014, 100, 522–526. [Google Scholar] [CrossRef]
- Sarwono, A.E.Y.; Mitsuhashi, S.; Kabir, M.H.B.; Shigetomi, K.; Okada, T.; Ohsaka, F.; Otsuguro, S.; Maenaka, K.; Igarashi, M.; Kato, K.; et al. Repurposing existing drugs: Identification of irreversible IMPDH inhibitors by high-throughput screening. J. Enzyme Inhib. Med. Chem. 2019, 34, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Juvale, K.; Shaik, A.; Kirubakaran, S. Inhibitors of inosine 5′-monophosphate dehydrogenase as emerging new generation antimicrobial agents. MedChemComm 2019, 10, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Juvale, K.; Purushothaman, G.; Singh, V.; Shaik, A.; Ravi, S.; Thiruvenkatam, V.; Kirubakaran, S. Identification of selective inhibitors of Helicobacter pylori IMPDH as a targeted therapy for the infection. Sci. Rep. 2019, 9, 190. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Wang, J.; Li, J.; Wang, Q.-H.; Wang, Y.; Cheng, M.-S. A three-dimensional pharmacophore model for IMPDH inhibitors. Chem. Biol. Drug Des. 2011, 78, 175–182. [Google Scholar] [CrossRef]
- Freedman, R.; Yu, R.; Sarkis, A.W.; Hedstrom, L. A structural determinant of mycophenolic acid resistance in eukaryotic inosine 5′-monophosphate dehydrogenases. Protein Sci. Publ. Protein Soc. 2020, 29, 686–694. [Google Scholar] [CrossRef]
- Sun, X.E.; Hansen, B.G.; Hedstrom, L. Kinetically controlled drug resistance: How Penicillium brevicompactum survives mycophenolic acid. J. Biol. Chem. 2011, 286, 40595–40600. [Google Scholar] [CrossRef]
- Minor, W.; Cymborowski, M.; Otwinowski, Z.; Chruszcz, M. HKL-3000: The integration of data reduction and structure solution—From diffraction images to an initial model in minutes. Acta Crystallograph. Sect. D Biol. Crystallograph. 2006, 62, 859–866. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallograph. Sect. D Biol. Crystallograph. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Macarthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck—A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallograph. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Eisenberg, D.; Luthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [PubMed]
- Wu, G.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Kerr, K.M.; Digits, J.A.; Kuperwasser, N.; Hedstrom, L. Asp338 controls hydride transfer in Escherichia coli IMP dehydrogenase. Biochemistry 2000, 39, 9804–9810. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CLas IMPDHΔ98-201 | |
|---|---|
| Beamline | SSRF BEAMLINE BL17U |
| wavelength (Å) | 0.9792 |
| Detector | ADSC QUANTUM 315r |
| resolution range (Å) | 42.47–2.55 |
| space group | C 1 2 1 |
| unit cell parameters (Å) | a = 143.13, b = 134.86, c = 85.62 |
| no. of residues/protein | 390 |
| Monomer molecular weight (kDa) | 41.0 |
| phasing method | MR |
| search model | chains A of 4R7J |
| Refinement resolution range (Å) | 42.46–2.55 |
| no. of reflections | 50928 |
| σ cutoff | 1.36 |
| Rwork | 0.222 |
| Rfree | 0.264 |
| mean B factor (Å2) | 69.3 |
| data completeness (%) | 98.2 |
| redundancy | 2.58 |
| Ramachandran plot [most favored/outliers (%)] | 95.2/0.5 |
| PDB entry | 6KCF |
| Name | Molecular Weight (g/mol) | -CDOCKER_ENERGY (kcal/mol) |
|---|---|---|
| Disulfiram | 296.54 | 25.0346 |
| Mercaptopurine | 152.18 | 16.6785 |
| Bronopol | 199.99 | 11.1913 |
| Ebselen | 274.18 | 6.24157 |
| Mycophenolic_acid | 320.34 | 5.38177 |
| Mizoribine | 259.22 | 4.50734 |
| Ribavirin | 244.20 | −5.62032 |
| Mycophenolate_mofetil | 433.50 | −43.3176 |
| Inhibitor | IMP Ki (µM) |
|---|---|
| Bronopol | 0.23 ± 0.01 |
| Disulfiram | 0.62 ± 0.04 |
| Ebselen | 4.13 ± 0.19 |
| Mycophenolic acid | 2.43 ± 0.10 |
| Mercaptopurine | 165 ± 9.89 |
| Mycophenolate mofetil | 24.42 ± 1.65 |
| Mizoribine | 307.7 |
| Ribavirin | >3500 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nan, J.; Zhang, S.; Zhan, P.; Jiang, L. Evaluation of Bronopol and Disulfiram as Potential Candidatus Liberibacter asiaticus Inosine 5′-Monophosphate Dehydrogenase Inhibitors by Using Molecular Docking and Enzyme Kinetic. Molecules 2020, 25, 2313. https://doi.org/10.3390/molecules25102313
Nan J, Zhang S, Zhan P, Jiang L. Evaluation of Bronopol and Disulfiram as Potential Candidatus Liberibacter asiaticus Inosine 5′-Monophosphate Dehydrogenase Inhibitors by Using Molecular Docking and Enzyme Kinetic. Molecules. 2020; 25(10):2313. https://doi.org/10.3390/molecules25102313
Chicago/Turabian StyleNan, Jing, Shaoran Zhang, Ping Zhan, and Ling Jiang. 2020. "Evaluation of Bronopol and Disulfiram as Potential Candidatus Liberibacter asiaticus Inosine 5′-Monophosphate Dehydrogenase Inhibitors by Using Molecular Docking and Enzyme Kinetic" Molecules 25, no. 10: 2313. https://doi.org/10.3390/molecules25102313
APA StyleNan, J., Zhang, S., Zhan, P., & Jiang, L. (2020). Evaluation of Bronopol and Disulfiram as Potential Candidatus Liberibacter asiaticus Inosine 5′-Monophosphate Dehydrogenase Inhibitors by Using Molecular Docking and Enzyme Kinetic. Molecules, 25(10), 2313. https://doi.org/10.3390/molecules25102313