1. Introduction
Pancreatic cancer is one of the most fatal and aggressive cancers and has a particularly low chance of survival. In fact, in the United States, the 5-year survival rate of patients diagnosed with pancreatic cancer is only 8%. Pancreatic cancers are very difficult to diagnose early and treat effectively because of their silent initial symptoms [
1]. Typically, the most problematic type of pancreatic cancer is pancreatic ductal adenocarcinoma (PDAC), which occurs in the duct area. Following lung cancer, PDAC is expected to be the second leading cause of cancer-related deaths [
2].
Unlike healthy cells, cancer cells have specific abilities that contribute to tumor formation and further cancer progression such as proliferation without growth factors, unlimited replication, evasion of apoptosis, and metastasis [
3]. Previous studies have reported that aberrant activation of signal transducers and activators of transcription 3 (STAT3) is involved in oncogenesis in diverse types of human cancer including pancreatic cancer cells [
4]. STAT3 phosphorylation can be activated by mutation of the Src protein, and phosphorylated STAT3 is associated with ERK activation. Moreover, because it is a transcription factor that regulates cellular proliferation, invasion, and migration, which are all critical for cancer progression, targeting STAT3 is an appealing anti-cancer strategy for inhibiting cellular oncogenic functions [
5].
The pharmaceutical industry has produced chemotherapy drugs that target and kill cancer cells using their unique characteristics compared to normal cells. However, such drugs are limited because they cause toxicity such as cardiotoxicity and myelotoxicity in the human body. Furthermore, radiation therapy accompanied by chemotherapeutic drugs is used to suppress the cellular replication system in cancer cells, but it has the problem of low selectivity [
6]. Thus, currently used treatments for cancer using these approaches have some obstacles in considering toxicity and drug resistance. In this situation, plants can be a safe and promising resource against cancer cells [
7]. Herbal-derived drugs such as polyphenols and taxols are already available natural medicines for cancer therapies. Some researchers have also suggested the conjugation of alternative treatments and conventional therapy to more effectively impact cancers. Plants with a high potential to provide newer drugs as anti-cancer agents can be an effective alternative treatment [
8].
Oxalis obtriangulata is a plant belonging to the
Oxalidaceae family, which is distributed in deep mountains throughout China, Korea, and Japan. The
Oxalidaceae family is characterized by a sour taste and is a long-established medicinal plant and is known to contain oxalic acid, malic acid, and tartaric acid [
9]. In Korea, its leaves and stems have been used for skin diseases such as atopic dermatitis by spraying the juice on the symptomatic area. It has been reported that medicinal herbs containing
Oxalis reduced acne caused by
Propionibaterium acnes in mice [
10]. In addition, it is reported that
Oxalidaceae has a detoxifying effect, and as a plant medicine, it has been used to treat various diseases including diarrhea, dysentery, jaundice, and prolapse.
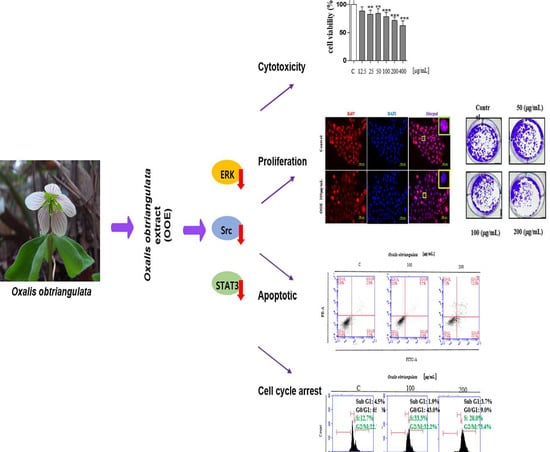
However, there is little data on the anti-cancer efficacy of Oxalidaceae. The present study aimed to examine the effect of the methanol extract of Oxalis obtriangulata on ERK/Src/STAT3 activation in a human PDAC cell line, BxPC3, by assessing the induction of apoptosis, cell cycle arrest, and anti-proliferative effects. These data will provide novel insight regarding Oxalis obtriangulata as a natural source of anti-cancer agents against pancreatic cancers.
3. Discussion
Pancreatic ductal adenocarcinoma (PDAC) has proven to be among the most unbending targets in the modern era in terms of cancer treatment. Achieving a breakthrough with pancreatic cancer is difficult because early diagnosis is not possible, diagnosis is inaccurate, and the few treatments are burdened with many patients. Therefore, it is necessary to identify efficacious and stable substances for treating PDAC [
2]. This research was performed to distinguish OOE as a novel source of potential anti-cancer agents based on the blockage of the STAT3 pathway in a human pancreatic ductal cancer cell line, BxPC3. Our data comprehensively showed that OOE inhibited proliferation, induced cell cycle arrest, and had a slight apoptotic effect in BxPC3 cells. OOE modulated the expression of ERK, Src, and STAT3, which are upregulated in BxPC3 cells as well as STAT3-targeted downstream genes including Cyclin B1, CDK1, CDK2, PARP, caspase-3, MMP-9, VEGF-1, Ki67, p27, c-Myc, and Survivin.
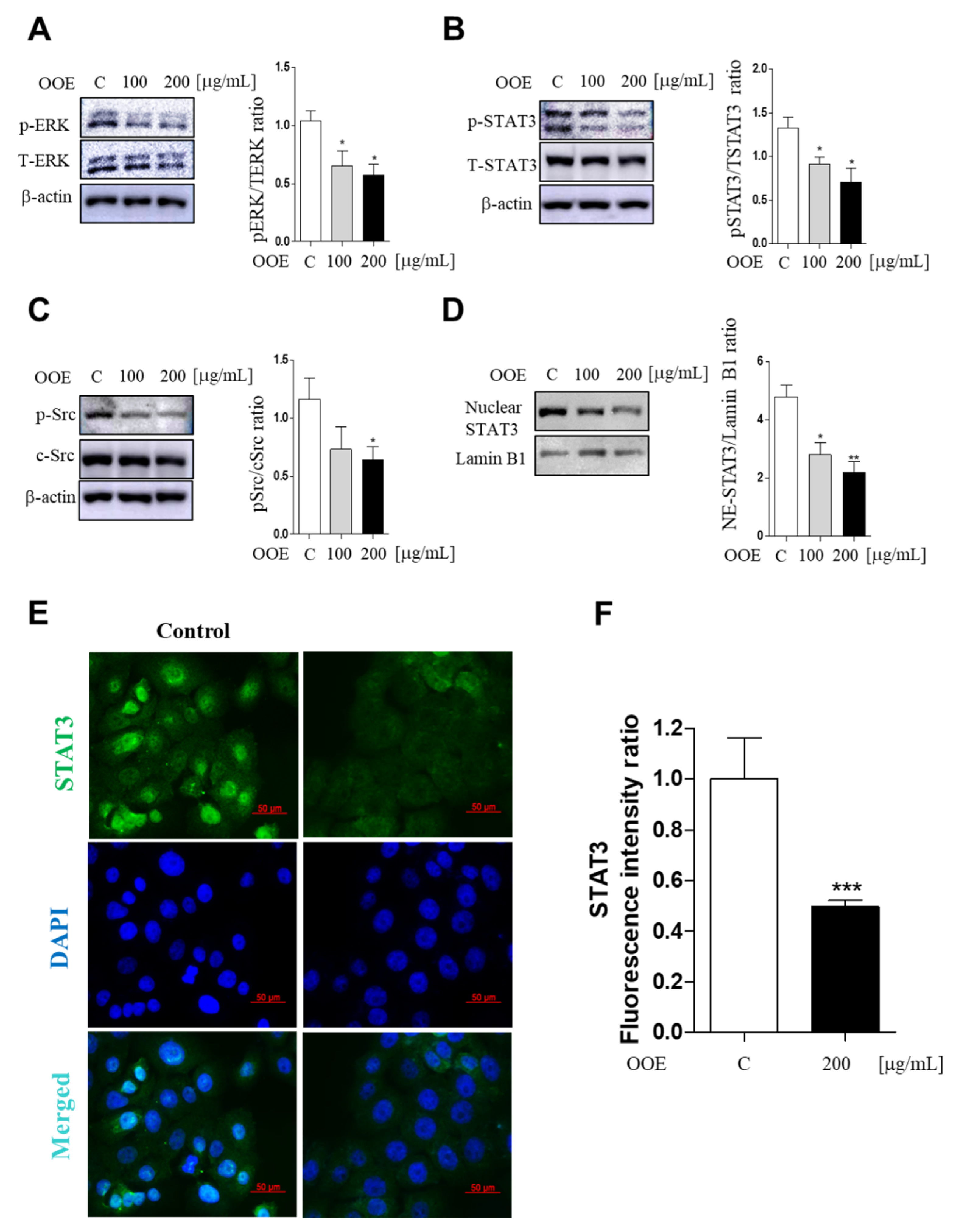
STAT3 is a signaling molecule that delivers signals detected, along with Src activation, through growth factors or cytokine receptors from the cell membrane to the nucleus [
11]. Previous study has shown that STAT3 plays a critical role in cell apoptosis, metastasis, proliferation, and immunomodulation in cancer cells including pancreatic cancer [
12]. For its activation, STAT3 has two important phosphorylation sites, Tyr705 and Ser727. In PDAC, constitutive phosphorylation of STAT3 at Tyr705 has been reported in 30–100% of human tumor specimens. Additionally, phosphorylation at Ser727 has been considered as a secondary event after Tyr705 phosphorylation [
13]. The functional differences between the two phosphorylation sites in pancreatic cancer have not been clearly identified. However, one study has shown that Ser727 phosphorylation is involved in cell survival and nuclear translocation of STAT3 regardless of Tyr705 phosphorylation in melanoma cells [
14]. In our study, OOE suppressed Src and STAT3 phosphorylation (at both Tyr705 and Ser727) as well as STAT3 transcriptional action through inhibiting nuclear translocation. Moreover, we speculated that the blockage of both phosphorylation sites may be due to the anti-cancer potential of OOE. Tyrosine phosphorylation of STAT3 can be stimulated by cytosolic kinases including Src, and the secondary phosphorylation of serine is modulated by ERK [
15]. The secondary phosphorylation is considered to enhance the capacity of tyrosine-phosphorylated STAT3, thus promoting the transcription of cell function-related factors in cancer. ERK also widely affects diverse cell processes including not only cancer proliferation, but also metastatic and angiogenic effects [
16]. Moreover, a previous study reported that in BxPC3 cells, chemotherapeutic agents such as gemcitabine could induce ERK1/2 activity [
17]. Hence, this ERK1/2 activation seriously contributes to chemotherapy resistance in pancreatic cells [
17]. In connection with this, it is also reported that the silencing of pERK1/2 sensitizes BxPC3 cells to gemcitabine-induced apoptosis [
18]. Accordingly, the suppressive effect of OOE on ERK indicates that OOE not only inhibits BxPC3 cell proliferation and growth, but also has a possibility of lowering activated ERK by chemotherapy such as gemcitabine (
Supplementary Materials Figure S2). Considering this, OOE may be an appropriate alternative treatment to enhance chemotherapy.
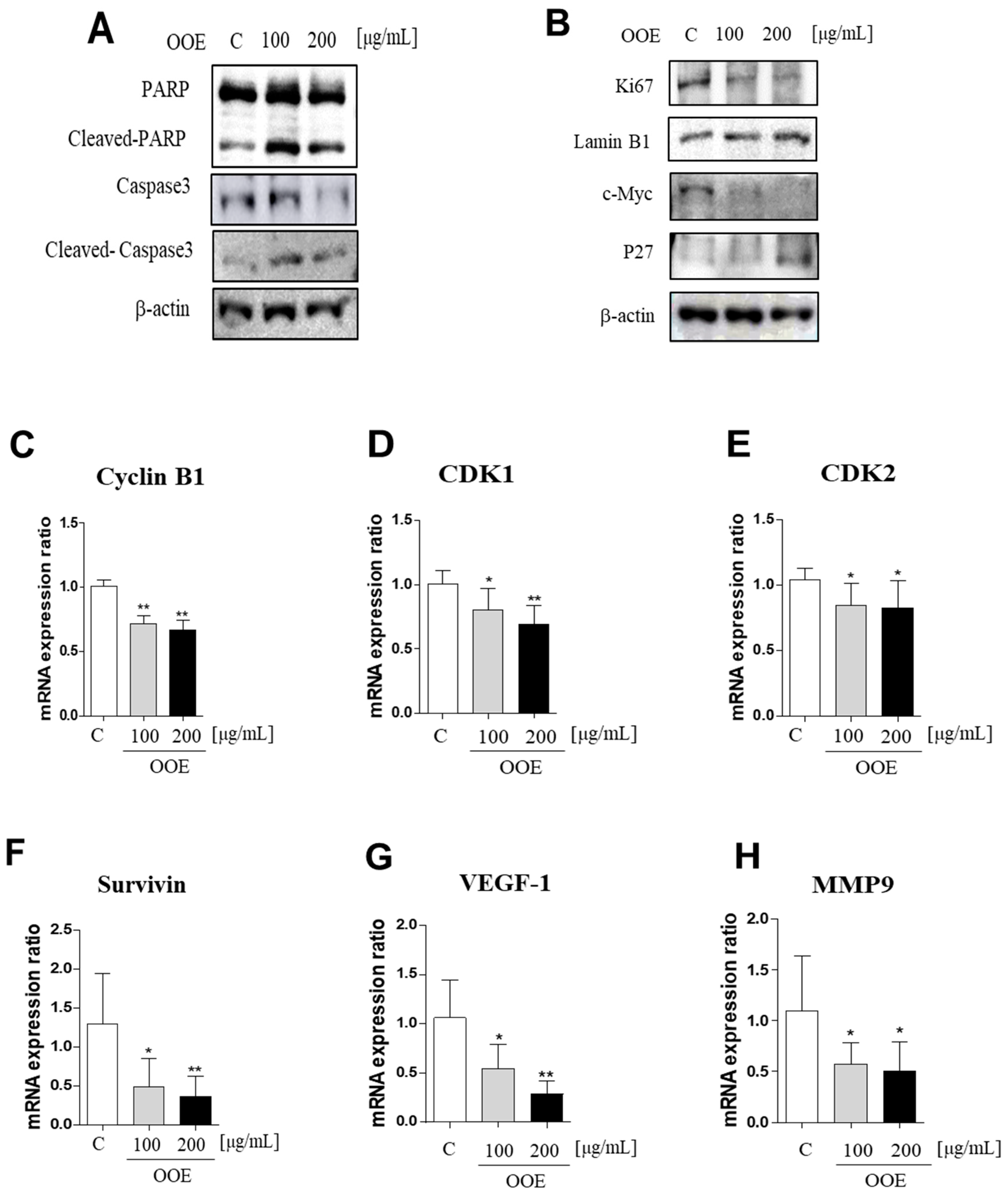
STAT3 coordinates the expression of genes involved in cell cycle regulation. Among them, CDK1 and the cyclin B1 complex are key modulators of the G2/M phase checkpoint [
19]. CDK2, which is well known as an S phase marker, is also reported to contribute to G2/M progression by facilitating cyclin B accumulation [
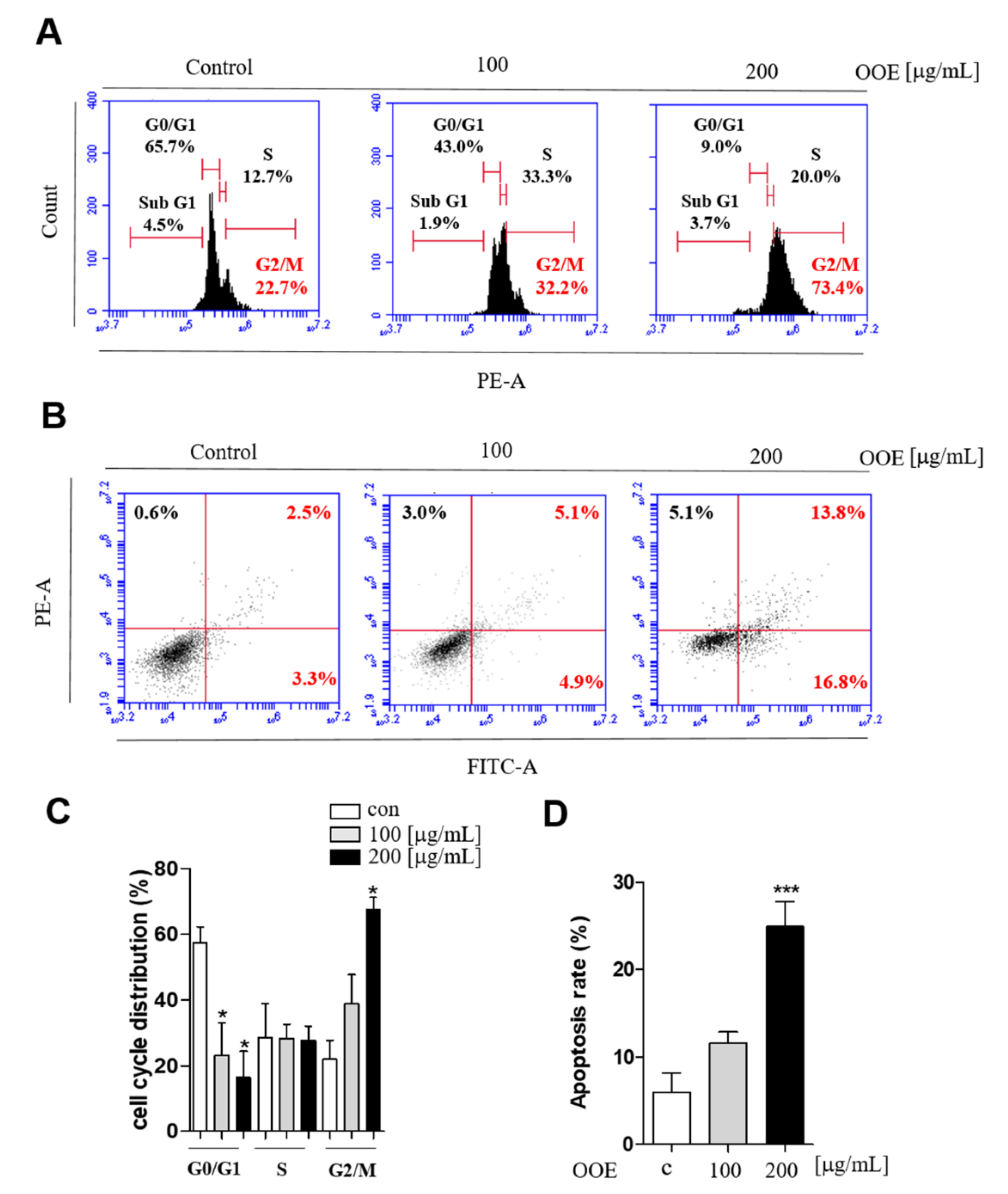
20]. Problems with the G2/M phase due to defects in these genes may cause damaged cells to enter mitosis and undergo apoptosis, therefore increasing the cytotoxicity of chemotherapy.
Activated c-Myc is a hallmark of cancers, which is associated with most human tumor types. c-Myc activation is involved in the growth of cancer cells including transcription and replication of DNA, and regulation of stemness and differentiation of cancer [
21]. One of the targets inhibited by myc is the P27 CDK inhibitor, which plays a major role in the cell cycle process. P27 is a tumor suppressor that binds to the cyclin-cdk complex to repress the activity and mitogenic signals in cancer cells. Decreased levels of P27 are related with tumor grade and progression stage in various human carcinoma including colorectal and breast cancers. In oncogenic cells, P27 inhibited the cell proliferation. Moreover, tumor development is prevented by P27 activation suppressing cell cycle process [
22,
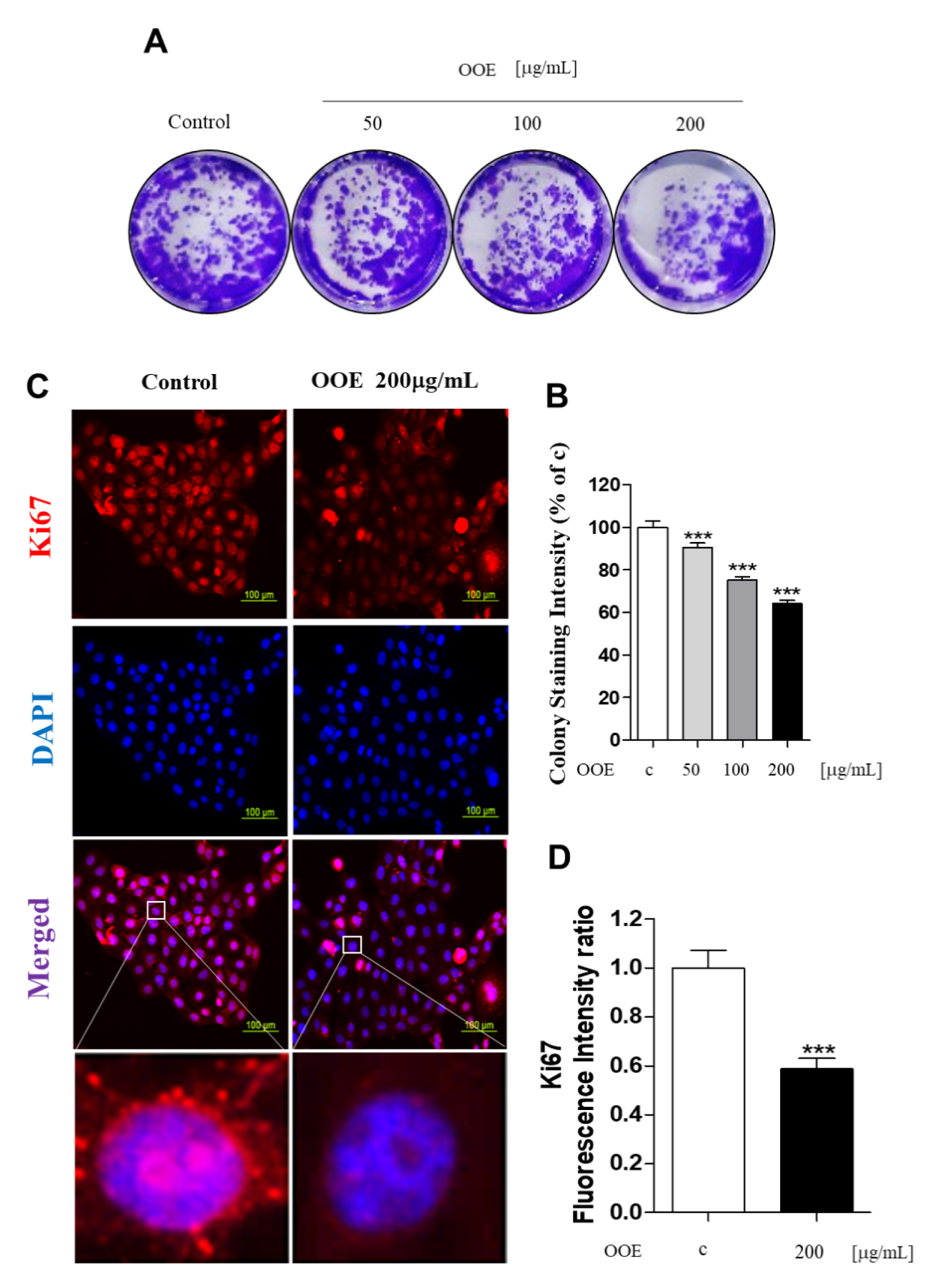
23]. In this study, OOE inhibited the mRNA expression levels of CDK1, CDK2, and cyclin B1. This indicated that OOE could induce G2/M phase arrest starting from the S phase, which is consistent with our cell cycle analysis data. The arrest effect of OOE also reduced the expression of Ki67, P27, and c-Myc, the markers of proliferation and cell cycle arrest, in BxPC3. Taken together, these data suggest that OOE induced delayed proliferation with potent cell cycle arrest through the inhibition of these multiple factors.
Meanwhile, the apoptotic pathway is another target in pancreatic cancer, and caspase-3 is a central effector caspase that initiates apoptosis signals. Activated caspase-3 cleaves PARP protein, and cleaved PARP is considered to be a hallmark of apoptosis [
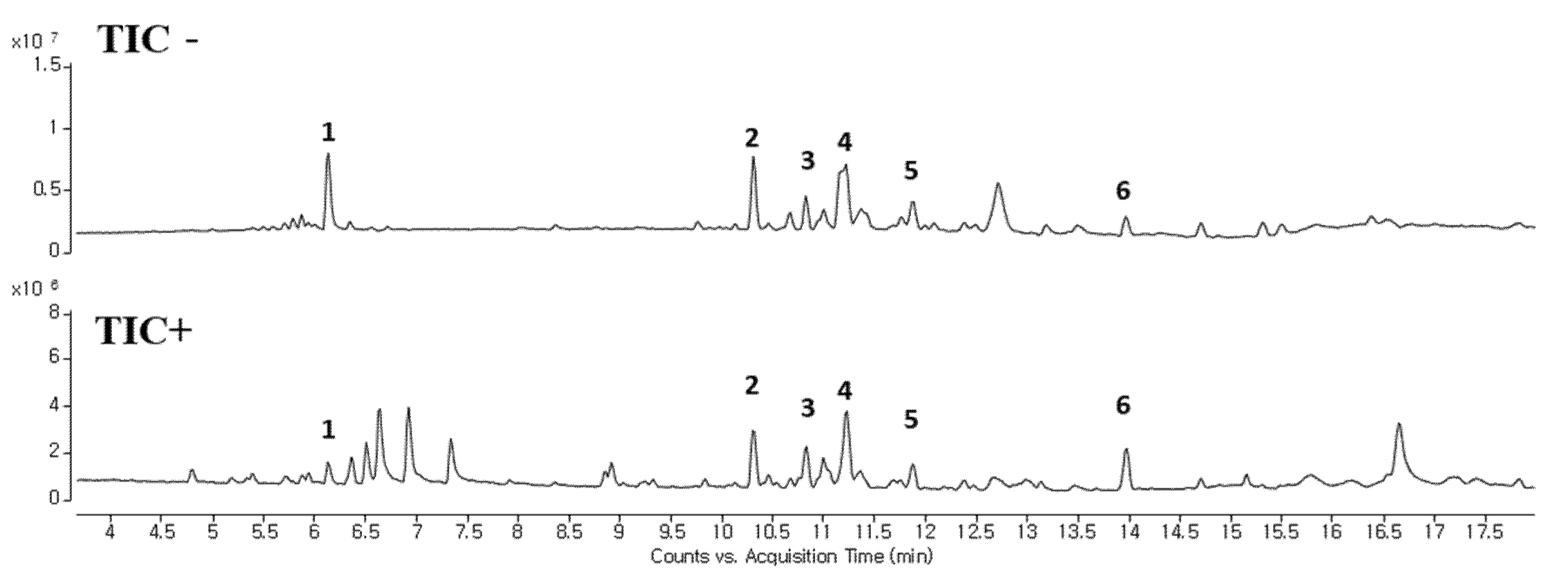
24]. According to our data, OOE showed increased cleaved caspase-3, cleaved PARP, and mRNA expression of Survivin, one of the apoptotic downstream genes of STAT3. OOE also inhibited the expression of metastasis-related factors such as MMP-9 and VEGF, which are targets of STAT3. The FACS data showed OOE significantly increased apoptosis rate. This result may be due to the inclusion of substances with other unexpected effects in OOE, and it supports the need for a component analysis study on OOE. As shown in
Figure 6 and
Table 1, we performed LC-MS to analyze OOE components and recorded the molecular weights of the substances with matched retention times. However, there is little previous analytical data on OOE to determine what components the peaks represent; thus, additional analytical research is required to confirm and identify the major compound that mainly contributed to the anti-cancer effect of OOE.
In this study, we confirmed the anti-cancer activity of OOE on BxPC3, a pancreatic cancer cell. OOE modulated ERK/Src/STAT3 activation and regulated STAT3-downstream genes related with tumor development. Moreover, OOE affected cell viability, proliferation, and induced apoptotic effect and accumulation at G2/M phase in BxPC3. Considering our results comprehensively, we showed the possibility of OOE as an anti-cancer agent. Additionally, this report suggests a new insight of OOE as a source of potential anti-cancer compound. It is necessary to carry out additional experiments to accurately analyze the components in OOE and evaluate the efficacy of each compound.
4. Materials and Methods
4.1. Cell Culture
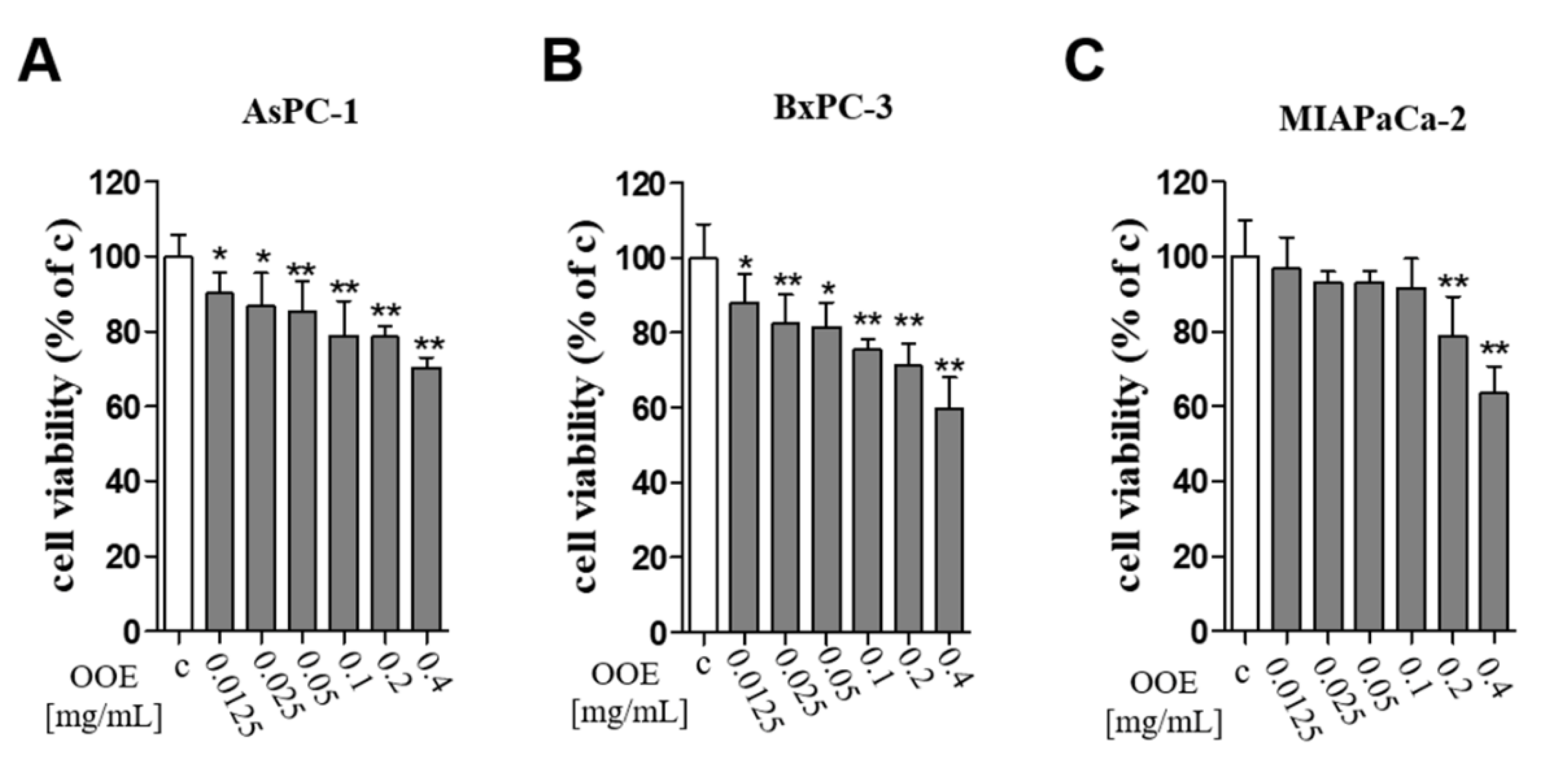
BxPC3, MIAPaCa2, AsPC1, and GES-1 were cultured in Roswell Park Memorial Institute 1640 (RPMI-1640) (Corning Inc., New York, NY, USA). A549 and HepG2 were cultured in Dulbecco Modified Eagle Medium (DMEM). All media contained 10% fetal bovine serum (Gibco, Grand Island, NY, USA) and 1 × antibiotic-antimycotic solution (Corning Inc., New York, NY, USA) and were incubated at 37 °C in 5% CO2.
4.2. Plant Materials
The plant extract (014–094) used in this research was obtained from the Korea Plant Extract Bank at the Korea Research Institute of Bioscience and Biotechnology (Daejeon, Korea). The plant was collected from Geoje-si, Gyeongsangnam-do, KOREA in 2002. A voucher specimen (KRIB 0000057) is kept in the herbarium of the Korea Research Institute of Bioscience and Biotechnology (Ochang, Korea). The plant (34 g), dried in the shade and powdered, was added to 1 L of methyl alcohol 99.9 % (HPLC grade) and extracted through 30 cycles (40 KHz, 1500 W, 15 min ultrasonication–120 min standing per cycle) at room temperature using an ultrasonic extractor (SDN-900H, SD-ULTRASONIC Co. Ltd, Seoul, Korea). After filtration and drying under reduced pressure, the
O. obtriangulata extract (4.0 g) was obtained. The dried sample (voucher specimen: 014–094) was dissolved in dimethyl sulfoxide for experimentation. All plant materials were deposited in the Plant Extract Bank of Korea Research Institute of Bioscience and Biotechnology (KRIBB) in Daejeon, Korea (Daejeon, Korea,
http://extract.kribb.re.kr/).
4.3. MTT Assay (Cell Viability Assay)
BxPC3 cells were seeded at 1 × 104 cells per well into 96-well plates and incubated overnight; then, they were treated with OOE (12.5–400 µg/mL) for 24 h to confirm the cytotoxicity of OOE. MTT (3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-[2H]-tetrazolium bromide) was added to the wells at a final concentration of 0.5 mg/mL and incubated for 2 h. The cell medium was removed, and formazan crystals were dissolved with DMSO to check absorbance. The plates were incubated for 5 min in a slow shaker, and the resulting formazan was detected using a microplate reader at a wavelength of 540 nm.
4.4. Colony Formation Assay
For the colony formation assay, 5000 cells were seeded in 6-well plates overnight. The medium was removed and replaced with fresh media with or without OOE once every three days for 14 days. When colonies were visible without a microscope, they were fixed with 4% paraformaldehyde and stained using 0.1% crystal violet solution. After observation, stained colonies were dissolved in DMSO, and the intensity was measured by a microplate reader at 540 nm [
25].
4.5. Immunofluorescence Assay
The cells were treated with or without OOE in 4-well confocal dishes. After treatment, cells were fixed with 10% formalin for 10 min, treated with 0.2% Triton X in phosphate-buffered saline (PBS) for 20 min, washed three times with PBS, and then blocked with 5% bovine serum albumin (BSA) solution in PBS for 1 h. Cells were incubated overnight with anti-pSTAT3, Ki67 antibody (1:500 in PBS) at 4 °C followed by Alexa-Fluor-488 antibody and Alexa Flour-594 antibody for 1 h at room temperature. Cell nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, St. Louis, MO, USA) for 3 min at room temperature. Cells stained with DAPI were washed with PBS for 30 min, and fluorescence was observed using a microscope.
4.6. Annexin V/Propidium Iodide Staining Assay and Analysis of Cell Cycle Distribution
Cells were seeded at 50 × 104 cells per well into 6-well plates and then treated with OOE for 24 h. To confirm apoptosis, cell cycle analysis was conducted to determine cell accumulation in the G2/M phase as described previously using flow cytometry [
26].
4.7. Immunoblotting
Cells were completely lysed using cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA). Cytosolic and nuclear fractions were divided using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, Waltham, MA, USA). Bio-Rad Protein Assay Reagent (Bio-Rad, Hercules, CA, USA) was used to measure protein concentrations for the Bradford assay. Each sample was separated in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel and then electro-transferred to a nitrocellulose membrane. The membranes were blocked with 3% BSA in tris-buffered saline/Tween 20 (TBS-T). Membranes were incubated at 4 °C overnight with the following antibodies: p-Src, c-Src, p-STAT3, T-STAT3, PARP, cleaved PARP, pro-caspase-3, cleaved caspase-3, p-ERK1/2, ERK1/2, Ki67, Lamin b1, and beta-actin, c-Myc, and P27 (All were 1:3000). Membranes were extensively washed with TBS-T for 1 h and then incubated with the following secondary antibodies: goat anti-rabbit IgG-HRP and goat anti-mouse IgG-HRP (all were 1:5000 in TBS-T).
4.8. RT-qPCR
Cells were seeded at 50 × 104 cells per well into 6-well plates and incubated with or without OOE for 24 h. Then, cells were treated with Ribo-Ex to extract RNA using the GeneAll Hybrid-R RNA Purification Kit (GeneAll, Seoul, Korea). RNA was quantified by Nano Drop (Thermo Fisher Scientific, Waltham, MA, USA). Amplification of cDNA was performed as follows: 45 °C for 60 min and 95 °C for 15 min at 4 °C using the Maxime RT premix (iNtRON Biotechnology, South Korea). Real-time quantitative PCR was performed using the Universal SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA). Real-time PCR was performed on an Applied Biosystems Step One System (Applied Biosystems, Foster City, CA, USA). In this study, relative target gene expression was quantified relative to that of GAPDH to determine mRNA expression levels. The used PCR primers are as follows:
CDK1: Forward; 5TGGAGAAGGTACCTATGGAGTT3, Reverse; 5AGGAACCCCTTCCTCTTCAC3
CDK2: Forward; 5AAAGCCAGAAACAAGTTGACG3, Reverse; 5GAGATCTCTCGGATGGCAGT3
VEGF: Forward; 5GGAGTGTGTGCACGAGTC3, Reverse; 5GGTCGACTGAGAGCT3
Cyclin B1: Forward; 5GAACAACTGCAGGCCAAAAT3, Reverse; 5CACTGGCACCAGCATAGG3
Survivin: Forward; 5TTCTGCACATCTGAGTCG3, Reverse; 5TGTCGAGAGCTCAGT3
MMP9: Forward; 5TTGACAGCGACAGAGTG3, Reverse; 5GCATTCACGTCGTCCTTAT3
4.9. Liquid Chromatography-Mass Spectrometry (LC-MS)
Chromatographic separation of the extract was performed using an Agilent 1290 Infinity LC System (Agilent Technologies Santa Clara, CA, USA) with a Walters C18 column (2.1 mm × 100 mm, 1.7 μm) at 30 °C, which employed a mobile phase comprising 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The gradient was programmed as follows: 0–15 min, 5–95% B in A; 15–20 min, 95% B; and 20–25 min, 95–5% B; and the column was equilibrium with 5% B. A sample of 1 μL was injected into the column using an autosampler. The HPLC system was interfaced to the MS system, an Agilent 6550 Accurate-Mass Q-TOF (Agilent Technologies, Santa Clara, CA, USA) equipped with a dual agilent jet stream technology electrospray ionization (AJS ESI )source operating in positive and negative ion modes. The ESI spray voltage was set to 3500 V (Vcap). Mass spectra were acquired at a scan rate of 1 spectra/s with a mass range of 100–1700 m/z. Data analysis was performed using the Mass Hunter Qualitative Analysis Software (version B.07.00, Agilent Technologies, Santa Clara, CA, USA) for compound profiling [
27].
4.10. Statistical Analysis
Data were expressed as mean ± SEM of at least different three experiments. Statistical analysis was performed using one-way analysis of variance in Graph Pad Prism 5, followed by Dunnett’s multiple comparison test. A p-value < 0.05 was considered statistically significant, and p < 0.01 and p < 0.001 were considered highly significant.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}