Molecular Mechanism Underlying Hypoxic Preconditioning-Promoted Mitochondrial Translocation of DJ-1 in Hypoxia/Reoxygenation H9c2 Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
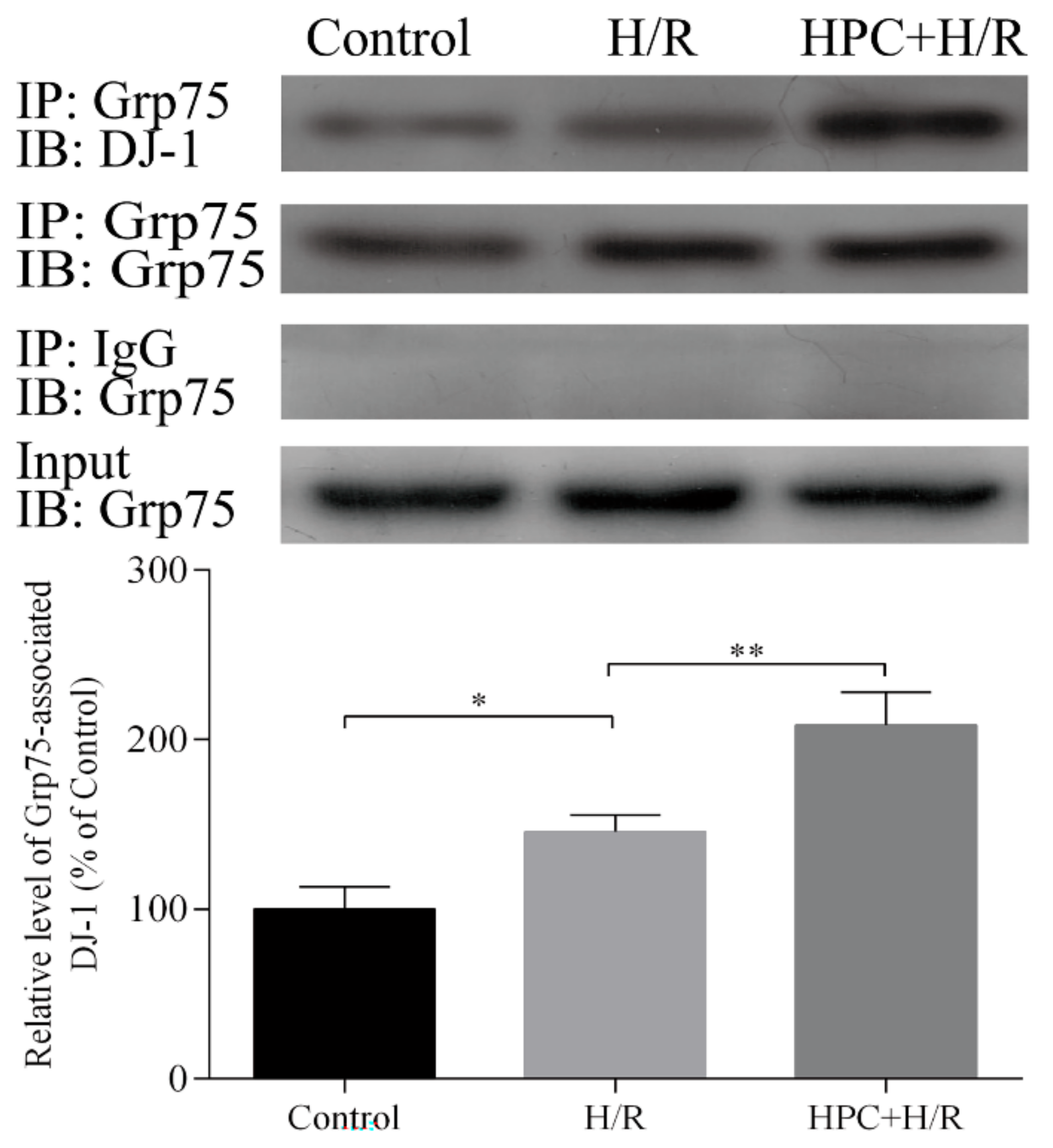
2.1. Effect of HPC on the Interaction between DJ-1 and Grp75 in H9c2 Cells Subjected to H/R
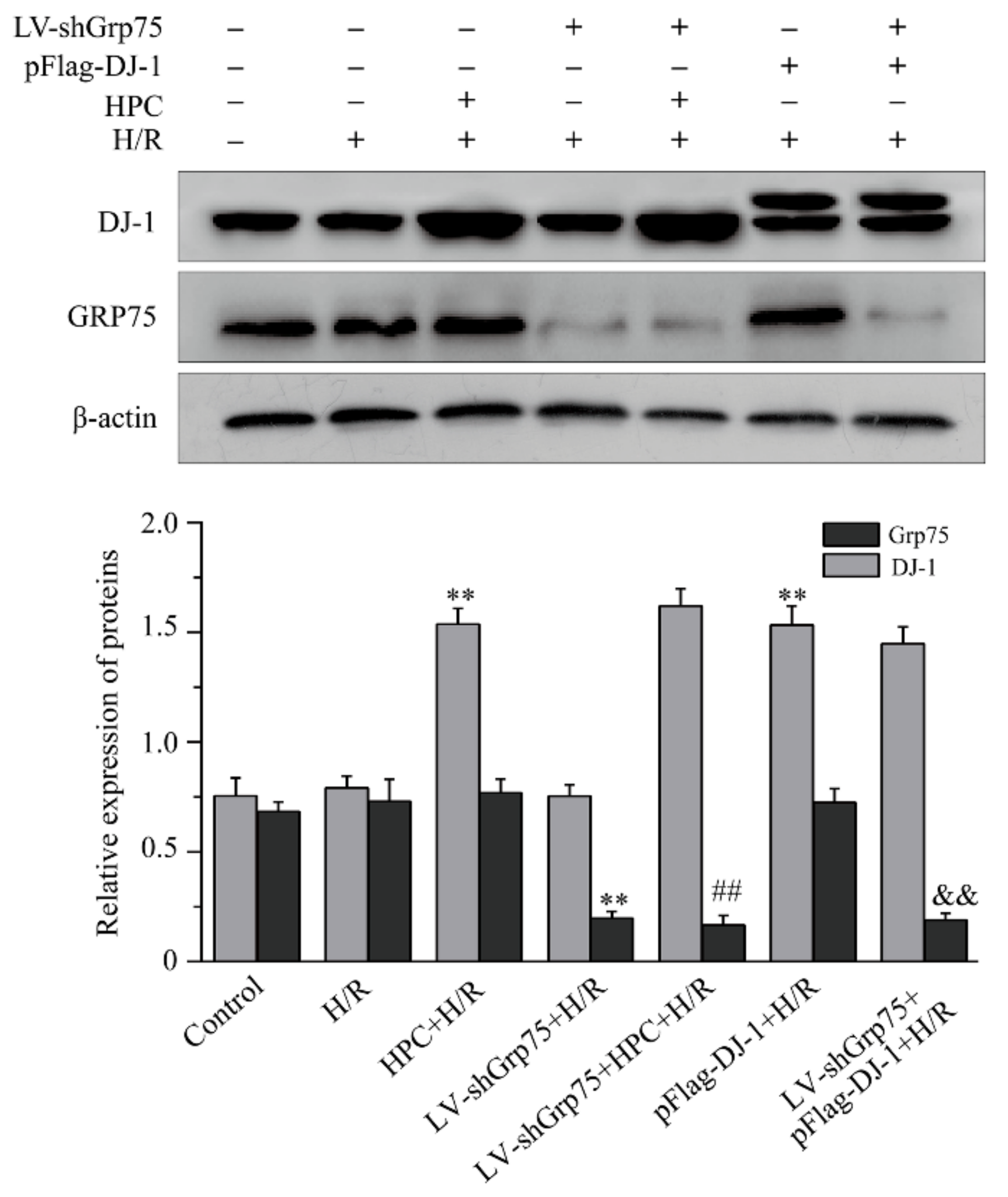
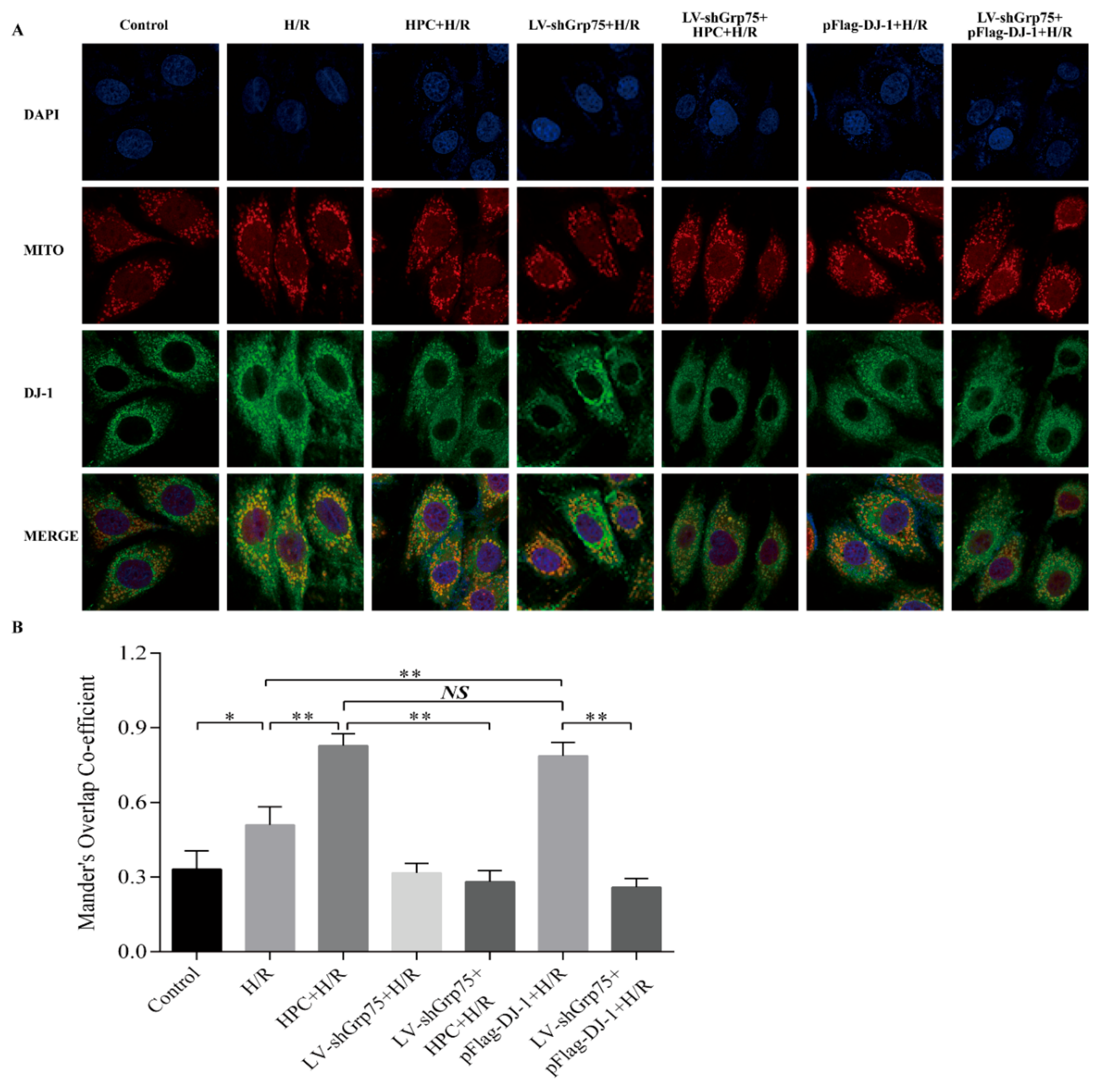
2.2. Effect of Grp75 Knockdown on HPC-Promoted Mitochondrial Translocation of DJ-1 in H9c2 Cells Subjected to H/R
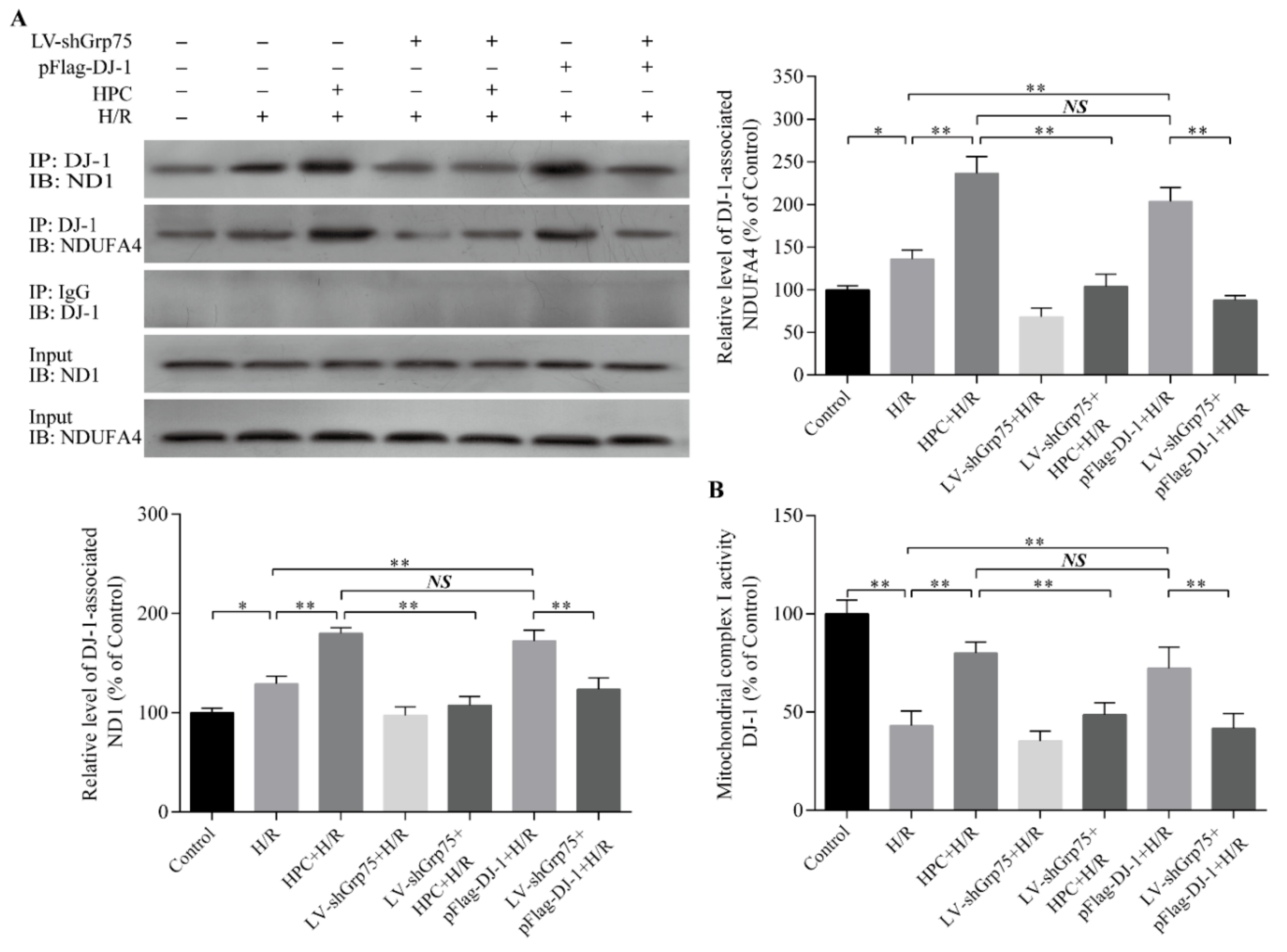
2.3. Effect of Grp75 Knockdown on DJ-1-Mediated Protection of Mitochondrial Complex I by HPC in H9c2 Cells Subjected to H/R
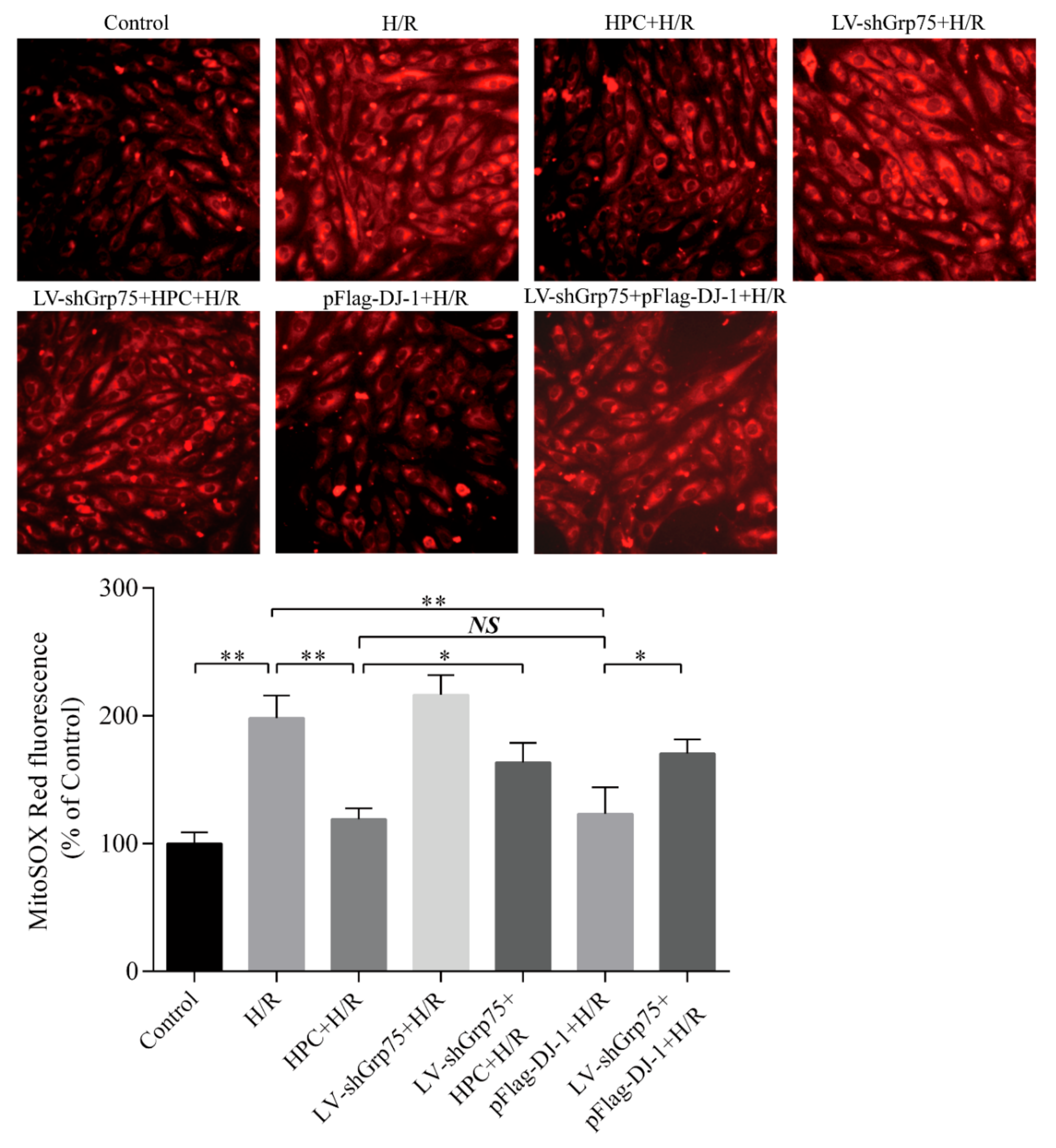
2.4. Effects of Grp75 Knockdown on DJ-1-Mediated Inhibition of Mitochondrial ROS Generation by HPC in H9c2 Cells Subjected to H/R
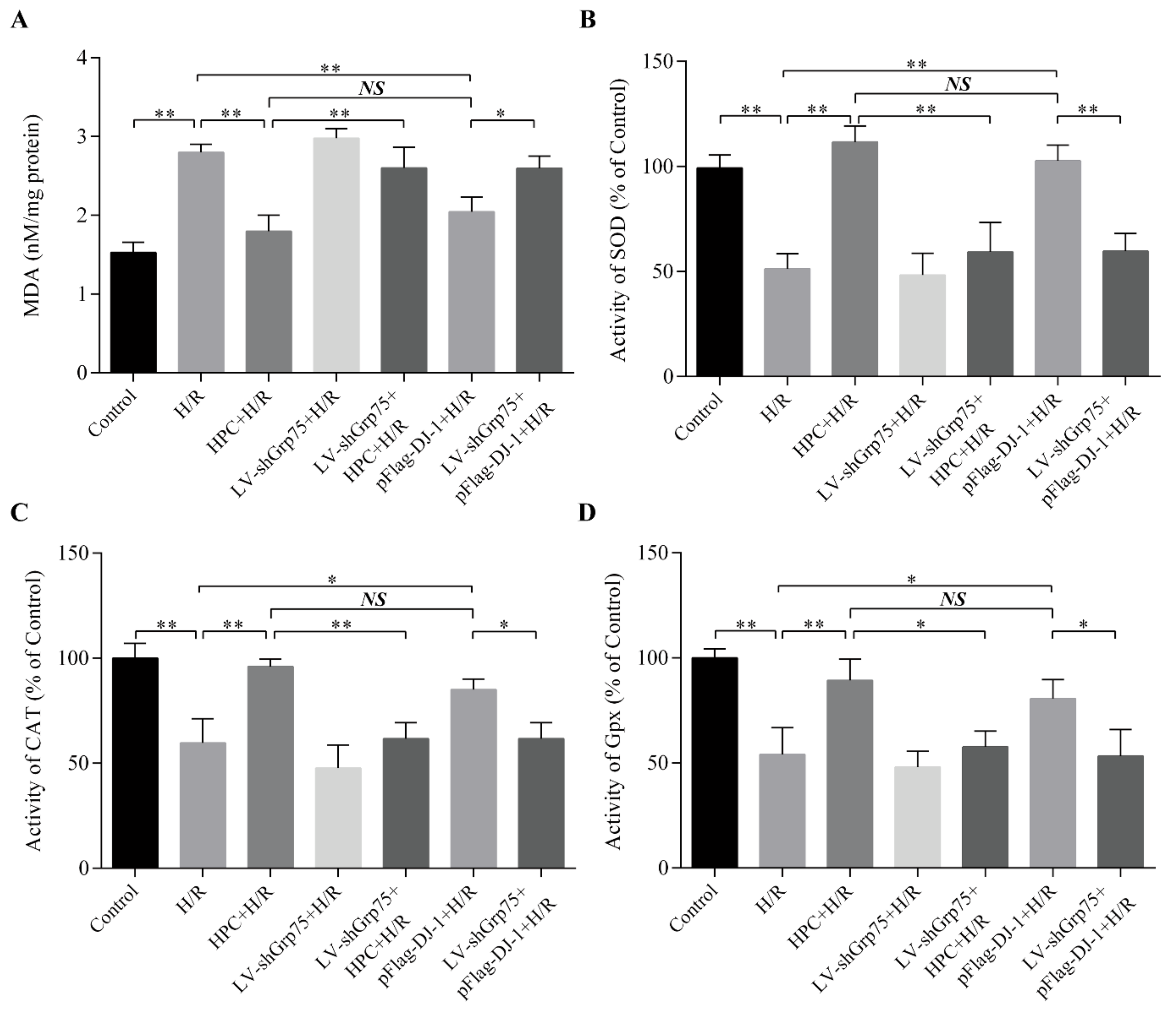
2.5. Effect of Grp75 Knockdown on DJ-1-Mediated Suppression of Oxidative Stress by HPC in H/R-Treated H9c2 Cells
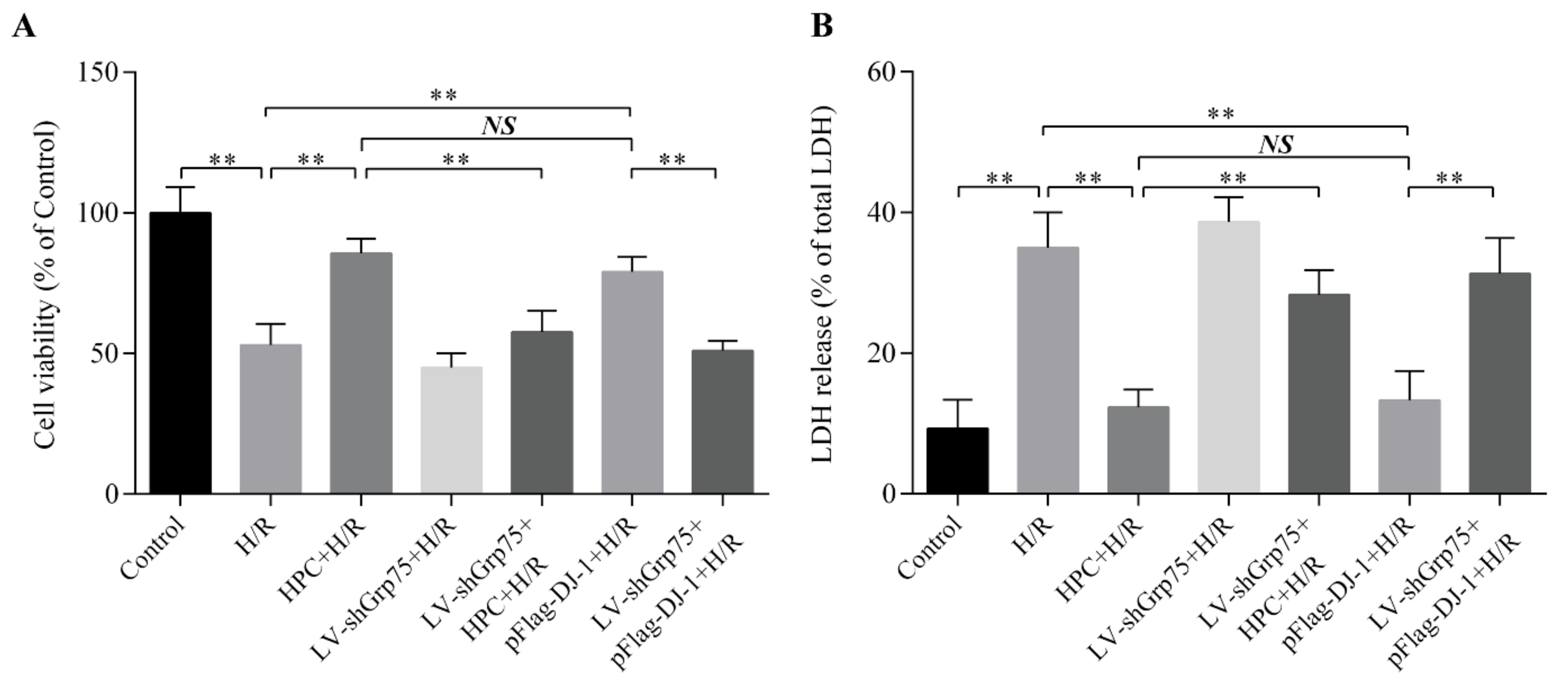
2.6. Effect of Grp75 Knockdown on DJ-1-Mediated Cardioprotection of HPC Against H/R Injury in H9c2 Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Cell Transfection
4.4. Establishment of Cellular Models of H/R and HPC
4.5. Immunofluorescence Microscopy
4.6. Western Blot Analysis
4.7. Co-Immunoprecipitation Assays
4.8. Measurement of Mitochondrial Complex I Activity
4.9. Measurement of Mitochondrial ROS Generation
4.10. Evaluation of Oxidative Stress
4.11. Assessment of Cell Injury
4.12. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef]
- Yellon, D.M.; Downey, J.M. Preconditioning the Myocardium: From Cellular Physiology to Clinical Cardiology. Physiol. Rev. 2003, 83, 1113–1151. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K.; Maulik, N. Cardiac genomic response following preconditioning stimulus. Cardiovasc. Res. 2006, 70, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Molecular basis of cardioprotection: Signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef] [PubMed]
- Sadat, U. Signaling pathways of cardioprotective ischemic preconditioning. Int. J. Surg. 2009, 7, 490–498. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.Y.; Qiu, L.Y.; Yan, Y.F.; Liao, Z.P.; Chen, H.P. Involvement of DJ1 in ischemic preconditioninginduced delayed cardioprotection in vivo. Mol. Med. Rep. 2017, 15, 995–1001. [Google Scholar] [CrossRef]
- Dongworth, R.K.; A Mukherjee, U.; Hall, A.R.; Astin, R.; Ong, S.-B.; Yao, Z.; Dyson, A.; Szabadkai, G.; Davidson, S.M.; Yellon, D.M.; et al. DJ-1 protects against cell death following acute cardiac ischemia-reperfusion injury. Cell Death Dis. 2014, 5, e1082. [Google Scholar] [CrossRef]
- Yan, Y.-F.; Chen, H.-P.; Huang, X.-S.; Qiu, L.-Y.; Liao, Z.-P.; Huang, Q.-R. DJ-1 Mediates the Delayed Cardioprotection of Hypoxic Preconditioning Through Activation of Nrf2 and Subsequent Upregulation of Antioxidative Enzymes. J. Cardiovasc. Pharmacol. 2015, 66, 148–158. [Google Scholar] [CrossRef]
- Nagakubo, D.; Taira, T.; Kitaura, H.; Ikeda, M.; Tamai, K.; Iguchi-Ariga, S.M.; Ariga, H. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem. Biophys. Res. Commun. 1997, 231, 509–513. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Cookson, M.R. Evolutionary and functional relationships within the DJ1 superfamily. BMC Evol. Boil. 2004, 4, 6. [Google Scholar]
- Ariga, H.; Takahashi-Niki, K.; Kato, I.; Maita, H.; Niki, T.; Iguchi-Ariga, S.M.M. Neuroprotective Function of DJ-1 in Parkinson’s Disease. Oxid. Med. Cell. Longev. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Taira, T.; Saito, Y.; Niki, T.; Iguchi-Ariga, S.M.M.; Takahashi, K.; Ariga, H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004, 5, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, N.; Kaneko, Y.; Pantcheva, P.; Elias, M.; Duncan, K.; Borlongan, C.V. The role of DJ-1 in the oxidative stress cell death cascade after stroke. Neural Regen. Res. 2014, 9, 1430–1433. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-K.; Lee, M.-S.; Bae, D.-W.; Lee, D.-H.; Cha, S.-S.; Chi, S.-W. Structural basis for the interaction between DJ-1 and Bcl-XL. Biochem. Biophys. Res. Commun. 2018, 495, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Xu, X.-W.; Wang, H.; Xiao, L.; Zhao, L.; Duan, G.-L.; Li, X.-R.; Ma, Z.-X.; Chen, H.-P. DJ-1 plays an obligatory role in the cardioprotection of delayed hypoxic preconditioning against hypoxia/reoxygenation-induced oxidative stress through maintaining mitochondrial complex I activity. Cell Biochem. Funct. 2018, 36, 147–154. [Google Scholar] [CrossRef]
- Wadhwa, R.; Taira, K.; Kaul, S.C. An Hsp70 family chaperone, mortalin/mthsp70/PBP74/Grp75: what, when, and where? Cell Stress Chaperon. 2002, 7, 309–316. [Google Scholar] [CrossRef]
- Deocaris, C.C.; Kaul, S.C.; Wadhwa, R. On the brotherhood of the mitochondrial chaperones mortalin and heat shock protein 60. Cell Stress Chaperon. 2006, 11, 116–128. [Google Scholar] [CrossRef]
- Kaul, S.C.; Deocaris, C.C.; Wadhwa, R. Three faces of mortalin: A housekeeper, guardian and killer. Exp. Gerontol. 2007, 42, 263–274. [Google Scholar] [CrossRef]
- Tai-Nagara, I.; Matsuoka, S.; Ariga, H.; Suda, T. Mortalin and DJ-1 coordinately regulate hematopoietic stem cell function through the control of oxidative stress. Blood 2014, 123, 41–50. [Google Scholar] [CrossRef]
- Chan, J.Y.; Chan, S.H. Activation of endogenous antioxidants as a common therapeutic strategy against cancer, neurodegeneration and cardiovascular diseases: A lesson learnt from DJ-1. Pharmacol. Ther. 2015, 156, 69–74. [Google Scholar] [CrossRef]
- Li, H.M.; Niki, T.; Taira, T.; Iguchi-Ariga, S.M.M.; Ariga, H. Association of DJ-1 with chaperones and enhanced association and colocalization with mitochondrial Hsp70 by oxidative stress. Free. Radic. Res. 2005, 39, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Jang, W.H.; Zhao, X.; Jeong, B.S.; Mouradian, M.M. Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J. Neurosci. Res. 2009, 87, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Lev, N.; Ickowicz, D.; Melamed, E.; Offen, D. Oxidative insults induce DJ-1 upregulation and redistribution: Implications for neuroprotection. NeuroToxicology 2008, 29, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-S.; Chen, H.-P.; Wang, S.; Yu, H.-H.; Huang, X.-S.; Huang, Q.-R.; He, M. Hypoxic preconditioning up-regulates DJ-1 protein expression in rat heart-derived H9c2 cells through the activation of extracellular-regulated kinase 1/2 pathway. Mol. Cell. Biochem. 2012, 370, 231–240. [Google Scholar] [CrossRef]
- Neupert, W.; Herrmann, J.M. Translocation of Proteins into Mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef]
- Yaguchi, T.; Aida, S.; Kaul, S.C.; Wadhwa, R. Involvement of Mortalin in Cellular Senescence from the Perspective of its Mitochondrial Import, Chaperone, and Oxidative Stress Management Functions. Ann. N. Y. Acad. Sci. 2007, 1100, 306–311. [Google Scholar] [CrossRef]
- Jin, J.; Li, G.J.; Davis, J.; Zhu, D.; Wang, Y.; Pan, C.; Zhang, J. Identification of novel proteins associated with both alpha-synuclein and DJ-1. Mol. Cell. Proteom. 2007, 6, 845–859. [Google Scholar] [CrossRef]
- Bolender, N.; Sickmann, A.; Wagner, R.; Meisinger, C.; Pfanner, N. Multiple pathways for sorting mitochondrial precursor proteins. EMBO Rep. 2008, 9, 42–49. [Google Scholar] [CrossRef]
- Psarra, A.-M.G.; Sekeris, C.E. Nuclear receptors and other nuclear transcription factors in mitochondria: Regulatory molecules in a new environment. Biochim. Biophys. Acta BBA Bioenerg. 2008, 1783, 1–11. [Google Scholar] [CrossRef]
- Hayashi, T.; Ishimori, C.; Takahashi-Niki, K.; Taira, T.; Kim, Y.-C.; Maita, H.; Maita, C.; Ariga, H.; Iguchi-Ariga, S.M. DJ-1 binds to mitochondrial complex I and maintains its activity. Biochem. Biophys. Res. Commun. 2009, 390, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.Y.; Park, J.H.; Kim, S.J.; Seo, K.S.; Han, J.S.; Lee, S.H.; Kim, J.M.; Park, J.I.; Park, S.K.; Lim, K.; et al. DJ-1 null dopaminergic neuronal cells exhibit defects in mitochondrial function and structure: involvement of mitochondrial complex I assembly. PLoS ONE 2012, 7, e32629. [Google Scholar] [CrossRef] [PubMed]
- Wirth, C.; Brandt, U.; Hunte, C.; Zickermann, V. Structure and function of mitochondrial complex I. Biochim. Biophys. Acta BBA Bioenerg. 2016, 1857, 902–914. [Google Scholar] [CrossRef]
- Vinogradov, A.D.; Grivennikova, V.G. Oxidation of NADH and ROS production by respiratory complex I. Biochim. Biophys. Acta BBA Gen. Subj. 2016, 1857, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Mimaki, M.; Wang, X.; McKenzie, M.; Thorburn, D.R.; Ryan, M.T. Understanding mitochondrial complex I assembly in health and disease. Biochim. Biophys. Acta BBA Bioenerg. 2012, 1817, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Giaime, E.; Yamaguchi, H.; Gautier, C.A.; Kitada, T.; Shen, J. Loss of DJ-1 Does Not Affect Mitochondrial Respiration but Increases ROS Production and Mitochondrial Permeability Transition Pore Opening. PLoS ONE 2012, 7, e40501. [Google Scholar] [CrossRef]
- Zhou, M.; Xia, Z.-Y.; Lei, S.-Q.; Leng, Y.; Xue, R. Role of mitophagy regulated by Parkin/DJ-1 in remote ischemic postconditioning-induced mitigation of focal cerebral ischemia-reperfusion. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4866–4871. [Google Scholar]
- Gao, H.; Yang, W.; Qi, Z.; Lu, L.; Duan, C.; Zhao, C.; Yang, H. DJ-1 Protects Dopaminergic Neurons against Rotenone-Induced Apoptosis by Enhancing ERK-Dependent Mitophagy. J. Mol. Boil. 2012, 423, 232–248. [Google Scholar] [CrossRef]
- Yu, H.-H.; Xu, Q.; Chen, H.-P.; Wang, S.; Huang, X.-S.; Huang, Q.-R.; He, M. Stable overexpression of DJ-1 protects H9c2 cells against oxidative stress under a hypoxia condition. Cell Biochem. Funct. 2013, 31, 643–651. [Google Scholar] [CrossRef]
- Huang, X.S.; Chen, H.P.; Yu, H.H.; Yan, Y.F.; Liao, Z.P.; Huang, Q.R. Nrf2-dependent upregulation of antioxidative enzymes: a novel pathway for hypoxic preconditioning-mediated delayed cardioprotection. Mol. Cell. Biochem. 2014, 385, 33–41. [Google Scholar] [CrossRef]
- Mizukami, Y.; Iwamatsu, A.; Aki, T.; Kimura, M.; Nakamura, K.; Nao, T.; Okusa, T.; Matsuzaki, M.; Yoshida, K.; Kobayashi, S. ERK1/2 regulates intracellular ATP levels through alpha-enolase expression in cardiomyocytes exposed to ischemic hypoxia and reoxygenation. J. Biol. Chem. 2004, 279, 50120–50131. [Google Scholar] [CrossRef]
- Jiao, J.D.; Garg, V.; Yang, B.; Hu, K. Novel functional role of heat shock protein 90 in ATP-sensitive K+ channel-mediated hypoxic preconditioning. Cardiovasc. Res. 2008, 77, 126–133. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, Y.-Z.; Xiao, L.; Zhao, L.; Qiu, L.-J.; Ma, Z.-X.; Xu, X.-W.; Liu, H.-Y.; Zhou, T.-T.; Wang, X.-Y.; Tang, L.; et al. Molecular Mechanism Underlying Hypoxic Preconditioning-Promoted Mitochondrial Translocation of DJ-1 in Hypoxia/Reoxygenation H9c2 Cells. Molecules 2020, 25, 71. https://doi.org/10.3390/molecules25010071
Deng Y-Z, Xiao L, Zhao L, Qiu L-J, Ma Z-X, Xu X-W, Liu H-Y, Zhou T-T, Wang X-Y, Tang L, et al. Molecular Mechanism Underlying Hypoxic Preconditioning-Promoted Mitochondrial Translocation of DJ-1 in Hypoxia/Reoxygenation H9c2 Cells. Molecules. 2020; 25(1):71. https://doi.org/10.3390/molecules25010071
Chicago/Turabian StyleDeng, Yi-Zhang, Lin Xiao, Le Zhao, Le-Jia Qiu, Zhao-Xia Ma, Xing-Wang Xu, Hao-Yue Liu, Ting-Ting Zhou, Xue-Ying Wang, Lei Tang, and et al. 2020. "Molecular Mechanism Underlying Hypoxic Preconditioning-Promoted Mitochondrial Translocation of DJ-1 in Hypoxia/Reoxygenation H9c2 Cells" Molecules 25, no. 1: 71. https://doi.org/10.3390/molecules25010071
APA StyleDeng, Y.-Z., Xiao, L., Zhao, L., Qiu, L.-J., Ma, Z.-X., Xu, X.-W., Liu, H.-Y., Zhou, T.-T., Wang, X.-Y., Tang, L., & Chen, H.-P. (2020). Molecular Mechanism Underlying Hypoxic Preconditioning-Promoted Mitochondrial Translocation of DJ-1 in Hypoxia/Reoxygenation H9c2 Cells. Molecules, 25(1), 71. https://doi.org/10.3390/molecules25010071