Astaxanthin Protects PC12 Cells against Homocysteine- and Glutamate-Induced Neurotoxicity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussions
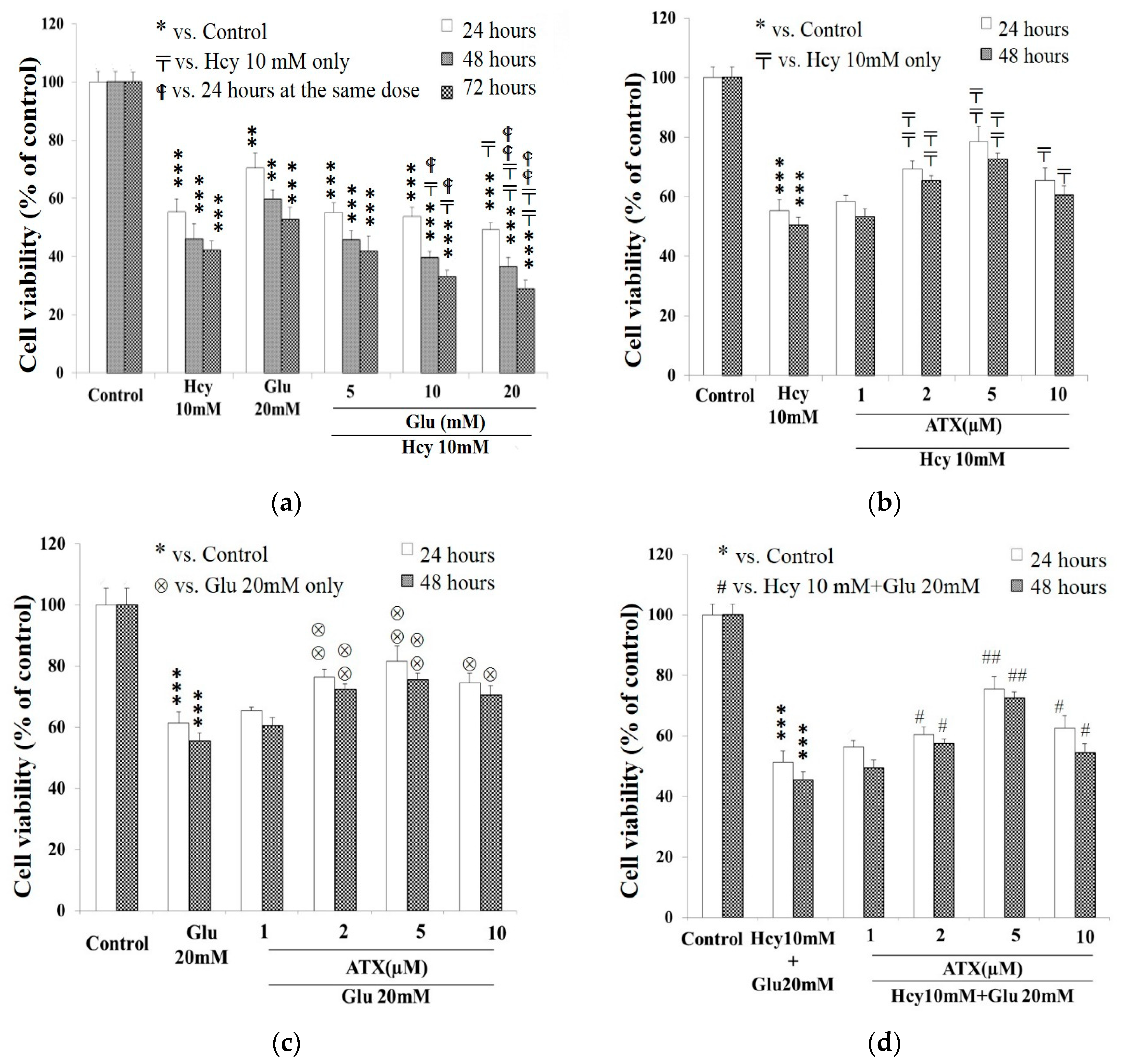
2.1. Cell Viability Affected by Astaxanthin, Homocysteine, and Glutamate
2.2. Potentiation of Glutamate on the Cytotoxicity of Homocysteine
2.3. Protective Effect of Astaxanthin against The Insult Exerted by Homocysteine and Glutamate
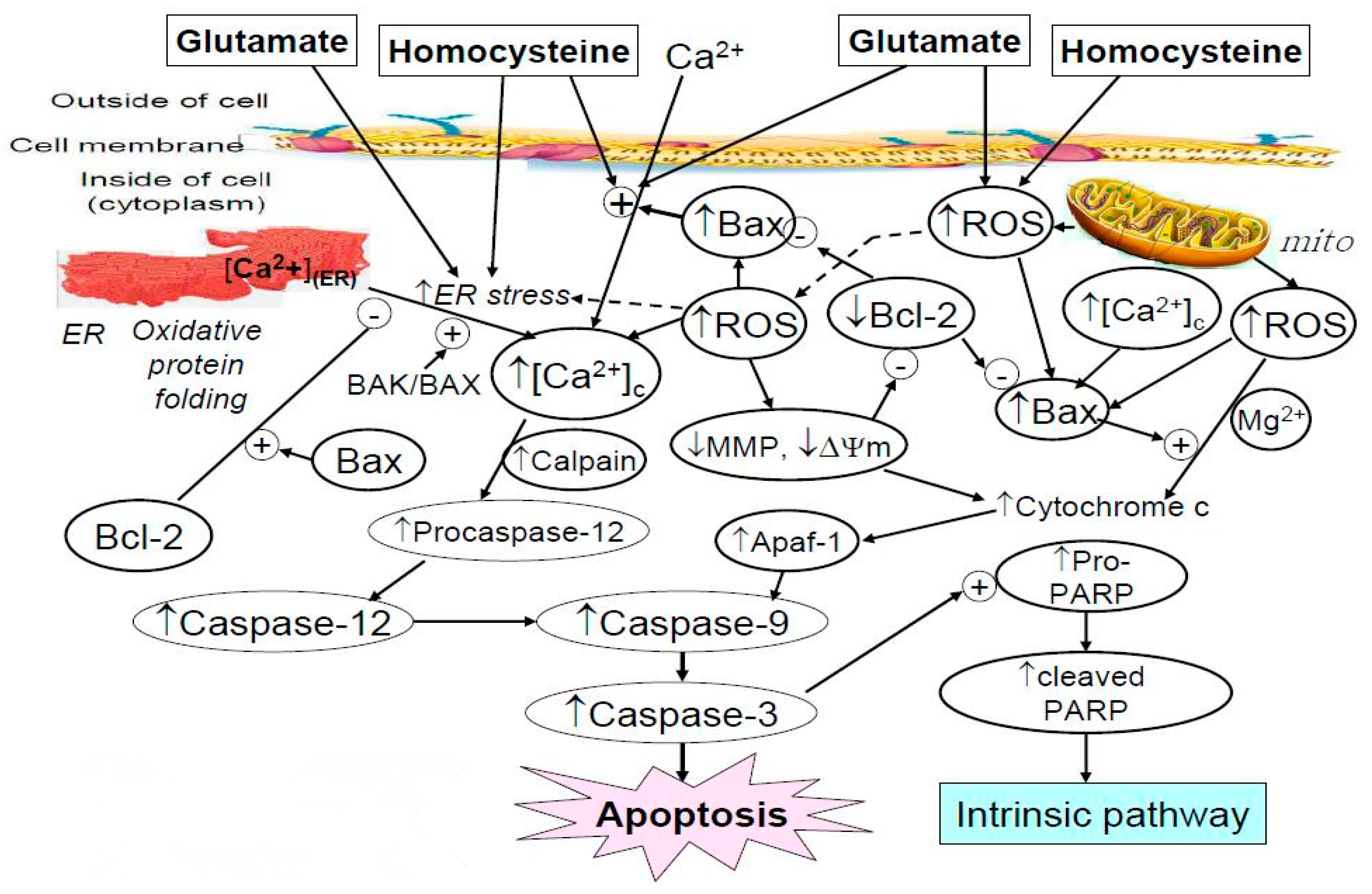
2.4. Effect of Astaxanthin on the Intracellular Calcium Ion Level Affected by Homocysteine and Glutamate
2.5. Effect of Astaxanthin on the ROS Production Caused by Homocysteine and Glutamate
2.6. Effect of Astaxanthin on the MDA Production Resulting from Insult of Homocysteine and Glutamate
2.7. Western Blot Analysis Indicated Astaxanthin Restored Bax and Bcl-2 Homeostasis
2.8. Effect of Astaxanthin on the Expression of Caspase-12 and Caspase-3 in PC12 Cells Treated with Homocysteine and Glutamate
3. Materials and Methods
3.1. Chemicals
3.2. Source of Cell Line
3.3. Cultivation of PC12 Cell Line
3.4. Preparation of Complete Medium
3.5. Preparation of Differentiating Medium
3.6. Preparation of Astaxanthin Solution
3.7. Preparation of Homocysteine and Glutamate Solutions
3.8. MTT Assay Cell Viability Test
3.9. The Cytotoxicity of Astaxanthin
3.10. Protective Effect of Astaxanthin against the Cytotoxicity of Homocysteine, Glutamate, or Homocysteine Plus Glutamate
3.11. Determination of Intracellular Calcium Ion Concentration
3.12. Determination of Intracellular Reactive Oxygen Species (ROS)
3.13. Protein Extraction
3.14. Assay for the Malondialdehyde (MDA) Concentration
3.15. Western Blot Analysis
3.16. Statistical Analysis
 ,〒 p < 0.05; **,⊗⊗,##,,〒〒 p < 0.01; *** ,⊗⊗⊗,###,,〒〒〒 p < 0.005.
,〒 p < 0.05; **,⊗⊗,##,,〒〒 p < 0.01; *** ,⊗⊗⊗,###,,〒〒〒 p < 0.005. 4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Meldrum, B.S. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J. Nutr. 2000, 130 (Suppl. S4), 1007S–1015S. [Google Scholar] [CrossRef]
- Boldyrev, A.A.; Johnson, P. Homocysteine and its derivatives as possible modulators of neuronal and non-neuronal cell glutamate receptors in Alzheimer’s disease. J. Alzheimers Dis. 2007, 11, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 1969, 164, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; An, S.; Hu, W.; Teng, M.; Wang, X.; Qu, Y.; Liu, Y.; Yuan, Y.; Wang, D. The Neuroprotective Properties of Hericium erinaceus in Glutamate-Damaged Differentiated PC12 Cells and an Alzheimer’s Disease Mouse Model. Int. J. Mol. Sci. 2016, 17, 1810. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [PubMed]
- Cheriyan, J.; Balsara, R.D.; Hansen, K.B.; Castellino, F.J. Pharmacology of triheteromeric N-Methyl-D-Aspartate Receptors. Neurosci. Lett. 2016, 617, 240–246. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Li, F.; Maiese, K. Stress in the brain: Novel cellular mechanisms of injury linked to Alzheimer’s disease. Brain Res. Brain Res. Rev. 2005, 49, 1–21. [Google Scholar] [CrossRef]
- Ho, P.I.; Ortiz, D.; Rogers, E.; Shea, T.B. Multiple aspects of homocysteine neurotoxicity: Glutamate excitotoxicity, kinase hyperactivation and DNA damage. J. Neurosci. Res. 2002, 70, 694–702. [Google Scholar] [CrossRef]
- Herrmann, W.; Lorenzl, S.; Obeid, R. [Review of the role of hyperhomocysteinemia and B-vitamin deficiency in neurological and psychiatric disorders--current evidence and preliminary recommendations]. Fortschr. Neurol. Psychiatr. 2007, 75, 515–527. [Google Scholar] [CrossRef]
- Lipton, S.A.; Rosenberg, P.A. Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.A.; Tischler, A.S. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar] [CrossRef] [PubMed]
- Westerink, R.H.; Ewing, A.G. The PC12 cell as model for neurosecretion. Acta Physiol. (Oxf.) 2008, 192, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Chen, H.X.; Yu, G.; Peng, C.C.; Peng, R.Y. Curcumin-Protected PC12 Cells Against Glutamate-Induced Oxidative Toxicity. Food Technol. Biotechnol. 2014, 52, 468–478. [Google Scholar] [CrossRef]
- Wang, D.; Hu, S.; Zhang, J.; Li, Q.; Liu, X.; Li, Y. Investigation of the neuroprotective effects of a novel synthetic compound via the mitochondrial pathway. Mol. Med. Rep. 2017, 16, 1133–1138. [Google Scholar] [CrossRef][Green Version]
- Gleichmann, M.; Mattson, M.P. Neuronal calcium homeostasis and dysregulation. Antioxid. Redox Signal. 2011, 14, 1261–1273. [Google Scholar] [CrossRef]
- Mattson, M.P.; Chan, S.L. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium 2003, 34, 385–397. [Google Scholar] [CrossRef]
- Obeid, R.; Herrmann, W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006, 580, 2994–3005. [Google Scholar] [CrossRef]
- Naguib, Y.M. Antioxidant activities of astaxanthin and related carotenoids. J. Agric. Food Chem. 2000, 48, 1150–1154. [Google Scholar] [CrossRef]
- Wu, H.; Niu, H.; Shao, A.; Wu, C.; Dixon, B.J.; Zhang, J.; Yang, S.; Wang, Y. Astaxanthin as a Potential Neuroprotective Agent for Neurological Diseases. Mar. Drugs 2015, 13, 5750–5766. [Google Scholar] [CrossRef]
- Ye, Q.; Zhang, X.; Huang, B.; Zhu, Y.; Chen, X. Astaxanthin suppresses MPP(+)-induced oxidative damage in PC12 cells through a Sp1/NR1 signaling pathway. Mar. Drugs 2013, 11, 1019–1034. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Zou, W.; Liang, X.Y.; Jiang, Z.S.; Li, X.; Wei, H.J.; Tang, Y.Y.; Zhang, P.; Tang, X.Q. Hydrogen sulfide prevents homocysteineinduced endoplasmic reticulum stress in PC12 cells by upregulating SIRT1. Mol. Med. Rep. 2017, 16, 3587–3593. [Google Scholar] [CrossRef] [PubMed]
- Olatunji, O.J.; Feng, Y.; Olatunji, O.O.; Tang, J.; Ouyang, Z.; Su, Z.; Wang, D.; Yu, X. Neuroprotective effects of adenosine isolated from Cordyceps cicadae against oxidative and ER stress damages induced by glutamate in PC12 cells. Environ. Toxicol. Pharmacol. 2016, 44, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhao, Y.; Li, S. Astaxanthin attenuates glutamate-induced apoptosis via inhibition of calcium influx and endoplasmic reticulum stress. Eur. J. Pharmacol. 2017, 806, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Broch, O.J.; Ueland, P.M. Regional distribution of homocysteine in the mammalian brain. J. Neurochem. 1984, 43, 1755–1757. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, A.; Brattstrom, L.; Norrving, B.; Hultberg, B.; Andersson, A.; Johansson, B.B. Plasma homocysteine in the acute and convalescent phases after stroke. Stroke 1995, 26, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Perry, I.J.; Refsum, H.; Morris, R.W.; Ebrahim, S.B.; Ueland, P.M.; Shaper, A.G. Prospective study of serum total homocysteine concentration and risk of stroke in middle-aged British men. Lancet 1995, 346, 1395–1398. [Google Scholar] [CrossRef]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate—Induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef]
- Froissard, P.; Duval, D. Cytotoxic effects of glutamic acid on PC12 cells. Neurochem. Int. 1994, 24, 485–493. [Google Scholar] [CrossRef]
- Wang, K.; Zhu, X.; Zhang, K.; Wu, Z.; Sun, S.; Zhou, F.; Zhu, L. Neuroprotective Effect of Puerarin on Glutamate-Induced Cytotoxicity in Differentiated Y-79 Cells via Inhibition of ROS Generation and Ca(2+) Influx. Int. J. Mol. Sci. 2016, 17, 1109. [Google Scholar] [CrossRef]
- Wang, D.; Tan, Q.R.; Zhang, Z.J. Neuroprotective effects of paeoniflorin, but not the isomer albiflorin, are associated with the suppression of intracellular calcium and calcium/calmodulin protein kinase II in PC12 cells. J. Mol. Neurosci. 2013, 51, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cheong, Y.K.; Wang, H.; Ren, G.; Yang, Z. Neuroprotective Effects of Etidronate and 2,3,3-Trisphosphonate Against Glutamate-Induced Toxicity in PC12 Cells. Neurochem. Res. 2016, 41, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Wang, D.; Zhang, J.; Du, M.; Cheng, Y.; Liu, Y.; Zhang, N.; Wang, D.; Wu, Y. Mitochondria Related Pathway Is Essential for Polysaccharides Purified from Sparassis crispa Mediated Neuro-Protection against Glutamate-Induced Toxicity in Differentiated PC12 Cells. Int. J. Mol. Sci. 2016, 17, 133. [Google Scholar] [CrossRef] [PubMed]
- Shea, T.B.; Lyons-Weiler, J.; Rogers, E. Homocysteine, folate deprivation and Alzheimer neuropathology. J. Alzheimers Dis. 2002, 4, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, C.L.; Chi, C.L.; Awobuluyi, M.; Sucher, N.J. Expression of N-methyl-D-aspartate receptor subunit mRNAs in the rat pheochromocytoma cell line PC12. Neurosci. Lett. 1995, 201, 103–106. [Google Scholar] [CrossRef]
- Sibarov, D.A.; Abushik, P.A.; Giniatullin, R.; Antonov, S.M. GluN2A Subunit-Containing NMDA Receptors Are the Preferential Neuronal Targets of Homocysteine. Front. Cell Neurosci. 2016, 10, 246. [Google Scholar] [CrossRef]
- Wang, X.J.; Chen, W.; Fu, X.T.; Ma, J.K.; Wang, M.H.; Hou, Y.J.; Tian, D.C.; Fu, X.Y.; Fan, C.D. Correction to: Reversal of homocysteine-induced neurotoxicity in rat hippocampal neurons by astaxanthin: Evidences for mitochondrial dysfunction and signaling crosstalk. Cell Death Discov. 2019, 5, 70. [Google Scholar] [CrossRef]
- Fan, C.D.; Sun, J.Y.; Fu, X.T.; Hou, Y.J.; Li, Y.; Yang, M.F.; Fu, X.Y.; Sun, B.L. Astaxanthin Attenuates Homocysteine-Induced Cardiotoxicity in Vitro and in Vivo by Inhibiting Mitochondrial Dysfunction and Oxidative Damage. Front. Physiol. 2017, 8, 1041. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W.; Hao, C.; Mao, X.; Zhang, L. Astaxanthin protects PC12 cells from glutamate-induced neurotoxicity through multiple signaling pathways. J. Funct. Foods 2015, 16, 137–151. [Google Scholar] [CrossRef]
- Qi, H.; Shuai, J. Alzheimer’s disease via enhanced calcium signaling caused by the decrease of endoplasmic reticulum-mitochondrial distance. Med. Hypotheses 2016, 89, 28–31. [Google Scholar] [CrossRef]
- Robert, K.; Pages, C.; Ledru, A.; Delabar, J.; Caboche, J.; Janel, N. Regulation of extracellular signal-regulated kinase by homocysteine in hippocampus. Neuroscience 2005, 133, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Magi, S.; Castaldo, P.; Macri, M.L.; Maiolino, M.; Matteucci, A.; Bastioli, G.; Gratteri, S.; Amoroso, S.; Lariccia, V. Intracellular Calcium Dysregulation: Implications for Alzheimer’s Disease. Biomed. Res. Int. 2016, 2016, 6701324. [Google Scholar] [CrossRef] [PubMed]
- Pierce, G.N. Advanced Bioactive Compounds Countering the Effects of Radiological, Chemical and Biological Agents: Strategies to Counter Biological Damage; Springer: New York, NY, USA, 2013. [Google Scholar]
- Kidd, P. Astaxanthin, cell membrane nutrient with diverse clinical benefits and anti-aging potential. Altern. Med. Rev. 2011, 16, 355–364. [Google Scholar] [PubMed]
- Chang, C.H.; Chen, C.Y.; Chiou, J.Y.; Peng, R.Y.; Peng, C.H. Astaxanthine secured apoptotic death of PC12 cells induced by beta-amyloid peptide 25–35: Its molecular action targets. J. Med. Food 2010, 13, 548–556. [Google Scholar] [CrossRef]
- Shen, H.; Kuo, C.C.; Chou, J.; Delvolve, A.; Jackson, S.N.; Post, J.; Woods, A.S.; Hoffer, B.J.; Wang, Y.; Harvey, B.K. Astaxanthin reduces ischemic brain injury in adult rats. FASEB J. 2009, 23, 1958–1968. [Google Scholar] [CrossRef]
- Yamagishi, R.; Aihara, M. Neuroprotective effect of astaxanthin against rat retinal ganglion cell death under various stresses that induce apoptosis and necrosis. Mol. Vis. 2014, 20, 1796–1805. [Google Scholar]
- Galasso, C.; Orefice, I.; Pellone, P.; Cirino, P.; Miele, R.; Ianora, A.; Brunet, C.; Sansone, C. On the Neuroprotective Role of Astaxanthin: New Perspectives? Mar. Drugs 2018, 16, 247. [Google Scholar] [CrossRef]
- Eskes, R.; Antonsson, B.; Osen-Sand, A.; Montessuit, S.; Richter, C.; Sadoul, R.; Mazzei, G.; Nichols, A.; Martinou, J.C. Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J. Cell Biol. 1998, 143, 217–224. [Google Scholar] [CrossRef]
- Gomez-Crisostomo, N.P.; Lopez-Marure, R.; Zapata, E.; Zazueta, C.; Martinez-Abundis, E. Bax induces cytochrome c release by multiple mechanisms in mitochondria from MCF7 cells. J. Bioenerg. Biomembr. 2013, 45, 441–448. [Google Scholar] [CrossRef]
- Jiang, J.M.; Wang, L.; Gu, H.F.; Wu, K.; Xiao, F.; Chen, Y.; Guo, R.M.; Tang, X.Q. Arecoline Induces Neurotoxicity to PC12 Cells: Involvement in ER Stress and Disturbance of Endogenous H2S Generation. Neurochem. Res. 2016, 41, 2140–2148. [Google Scholar] [CrossRef]
- Garcia de la Cadena, S.; Massieu, L. Caspases and their role in inflammation and ischemic neuronal death. Focus on caspase-12. Apoptosis 2016, 21, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, Z.; Nowicki, M.J. Caspase-12 mediates carbon tetrachloride-induced hepatocyte apoptosis in mice. World J. Gastroenterol. 2014, 20, 18189–18198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, J.; Chen, S.; Liu, J.; Liu, L.; Liu, G.; Wang, F.; Jiang, W.; Zhang, C.; Wang, S.; et al. Caspase-12 is involved in stretch-induced apoptosis mediated endoplasmic reticulum stress. Apoptosis 2016, 21, 432–442. [Google Scholar] [CrossRef]
- Matsui, A.; Kaneko, H.; Kachi, S.; Ye, F.; Hwang, S.J.; Takayama, K.; Nagasaka, Y.; Sugita, T.; Terasaki, H. Expression of Vascular Endothelial Growth Factor by Retinal Pigment Epithelial Cells Induced by Amyloid-beta Is Depressed by an Endoplasmic Reticulum Stress Inhibitor. Ophthalmic Res. 2015, 55, 37–44. [Google Scholar] [CrossRef]
- Cultured for Neuronal PC12 Cells: A Model Function, Differentiation, and Survival. p. Available online: https://biocyclopedia.com/index/cell_biology_methods/cultured_for_neuronal_pc12_cells.php (accessed on 5 December 2019).
- Bolin, A.P.; Macedo, R.C.; Marin, D.P.; Barros, M.P.; Otton, R. Astaxanthin prevents in vitro auto-oxidative injury in human lymphocytes. Cell Biol. Toxicol. 2010, 26, 457–467. [Google Scholar] [CrossRef]
- Zhou, W.; Chai, H.; Lin, P.H.; Lumsden, A.B.; Yao, Q.; Chen, C. Ginsenoside Rb1 blocks homocysteine-induced endothelial dysfunction in porcine coronary arteries. J. Vasc. Surg. 2005, 41, 861–868. [Google Scholar] [CrossRef]
- Kawakami, Z.; Kanno, H.; Ueki, T.; Terawaki, K.; Tabuchi, M.; Ikarashi, Y.; Kase, Y. Neuroprotective effects of yokukansan, a traditional Japanese medicine, on glutamate-mediated excitotoxicity in cultured cells. Neuroscience 2009, 159, 1397–1407. [Google Scholar] [CrossRef]
- Sul, D.; Kim, H.S.; Lee, D.; Joo, S.S.; Hwang, K.W.; Park, S.Y. Protective effect of caffeic acid against beta-amyloid-induced neurotoxicity by the inhibition of calcium influx and tau phosphorylation. Life Sci. 2009, 84, 257–262. [Google Scholar] [CrossRef]
- Zhang, Y.; Peng, F.; Gao, B.; Ingram, A.J.; Krepinsky, J.C. High glucose-induced RhoA activation requires caveolae and PKCbeta1-mediated ROS generation. Am. J. Physiol. Renal. Physiol. 2012, 302, F159–F172. [Google Scholar] [CrossRef][Green Version]
- Chang, C.H.; Chen, Y.; Yew, X.X.; Chen, H.X.; Kim, J.X.; Chang, C.C.; Peng, C.C.; Peng, R.Y. Improvement of erinacine A productivity in Hericium erinaceus mycelia and its neuroprotective bioactivity against the glutamateinsulted apoptosis. LWT Food Sci. Technol. 2016, 65, 1100–1108. [Google Scholar] [CrossRef]
- Chang, X.; Wang, J.; Yang, S.; Chen, S.; Song, Y. Antioxidative, antibrowning and antibacterial activities of sixteen floral honeys. Food Funct. 2011, 2, 541–546. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not Available. |
 : vs. 24 h at the same dose; ⊗: vs. Glu 20 mM at the same time; #: vs. Hcy 10 mM + Glu 20 mM at the same time. The significance of the difference was judged by confidence levels of * p < 0.05; # p < 0.05; 〒 p < 0.05; ⊗ p < 0.05; p < 0.05; ** p < 0.01; ## p < 0.01; ⊗⊗ p < 0.01; p < 0.01; *** p < 0.005.
: vs. 24 h at the same dose; ⊗: vs. Glu 20 mM at the same time; #: vs. Hcy 10 mM + Glu 20 mM at the same time. The significance of the difference was judged by confidence levels of * p < 0.05; # p < 0.05; 〒 p < 0.05; ⊗ p < 0.05; p < 0.05; ** p < 0.01; ## p < 0.01; ⊗⊗ p < 0.01; p < 0.01; *** p < 0.005.
: vs. 24 h at the same dose; ⊗: vs. Glu 20 mM at the same time; #: vs. Hcy 10 mM + Glu 20 mM at the same time. The significance of the difference was judged by confidence levels of * p < 0.05; # p < 0.05; 〒 p < 0.05; ⊗ p < 0.05; p < 0.05; ** p < 0.01; ## p < 0.01; ⊗⊗ p < 0.01; p < 0.01; *** p < 0.005.
: vs. 24 h at the same dose; ⊗: vs. Glu 20 mM at the same time; #: vs. Hcy 10 mM + Glu 20 mM at the same time. The significance of the difference was judged by confidence levels of * p < 0.05; # p < 0.05; 〒 p < 0.05; ⊗ p < 0.05; p < 0.05; ** p < 0.01; ## p < 0.01; ⊗⊗ p < 0.01; p < 0.01; *** p < 0.005.





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-H.; Chen, K.-C.; Liaw, K.-C.; Peng, C.-C.; Peng, R.Y. Astaxanthin Protects PC12 Cells against Homocysteine- and Glutamate-Induced Neurotoxicity. Molecules 2020, 25, 214. https://doi.org/10.3390/molecules25010214
Chang C-H, Chen K-C, Liaw K-C, Peng C-C, Peng RY. Astaxanthin Protects PC12 Cells against Homocysteine- and Glutamate-Induced Neurotoxicity. Molecules. 2020; 25(1):214. https://doi.org/10.3390/molecules25010214
Chicago/Turabian StyleChang, Chi-Huang, Kuan-Chou Chen, Kuo-Chun Liaw, Chiung-Chi Peng, and Robert Y. Peng. 2020. "Astaxanthin Protects PC12 Cells against Homocysteine- and Glutamate-Induced Neurotoxicity" Molecules 25, no. 1: 214. https://doi.org/10.3390/molecules25010214
APA StyleChang, C.-H., Chen, K.-C., Liaw, K.-C., Peng, C.-C., & Peng, R. Y. (2020). Astaxanthin Protects PC12 Cells against Homocysteine- and Glutamate-Induced Neurotoxicity. Molecules, 25(1), 214. https://doi.org/10.3390/molecules25010214