Metabolome and Transcriptome Analysis Reveals Putative Genes Involved in Anthocyanin Accumulation and Coloration in White and Pink Tea (Camellia sinensis) Flower

 , , ,
, , ,
Abstract
1. Introduction
2. Results
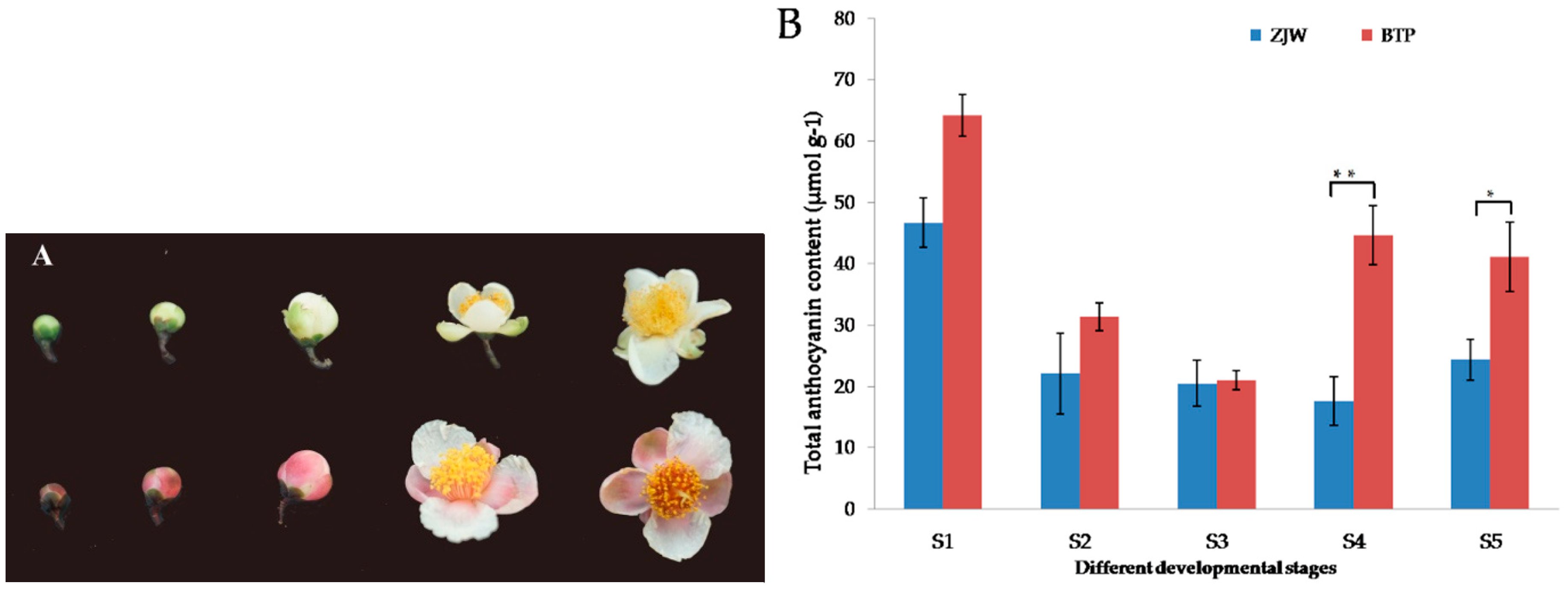
2.1. Anthocyanin Content in White and Pink Tea Flower
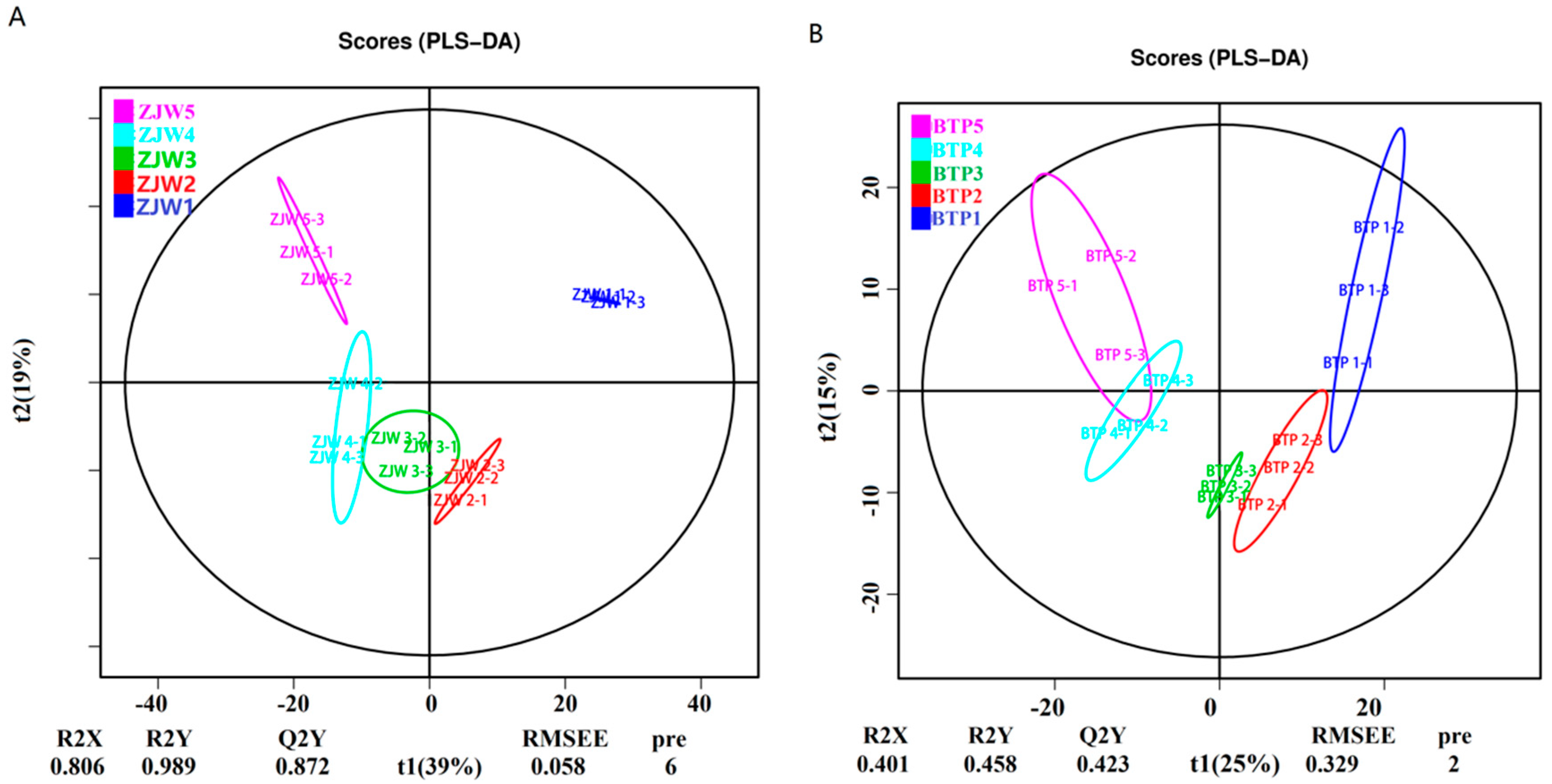
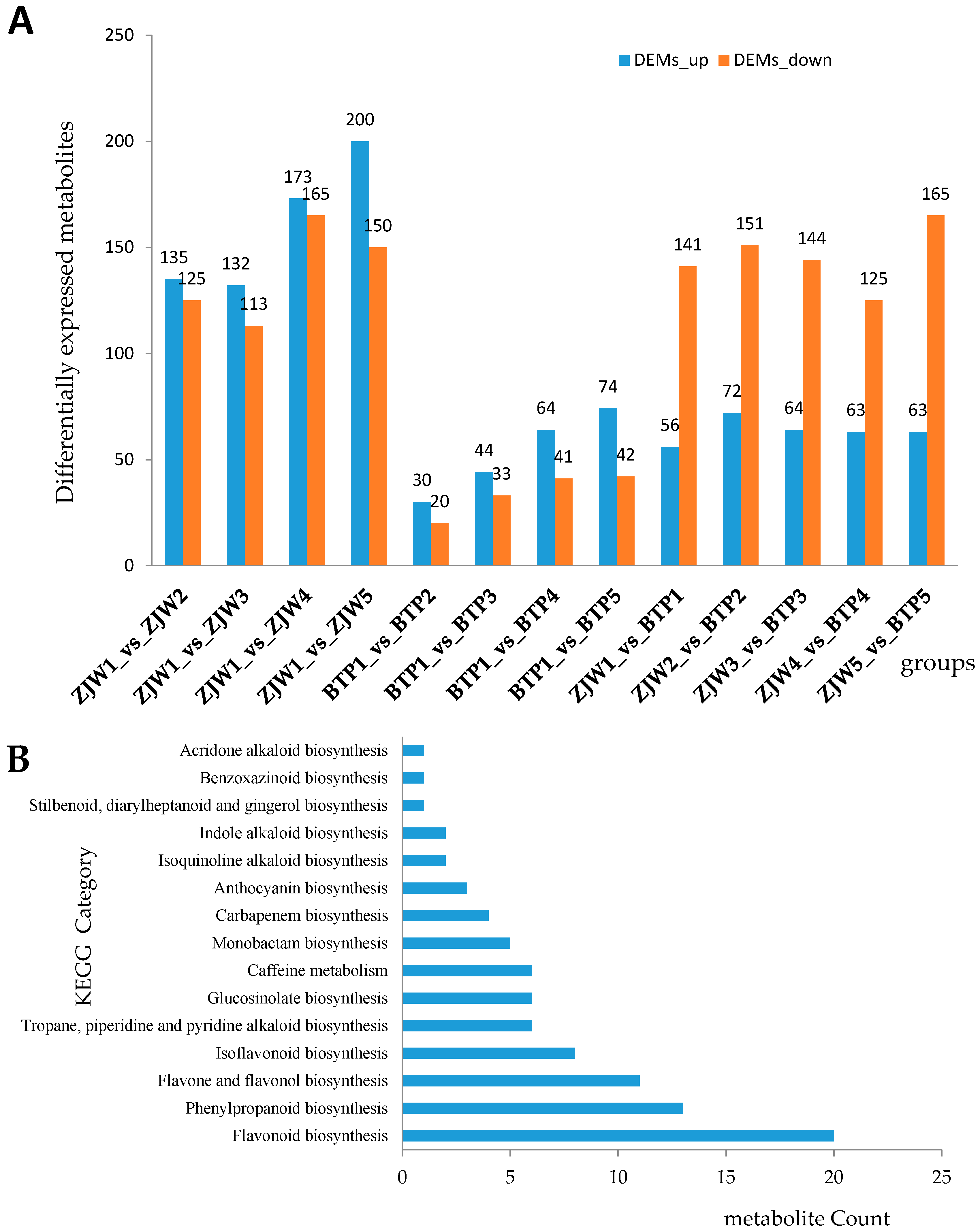
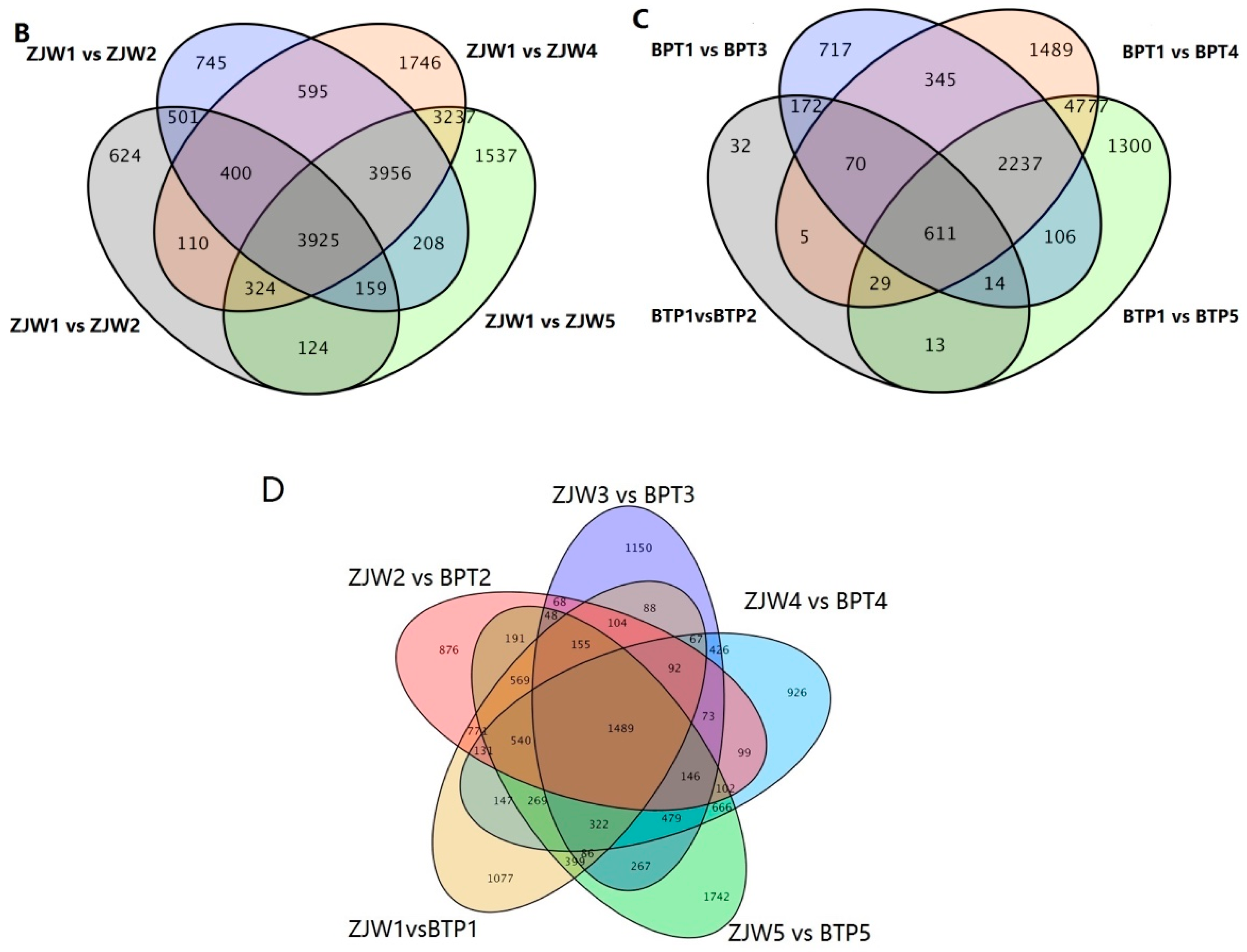
2.2. Metabolomics Analysis of Tea Flower Development Stages
2.3. RNA Sequencing and Sample Transcriptome Mapping
2.4. Differentially Expressed Genes (DEGs) in BTP and ZJW Flower Development
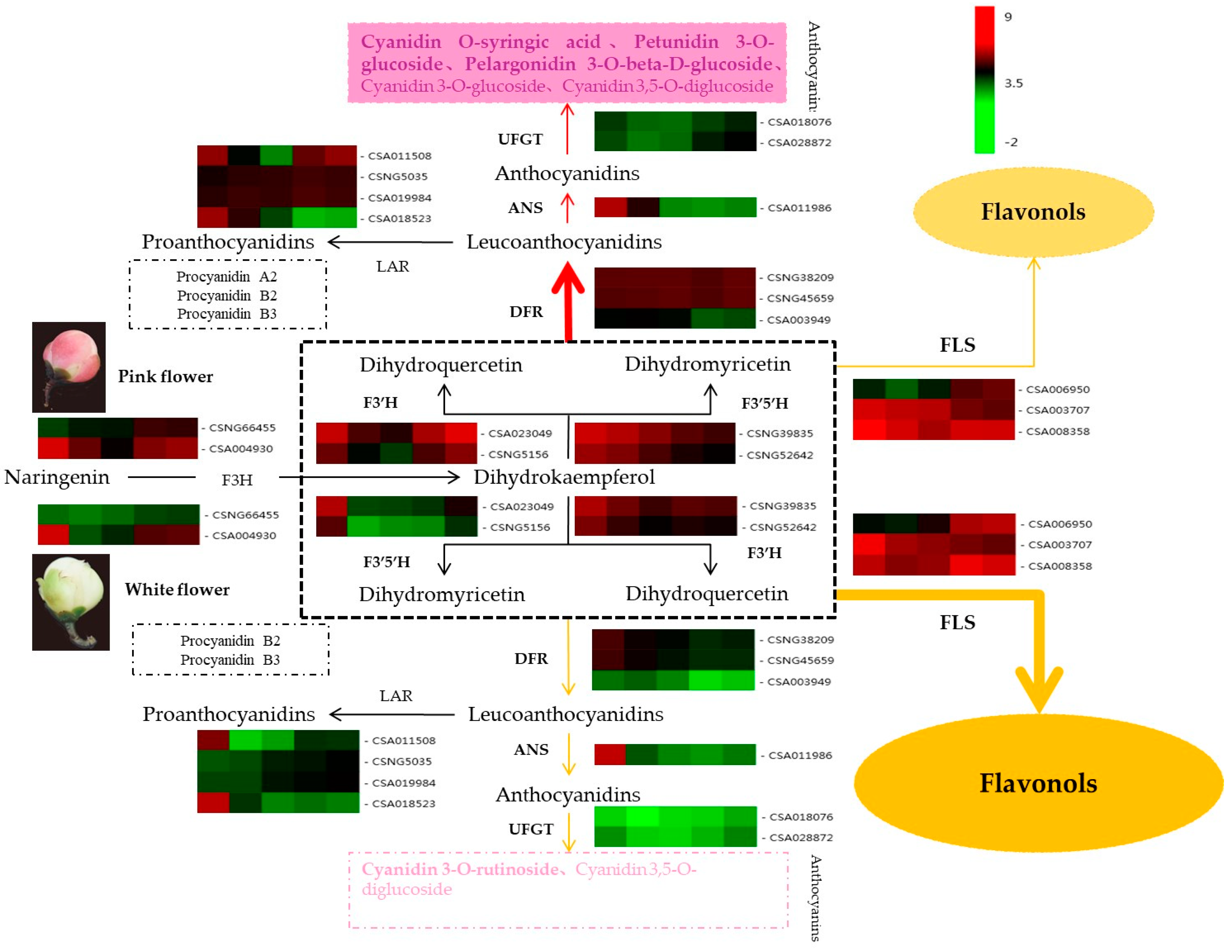
2.5. Co-Expression Analysis of Genes Related to the Flavonoid Biosynthesis Pathway
2.6. Verification of Anthocyanin Biosynthesis-Related Genes
3. Discussion
3.1. Effect of Content and Types of Anthocyanins on Coloration in White and Pink Tea Flowers
3.2. Key Genes in the Anthocyanin Pathway Affect Coloration in White and Pink Tea Flowers
3.3. Hub Genes Involved In Flavonoid Accumulation during Flower Development
4. Materials and Methods
4.1. Plant Materials
4.2. Extraction and Separation of Secondary Metabolites
4.2.1. Total Anthocyanin Measurements
4.2.2. Sample Extraction
4.3. RNA Extraction and Transcriptome Sequencing
4.4. Gene Functional Annotation and Expression Level Analysis
4.5. Identification of Hub Genes Using Weighted Gene Co-Expression Network Analysis
4.6. Quantitative Real Time PCR and Expression Validation
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Accessibility
References
- Rothenberg, D.O.; Zhou, C.B.; Zhang, L.Y. A Review on the Weight-Loss Effects of Oxidized Tea Polyphenols. Molecules 2018, 23, 1176. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, D.O.; Zhang, L.Y. Mechanisms Underlying the Anti-Depressive Effects of Regular Tea Consumption. Nutrients 2019, 11, 1361. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Zhang, Y.; Wu, L.; Li, H.; Ruan, L.; Bai, P.; Zhang, C.; Zhang, F.; Xu, L.; Wang, L.; et al. Gene expression analysis of bud and leaf color in tea. Plant. Physiol. Biochem. 2016, 107, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhu, Z.; Cao, P.; Chen, H.; Chen, C.; Zhou, X.; Mao, Y.; Lei, J.; Jiang, Y.; Meng, W.; et al. Purple foliage coloration in tea (Camellia sinensis L.) arises from activation of the R2R3-MYB transcription factor CsAN1. Sci. Rep. 2016, 6, 32534. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.X.; Pan, D.Z.; Liang, M.; Abubakar, Y.S.; Li, J.; Lin, J.K.; Chen, S.P.; Chen, W. Regulation of Anthocyanin Biosynthesis in Purple Leaves of Zijuan Tea (Camellia sinensis var. kitamura). Int. J. Mol. Sci. 2017, 18, 833. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Sun, W.; Lai, Z. Differential expression of genes in purple-shoot tea tender leaves and mature leaves during leaf growth. J. Sci. Food Agric. 2016, 96, 1982–1989. [Google Scholar] [CrossRef]
- Ko, H.J.; Lo, C.Y.; Wang, B.J.; Chiou, R.Y.Y.; Lin, S.M. Theaflavin-3,3‘-digallate, a black tea polyphenol, attenuates adipocyte-activated inflammatory response of macrophage associated with the switch of M1/M2-like phenotype. J. Funct. Foods 2014, 11, 36–48. [Google Scholar] [CrossRef]
- Yang, Z.; Xu, Y.; Jie, G.; He, P.; Tu, Y. Study on the antioxidant activity of tea flowers (Camellia sinensis). Asia Pac. J. Clin. Nutr. 2007, 16, 148–152. [Google Scholar]
- Zhao, D.; Tao, J. Recent advances on the development and regulation of flower color in ornamental plants. Front. Plant. Sci. 2015, 6, 261. [Google Scholar] [CrossRef]
- Wang, L.S.; Shiraishi, A.; Hashimoto, F.; Aoki, N.; Sakata, Y. Analysis of Petal Anthocyanins to Investigate Flower Coloration of Zhongyuan (Chinese) and Daikon Island (Japanese) Tree Peony Cultivars. J. Plant. Res. 2001, 114, 33–43. [Google Scholar] [CrossRef]
- Ma, K.F.; Zhang, Q.X.; Cheng, T.R.; Yan, X.L.; Pan, H.T.; Wang, J. Substantial Epigenetic Variation Causing Flower Color Chimerism in the Ornamental Tree Prunus mume Revealed by Single Base Resolution Methylome Detection and Transcriptome Sequencing. Int. J. Mol. Sci. 2018, 19, 2315. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.H.; Yang, J.X.; Xiao, C.H.; Mao, T.Y.; Zhang, J.; Zhang, H.Y. Differences in flavonoid pathway metabolites and transcripts affect yellow petal colouration in the aquatic plant Nelumbo nucifera. Bmc Plant. Biol. 2019, 19, 277. [Google Scholar] [CrossRef] [PubMed]
- Camara, B. Carotenoids Handbook; Britton, G., Liaaen-Jensen, S., Pfander, H., Eds.; Birkhäuser Basel: Basel, Switzerland, 2003; Volume 167. [Google Scholar]
- Zhou, X.; Li, J.; Zhu, Y.; Ni, S.; Chen, J.; Feng, X.; Zhang, Y.; Li, S.; Zhu, H.; Wen, Y. De novo Assembly of the Camellia nitidissima Transcriptome Reveals Key Genes of Flower Pigment Biosynthesis. Front. Plant. Sci. 2017, 8, 1545. [Google Scholar] [CrossRef] [PubMed]
- Li, J.B.; Hashimoto, F.; Shimizu, K.; Sakata, Y. Chemical taxonomy of red-flowered wild Camellia species based on floral anthocyanins. Phytochemistry 2013, 85, 99–106. [Google Scholar] [CrossRef]
- Terahara, N.; Takeda, Y.; Nesumi, A.; Honda, T. Anthocyanins from red flower tea (Benibana-cha), Camellia sinensis. Phytochemistry 2001, 56, 359–361. [Google Scholar] [CrossRef]
- Chen, S.M.; Li, C.H.; Zhu, X.R.; Deng, Y.M.; Sun, W.; Wang, L.S.; Chen, F.-D.; Zhang, Z. The identification of flavonoids and the expression of genes of anthocyanin biosynthesis in the chrysanthemum flowers. Biol. Plant. 2012, 56, 458–464. [Google Scholar] [CrossRef]
- Lou, Q.; Liu, Y.; Qi, Y.; Jiao, S.; Tian, F.; Jiang, L.; Wang, Y. Transcriptome sequencing and metabolite analysis reveals the role of delphinidin metabolism in flower colour in grape hyacinth. J. Exp. Bot. 2014, 65, 3157–3164. [Google Scholar] [CrossRef]
- Joshi, R.; Poonam; Gulati, A. Biochemical attributes of tea flowers (Camellia sinensis) at different developmental stages in the Kangra region of India. Sci. Hortic. 2011, 130, 266–274. [Google Scholar] [CrossRef]
- Shen, J.; Zou, Z.; Zhang, X.; Zhou, L.; Wang, Y.; Fang, W.; Zhu, X. Metabolic analyses reveal different mechanisms of leaf color change in two purple-leaf tea plant (Camellia sinensis L.) cultivars. Hortic. Res. 2018, 5, 7. [Google Scholar] [CrossRef]
- Zheng, C.; Ma, J.Q.; Chen, J.D.; Ma, C.L.; Chen, W.; Yao, M.Z.; Chen, L. Gene Coexpression Networks Reveal Key Drivers of Flavonoid Variation in Eleven Tea Cultivars (Camellia sinensis). J. Agric Food Chem 2019, 67, 9967–9978. [Google Scholar] [CrossRef]
- Saito, T.; Honma, D.; Tagashira, M.; Kanda, T.; Maeda-Yamamoto, M. Anthocyanins from New Red Leaf Tea ‘Sunrouge’. J. Agric. Food Chem. 2011, 59, 4779–4782. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape: Software for visualization and analysis of biological networks. Methods Mol. Biol. 2011, 696, 291–303. [Google Scholar] [PubMed]
- Tilbrook, K.; Arongaus, A.B.; Binkert, M.; Heijde, M.; Yin, R.; Ulm, R. The UVR8 UV-B Photoreceptor: Perception, Signaling and Response. Arab. Book 2013, 11, e0164. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, L.Y.; Wang, S.L.; Niu, X.Y. Analysis of anthocyanins and flavonols in petals of 10 Rhododendron species from the Sygera Mountains in Southeast Tibet. Plant. Physiol. Biochem. 2016, 104, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Wang, Z.G.; Zhang, W.; Li, Y.X.; Wang, J.; Lei, J.J. Flower pigment inheritance and anthocyanin characterization of hybrids from pink-flowered and white-flowered strawberry. Sci. Hortic. 2016, 200, 143–150. [Google Scholar] [CrossRef]
- Li, J.B.; Hashimoto, F.; Shimizu, K.; Sakata, Y. Anthocyanins from red flowers of Camellia cultivar ‘Dalicha’. Phytochemistry 2008, 69, 3166–3171. [Google Scholar] [CrossRef]
- Li, J.B.; Hashimoto, F.; Shimizu, K.; Sakata, Y. A new acylated anthocyanin from the red flowers of Camellia hongkongensis and characterization of anthocyanins in the section Camellia species. J. Integr. Plant. Biol. 2009, 51, 545–552. [Google Scholar] [CrossRef]
- Tanaka, Y.; Brugliera, F.; Chandler, S. Recent progress of flower colour modification by biotechnology. Int. J. Mol. Sci. 2009, 10, 5350–5369. [Google Scholar] [CrossRef]
- Zhu, M.; Zheng, X.; Shu, Q.; Li, H.; Zhong, P.; Zhang, H.; Xu, Y.; Wang, L.; Wang, L. Relationship between the composition of flavonoids and flower colors variation in tropical water lily (Nymphaea) cultivars. PloS ONE 2012, 7, e34335. [Google Scholar] [CrossRef]
- Khoo, H.E.; Azlan, A.; Tang, S.T.; Lim, S.M. Anthocyanidins and anthocyanins: Colored pigments as food, pharmaceutical ingredients, and the potential health benefits. Food Nutr. Res. 2017, 61, 1361779. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Shen, Y.; Wang, M.; Huang, M.; Yang, R.; Zhu, S.; Wang, L.; Xu, Y.; Wu, R. Natural Variation in Petal Color in Lycoris longituba Revealed by Anthocyanin Components. PloS ONE 2011, 6, e22098. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.W.; Berardi, A.E.; Smith, S.D.; Litt, A. Related allopolyploids display distinct floral pigment profiles and transgressive pigments. Am. J. Bot. 2017, 104, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Gou, J.; Jia, Z.; Yang, L.; Sun, Y.; Xiao, X.; Song, F.; Luo, K. Molecular cloning and characterization of two genes encoding dihydroflavonol-4-reductase from Populus trichocarpa. PloS ONE 2012, 7, e30364. [Google Scholar] [CrossRef]
- Martens, S.; Preuss, A.; Matern, U. Multifunctional flavonoid dioxygenases: Flavonol and anthocyanin biosynthesis in Arabidopsis thaliana L. Phytochemistry 2010, 71, 1040–1049. [Google Scholar] [CrossRef]
- Luo, P.; Ning, G.; Wang, Z.; Shen, Y.; Jin, H.; Li, P.; Huang, S.; Zhao, J.; Bao, M. Disequilibrium of Flavonol Synthase and Dihydroflavonol-4-Reductase Expression Associated Tightly to White vs. Red Color Flower Formation in Plants. Front. Plant. Sci. 2015, 6, 1257. [Google Scholar] [CrossRef]
- Han, Y.; Vimolmangkang, S.; Soria-Guerra, R.E.; Korban, S.S. Introduction of apple ANR genes into tobacco inhibits expression of both CHI and DFR genes in flowers, leading to loss of anthocyanin. J. Exp. Bot. 2012, 63, 2437–2447. [Google Scholar] [CrossRef]
- Huang, H.; Xia, E.H.; Zhang, H.B.; Yao, Q.Y.; Gao, L.Z. De novo transcriptome sequencing of Camellia sasanqua and the analysis of major candidate genes related to floral traits. Plant. Physiol. Biochem. 2017, 120, 103–111. [Google Scholar] [CrossRef]
- Beld, M.; Martin, C.; Huits, H.; Stuitje, A.R.; Gerats, A.G. Flavonoid synthesis in Petunia hybrida: Partial characterization of dihydroflavonol-4-reductase genes. Plant. Mol. Biol. 1989, 13, 491–502. [Google Scholar] [CrossRef]
- Yan, Y.; Chemler, J.; Huang, L.; Martens, S.; Koffas, M.A. Metabolic engineering of anthocyanin biosynthesis in Escherichia coli. Appl. Environ. Microbiol. 2005, 71, 3617–3623. [Google Scholar] [CrossRef]
- Boase, M.R.; Lewis, D.H.; Davies, K.M.; Marshall, G.B.; Patel, D.; Schwinn, K.E.; Deroles, S.C. Isolation and antisense suppression of flavonoid 3′,5′-hydroxylase modifies flower pigments and colour in cyclamen. Bmc Plant. Biol. 2010, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Nakano-Shimada, R.; Ohbayashi, M.; Okinaka, Y.; Kiyokawa, S.; Kikuchi, Y. Expression of chimeric P450 genes encoding flavonoid-3′, 5′-hydroxylase in transgenic tobacco and petunia plants(1). Febs. Lett. 1999, 461, 241–245. [Google Scholar] [CrossRef]
- Li, Z.; Vickrey, T.L.; McNally, M.G.; Sato, S.J.; Clemente, T.E.; Mower, J.P. Assessing Anthocyanin Biosynthesis in Solanaceae as a Model Pathway for Secondary Metabolism. Genes 2019, 10, 559. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, Y.; Katsumoto, Y.; Fukui, Y.; Fukuchi-Mizutani, M.; Ohkawa, H. Molecular characterization of the flavonoid biosynthetic pathway and flower color modification of Nierembergia sp. Plant. Biotechnol. 2006, 23, 19–24. [Google Scholar] [CrossRef]
- Okinaka, Y.; Shimada, Y.; Nakano-Shimada, R.; Ohbayashi, M.; Kiyokawa, S. Selective Accumulation of Delphinidin Derivatives in Tobacco Using a Putative Flavonoid 3′,5′-Hydroxylase cDNA from Campanula medium. J. Agric. Chem. Soc. Jpn. 2003, 67, 161–165. [Google Scholar]
- Brugliera, F.; Tanaka, Y.; Mason, J. Flavonoid 3′,5′ hydroxylase gene sequences and uses therefor. US8445748B2, 21 May 2013. [Google Scholar]
- Brugliera, F. Genetic sequences having methyltransferase activity and uses therefor. US7807877, 5 October 2010. [Google Scholar]
- Tanaka, Y.; Katsumoto, Y.; Brugliera, F.; Mason, J. Genetic engineering in floriculture. Plant. Cell Tissue Organ. Cult. 2009, 80, 1–24. [Google Scholar] [CrossRef]
- Wilmouth, R.C.; Turnbull, J.J.; Welford, R.W.D.; Clifton, I.J.; Prescott, A.G.; Schofield, C.J. Structure and Mechanism of Anthocyanidin Synthase from Arabidopsis thaliana. Structure 2002, 10, 93–103. [Google Scholar] [CrossRef]
- Liu, C.; Wang, X.; Shulaev, V.; Dixon, R.A. A role for leucoanthocyanidin reductase in the extension of proanthocyanidins. Nat. Plants 2016, 2, 16182. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4. Article 17. [Google Scholar] [CrossRef]
- Wang, L.Y.; Li, D.; Deng, Y.-S.; Lv, W.; Meng, Q.-W. Antisense-mediated depletion of tomato GDP-l-galactose phosphorylase increases susceptibility to chilling stress. J. Plant. Physiol. 2013, 170, 303–314. [Google Scholar] [CrossRef]
- Bulley, S.; Wright, M.; Rommens, C.; Yan, H.; Rassam, M.; Lin-Wang, K.; Andre, C.; Brewster, D.; Karunairetnam, S.; Allan, A.C.; et al. Enhancing ascorbate in fruits and tubers through over-expression of the l-galactose pathway gene GDP-l-galactose phosphorylase. Plant. Biotechnol. J. 2012, 10, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Fatima, T.; Kesari, V.; Watt, I.; Wishart, D.; Todd, J.F.; Schroeder, W.R.; Paliyath, G.; Krishna, P. Metabolite profiling and expression analysis of flavonoid, vitamin C and tocopherol biosynthesis genes in the antioxidant-rich sea buckthorn (Hippophae rhamnoides L.). Phytochemistry 2015, 118, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Gallego, A.M.; Rojas, L.F.; Parra, O.; Rodriguez, H.A.; Mazo Rivas, J.C.; Urrea, A.I.; Atehortua, L.; Fister, A.S.; Guiltinan, M.J.; Maximova, S.N.; et al. Transcriptomic analyses of cacao cell suspensions in light and dark provide target genes for controlled flavonoid production. Sci. Rep. 2018, 8, 13575. [Google Scholar] [CrossRef] [PubMed]
- Petridis, A.; Doll, S.; Nichelmann, L.; Bilger, W.; Mock, H.-P. Arabidopsis thaliana G2-LIKE FLAVONOID REGULATOR and BRASSINOSTEROID ENHANCED EXPRESSION1 are low-temperature regulators of flavonoid accumulation. New Phytol. 2016, 211, 912–925. [Google Scholar] [CrossRef]
- Luo, X.M.; Lin, W.H.; Zhu, S.W.; Zhu, J.Y.; Sun, Y.; Fan, X.Y.; Cheng, M.L.; Hao, Y.Q.; Oh, E.; Tian, M.M.; et al. Integration of Light- and Brassinosteroid-Signaling Pathways by a GATA Transcription Factor in Arabidopsis. Dev. Cell 2010, 19, 872–883. [Google Scholar] [CrossRef]
- Sun, R.-Z.; Pan, Q.-H.; Duan, C.-Q.; Wang, J. Light response and potential interacting proteins of a grape flavonoid 3′-hydroxylase gene promoter. Plant. Physiol. Biochem. Ppb 2015, 97, 70–81. [Google Scholar] [CrossRef]
- Xu, W.J.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant. Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef]
- Tingting, D.; Rongpeng, H.; Jiawen, Y.; Mingku, Z.; Yi, Z.; Ying, G.; Zongyun, L. Anthocyanins accumulation and molecular analysis of correlated genes by metabolome and transcriptome in green and purple asparaguses (Asparagus officinalis, L.). Food Chem. 2019, 271, 18–28. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Wang, L.K.; Feng, Z.X.; Wang, X.; Wang, X.W.; Zhang, X.G. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, D.; Yang, H.; Chen, M.; Zhang, W.; Zhang, L. Metabolome and Transcriptome Sequencing Analysis Reveals Anthocyanin Metabolism in Pink Flowers of Anthocyanin-Rich Tea (Camellia sinensis). Molecules 2019, 24, 1064. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CPS (Count Per Second) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compounds | ZJW1 | SE | ZJW2 | SE | ZJW3 | SE | ZJW4 | SE | ZJW5 | SE |
| Cyanidin 3-O-glucoside | 1.11 × 106 a | 6.56 × 105 | 6.89 × 105 a | 7.57 × 105 | 5.87 × 105 a | 6.09 × 105 | 3.17 × 106 a | 5.38 × 106 | 1.38 × 106 a | 9.29 × 105 |
| Peonidin | 4.28 × 104 a | 8.24 × 103 | 4.92 × 104 a | 5.69 × 103 | 6.32 × 104 a | 2.12 × 104 | 5.07 × 104 a | 1.85 × 104 | 5.10 × 104 a | 3.56 × 103 |
| Cyanidin O-syringic acid | NA | - | NA | - | NA | - | NA | - | NA | - |
| Cyanidin 3-O-rutinoside | 1.21 × 107 a | 2.60 × 106 | 1.33 × 107 a | 3.79 × 105 | 1.28 × 107 a | 2.65 × 105 | 1.16 × 107 a | 1.26 × 106 | 1.27 × 107 a | 8.96 × 105 |
| Cyanidin 3,5-O-diglucoside | 2.08 × 107 a | 3.79 × 105 | 1.81 × 107 a | 9.00 × 105 | 1.84 × 107 a | 5.29 × 105 | 1.68 × 107 a | 6.66 × 105 | 1.72 × 107 a | 1.47 × 106 |
| Malvidin 3,5-diglucoside | 1.23 × 104 a | 2.81 × 103 | 1.26 × 104 a | 3.16 × 103 | 1.24 × 104 a | 1.47 × 103 | 1.01 × 104 a | 1.72 × 103 | 1.10 × 104 a | 2.57 × 103 |
| Pelargonin | 5.42 × 105 a | 1.31 × 104 | 4.28 × 105 a | 5.54 × 104 | 4.55 × 105 a | 5.85 × 104 | 3.78 × 105 a | 5.16 × 104 | 3.86 × 105 a | 2.30 × 104 |
| Petunidin 3-O-glucoside | NA | - | NA | - | NA | - | NA | - | NA | - |
| Pelargonidin 3-O-β-d-glucoside | NA | - | NA | - | NA | - | NA | - | NA | - |
| Cyanidin | 1.04 × 107 a | 1.22 × 106 | 1.20 × 107 a | 1.53 × 105 | 1.28 × 107 a | 2.97 × 106 | 1.20 × 107a | 8.50 × 105 | 1.09 × 107 a | 5.03 × 105 |
| Procyanidin A2 | NA | - | NA | - | NA | - | NA | - | NA | - |
| Procyanidin B2 | 5.50 × 107 a | 6.51 × 105 | 5.45 × 107 a | 2.15 × 106 | 5.42 × 107 a | 2.17 × 106 | 5.00 × 107 a | 1.95 × 106 | 5.25 × 107 a | 2.00 × 106 |
| Procyanidin B3 | 5.83 × 107 a | 6.43 × 105 | 5.64 × 107 a | 2.61 × 106 | 5.60 × 107 a | 2.15 × 106 | 5.09 × 107 a | 9.29 × 105 | 5.42 × 107 a | 2.55 × 106 |
| Compounds | BTP1 | SE | BTP2 | SE | BTP3 | SE | BTP4 | SE | BTP5 | SE |
| Cyanidin 3-O-glucoside | 7.04 × 107 b | 5.01 × 106 | 5.54 × 107 b | 5.85 × 106 | 5.55 × 107 b | 8.66 × 106 | 4.33 × 107 b | 5.74 × 106 | 5.48 × 107 b | 9.47 × 106 |
| Peonidin | 2.43 × 104 a | 8.66 × 103 | 1.74 × 104 a | 1.27 × 104 | 1.64 × 104 a | 9.47 × 103 | 1.46 × 104 a | 5.74 × 103 | 2.37 × 104 a | 1.59 × 104 |
| Cyanidin O-syringic acid | 1.60 × 107 a | 1.81 × 106 | 1.29 × 107 a | 1.16 × 106 | 1.37 × 107 a | 2.79 × 106 | 1.05 × 107 a | 3.85 × 106 | 1.53 × 107 a | 2.10 × 106 |
| Cyanidin 3-O-rutinoside | 7.61 × 106 a | 4.59 × 106 | 6.31 × 106 a | 3.53 × 106 | 6.07 × 106 a | 3.59 × 106 | 6.15 × 106 a | 2.23 × 106 | 6.64 × 106 a | 1.39 × 106 |
| Cyanidin 3,5-O-diglucoside | 1.54 × 107 a | 4.92 × 106 | 1.21 × 107 a | 4.84 × 106 | 1.16 × 107 a | 3.16 × 106 | 1.03 × 107 a | 4.04 × 106 | 9.06 × 106 a | 1.73 × 106 |
| Malvidin 3,5-diglucoside | 2.62 × 104 a | 1.16 × 104 | 2.21 × 104 a | 7.04 × 103 | 2.36 × 104 a | 7.97 × 103 | 1.86 × 104 a | 8.21 × 103 | 2.06 × 104 a | 5.60 × 103 |
| Pelargonin | 5.41 × 105 a | 2.99 × 105 | 4.35 × 105 a | 1.62 × 105 | 4.13 × 105 a | 7.31 × 104 | 3.88 × 105 a | 2.06 × 105 | 2.36 × 105 a | 5.64 × 104 |
| Petunidin 3-O-glucoside | 5.46 × 107 a | 1.38 × 107 | 2.55 × 107 a | 3.33 × 106 | 2.23 × 107 a | 5.05 × 106 | 1.41 × 107 a | 5.33 × 106 | 1.51 × 107 a | 4.84 × 106 |
| Pelargonidin 3-O-β-d-glucoside | 2.33 × 106 a | 1.10 × 106 | 1.52 × 106 a | 5.74 × 105 | 1.33 × 106 a | 2.41 × 105 | 1.04 × 106 a | 5.08 × 105 | 1.44 × 106 a | 3.79 × 105 |
| Cyanidin | 1.28 × 107 a | 6.05 × 106 | 1.32 × 107 a | 4.84 × 106 | 1.38 × 107 a | 4.40 × 106 | 1.38 × 107 a | 4.91 × 106 | 1.21 × 107 a | 5.45 × 106 |
| Procyanidin A2 | NA | - | 8.02 × 104 a | 8.00 × 104 | 1.61 × 105 a | 1.34 × 105 | 2.10 × 105 a | 9.82 × 104 | 1.51 × 105 a | 5.42 × 104 |
| Procyanidin B2 | 3.73 × 107 a | 8.26 × 106 | 3.62 × 107 a | 6.37 × 106 | 3.73 × 107 a | 8.65 × 106 | 4.08 × 107 a | 3.07 × 106 | 4.53 × 107 a | 3.53 × 106 |
| Procyanidin B3 | 3.98 × 107 a | 9.33 × 106 | 3.87 × 107 a | 6.69 × 106 | 3.86 × 107 a | 8.06 × 106 | 4.21 × 107 a | 3.40 × 106 | 4.54 × 107 a | 2.64 × 106 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, C.; Mei, X.; Rothenberg, D.O.; Yang, Z.; Zhang, W.; Wan, S.; Yang, H.; Zhang, L. Metabolome and Transcriptome Analysis Reveals Putative Genes Involved in Anthocyanin Accumulation and Coloration in White and Pink Tea (Camellia sinensis) Flower. Molecules 2020, 25, 190. https://doi.org/10.3390/molecules25010190
Zhou C, Mei X, Rothenberg DO, Yang Z, Zhang W, Wan S, Yang H, Zhang L. Metabolome and Transcriptome Analysis Reveals Putative Genes Involved in Anthocyanin Accumulation and Coloration in White and Pink Tea (Camellia sinensis) Flower. Molecules. 2020; 25(1):190. https://doi.org/10.3390/molecules25010190
Chicago/Turabian StyleZhou, Caibi, Xin Mei, Dylan O’Neill Rothenberg, Zaibo Yang, Wenting Zhang, Shihua Wan, Haijun Yang, and Lingyun Zhang. 2020. "Metabolome and Transcriptome Analysis Reveals Putative Genes Involved in Anthocyanin Accumulation and Coloration in White and Pink Tea (Camellia sinensis) Flower" Molecules 25, no. 1: 190. https://doi.org/10.3390/molecules25010190
APA StyleZhou, C., Mei, X., Rothenberg, D. O., Yang, Z., Zhang, W., Wan, S., Yang, H., & Zhang, L. (2020). Metabolome and Transcriptome Analysis Reveals Putative Genes Involved in Anthocyanin Accumulation and Coloration in White and Pink Tea (Camellia sinensis) Flower. Molecules, 25(1), 190. https://doi.org/10.3390/molecules25010190