
Corrections of Molecular Morphology and Hydrogen Bond for Improved Crystal Density Prediction

 and
and
Abstract
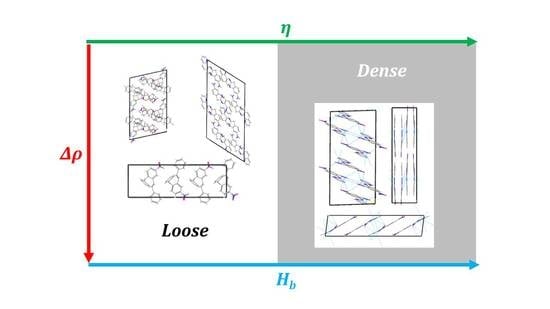
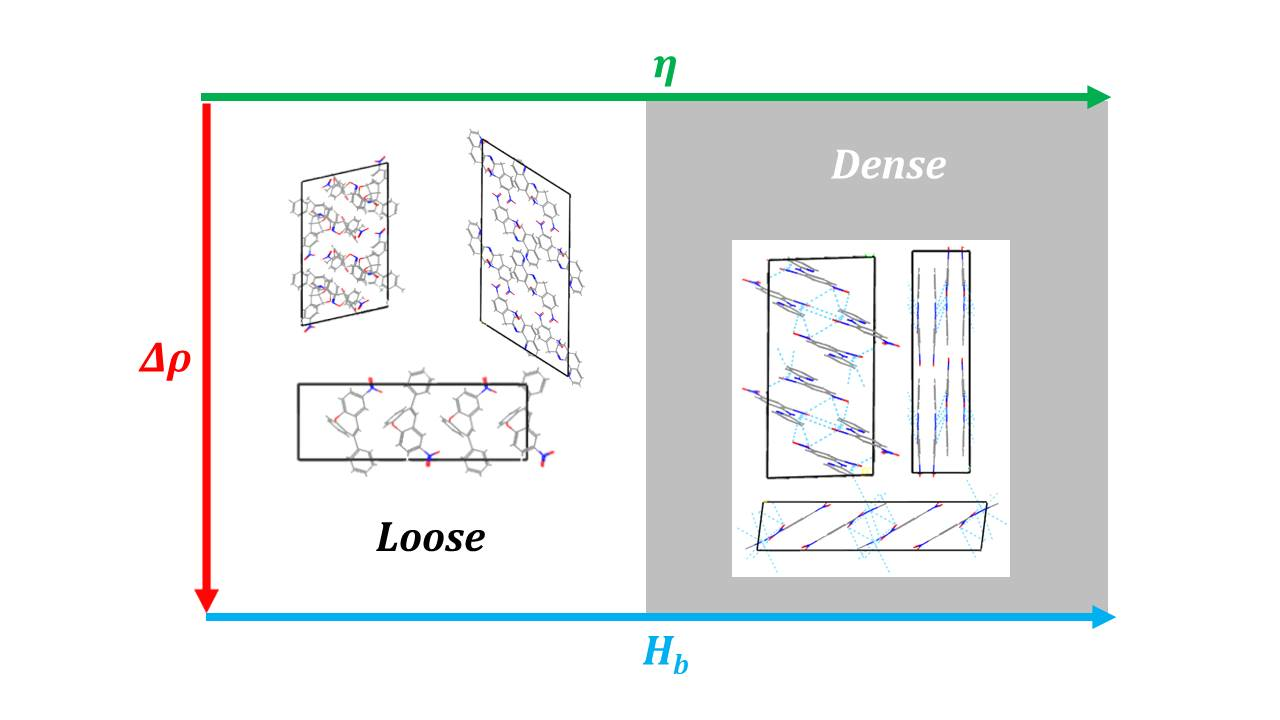
1. Introduction
2. Results and Discussion
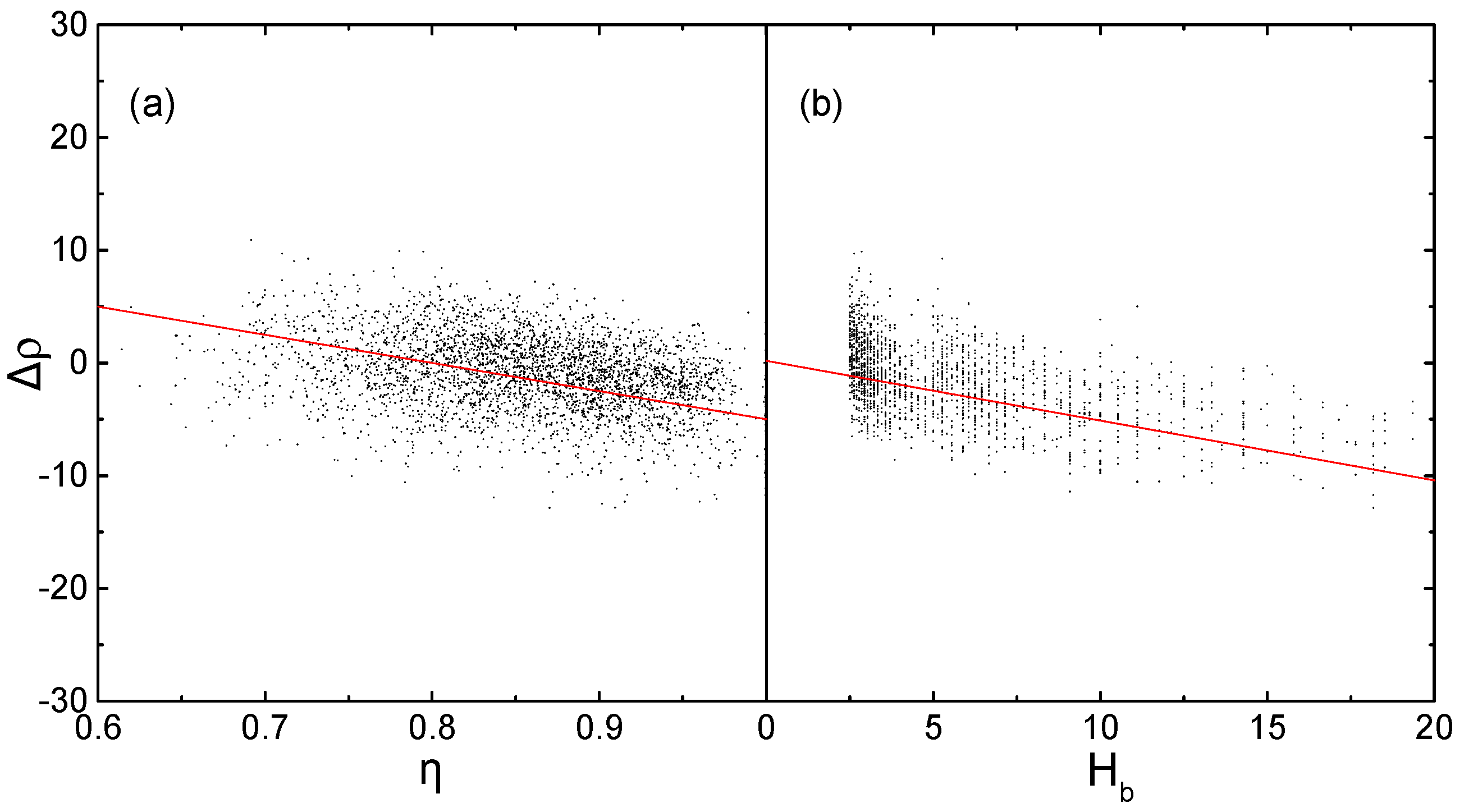
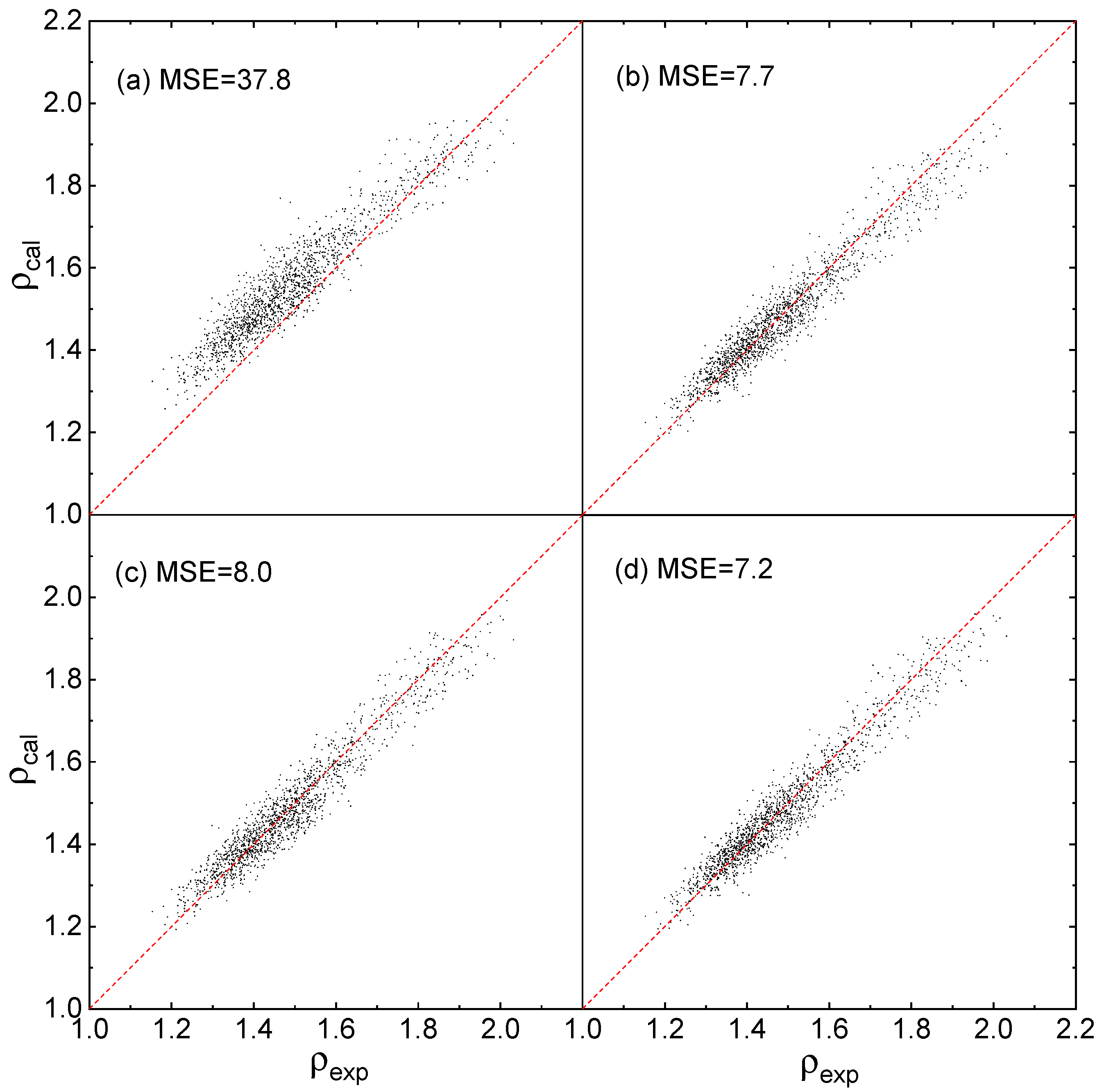
2.1. Corrections Ananysis on Prediction Error
2.2. Construction of Correction Formulas
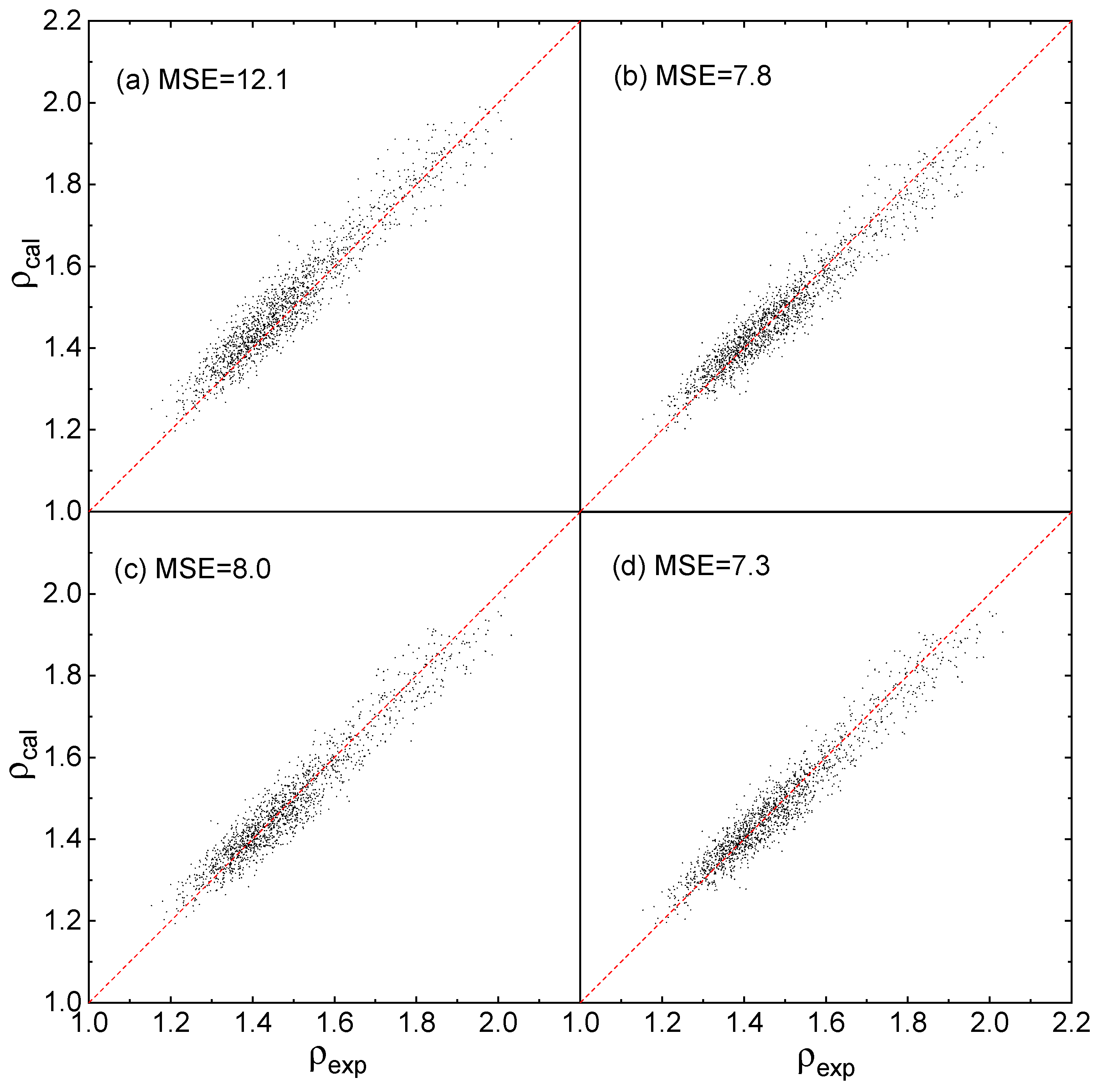
2.3. Fitting Results
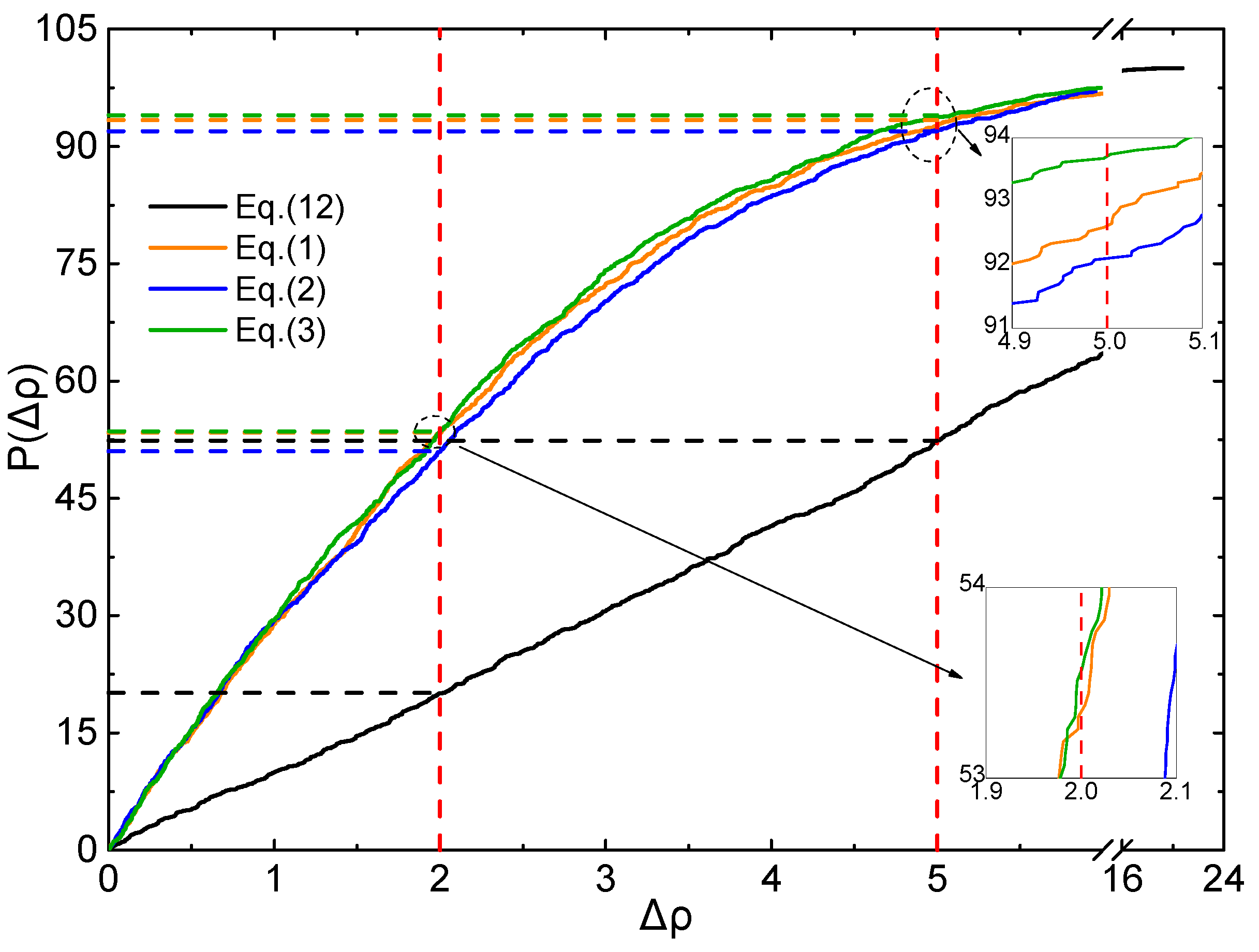
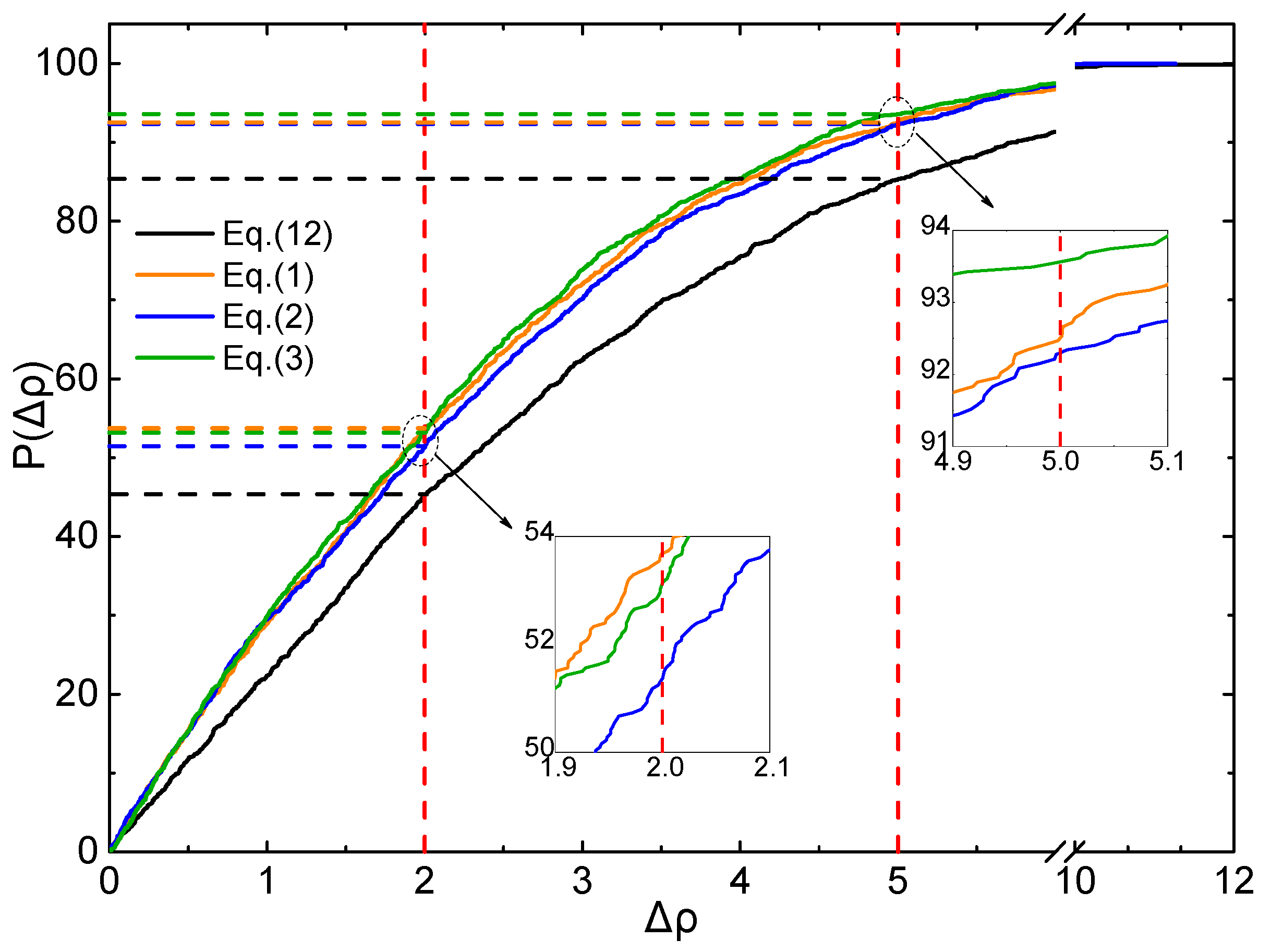
2.4. Evaluation of Accuracy and Generalization
3. Theory and Method
3.1. Preparation of Data Set
3.2. Morphology Descriptor and Calculation Method
3.3. Hydrogen Bond Descriptor and Its Calculation Method
3.4. Functional Forms
3.5. Accuracy Evaluatiom
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Politzer, P.; Murray, J.S. Impact sensitivity and crystal lattice compressibility/free space. J. Mol. Model. 2014, 20, 2223–2231. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, M.H. Simple Determination of Performance of Explosives without Using Any Experimental Data. J. Hazard. Mater. 2005, 119, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, M.H. Novel Method for Predicting Densities of Polynitro Arene and Polynitro Heteroarene Explosives in order to Evaluate Their Detonation Performance. J. Hazard. Mater. 2009, 165, 579–588. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Jacobs, S.J.J. Chemistry of Detonations. I. A Simple Method for Calculating Detonation Properties of C-H-N-O Explosives. Chem. Phys. 1968, 48, 23–35. [Google Scholar] [CrossRef]
- Zhang, Q.-H.; Zhang, J.-H.; Qi, X.-J.; Shreeve, J.M. Molecular Design and Property Prediction of High Density Polynitro[3.3.3]-Propellane-Derivatized Frameworks as Potential High Explosives. J. Phys. Chem. A 2014, 118, 10857–10865. [Google Scholar] [CrossRef]
- Fei, T.; Du, Y.; Pang, S.-P. Theoretical Design and Prediction of Properties for Dinitromethyl, Fluorodinitromethyl, and (Difluoroamino)dinitromethyl Derivatives of Triazole and Tetrazole. RSC. Adv. 2018, 8, 10215–10227. [Google Scholar] [CrossRef]
- Pan, Y.; Li, J.-S.; Cheng, B.-B.; Zhu, W.-H.; Xiao, H.-M. Computational Studies on The Heats of Formation, Energetic Properties, and Thermal Stability of Energetic Nitrogen-rich Furazano[3,4-b]pyrazine-based Derivatives. Comput. Theor. Chem. 2012, 992, 110–119. [Google Scholar] [CrossRef]
- Zhang, X.-W.; Xiao, H.-M. Theoretical Studies on Heats of Formation, Detonation Properties, and Bond Dissociation Energies of Monofurazan Derivatives. Int. J. Quant. Chem. 2010, 110, 1549–1558. [Google Scholar] [CrossRef]
- Zhang, X.-W.; Zhu, W.-H.; Xiao, H.-M. Comparative Theoretical Studies of Energetic Carbon-and Nireogen-bridged Difurazans. J. Phys. Chem. A 2010, 114, 603–612. [Google Scholar] [CrossRef]
- Liu, Z.-C.; Wu, Q.; Zhu, W.-H.; Xiao, H.-M. Theoretical Study of Energetic Trinitromethylsubstituted Tetrazole and Tetrazine Derivatives. J. Phys. Org. Chem. 2013, 26, 939–947. [Google Scholar] [CrossRef]
- Wang, F.; Wang, G.-X.; Du, H.-C.; Zhang, J.-Y.; Gong, X.-D. Theoretical Studies on the Heats of Formation, Detonation Properties, and Pyrolysis Mechanisms of Energetic Cyclic Nitramines. J. Phys. Chem. A 2011, 115, 13858–13864. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Wu, J.-Z.; Zhu, W.-H.; Zhang, C.-C.; Xiao, H.-M. Characterization of Nitrogen-bridged 1,2,4,5-tetrazine-, furazan-, and 1H-tetrazole-based Polyheterocyclic Compounds: Heats of Formation, Thermal Stability, and Detonation Properties. J. Mol. Model. 2012, 18, 3467–3479. [Google Scholar] [CrossRef]
- Somayazulu, M.; Ahart, M.; Mishra, A.K.; Geballe, Z.M.; Baldini, M.; Meng, Y.; Struzhkin, V.V.; Hemley, R. Evidence for Superconductivity above 260 K in Lanthanum Superhydride at Megabar Pressures. J. Phys. Rev. Lett. 2019, 122, 027001. [Google Scholar] [CrossRef] [PubMed]
- Tarver, C.M. Density Estimations for Explosives and Related Compounds Using the Group Sdditivity Spproach. J. Chem. Eng. Data 1979, 24, 136–145. [Google Scholar] [CrossRef]
- Exner, O. Additive Physical Properties. II. Molar Volume as an Additive Property. Collect. Czech. Chem. Commun. 1967, 32, 1–23. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Thiel, W.T. Group States of Molecules. 38. The MNDD Method. Approximations and Parameters. J. Am. Chem. Soc. 1977, 99, 4899–4907. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Brinck, T.; Lane, P. ACS Symp. Ser. 586; Nelson, J.O., Karu, A.E., Wong, R.B., Eds.; American Chemical Society: Washington, DC, USA, 1994. [Google Scholar]
- Wang, G.-X.; Xiao, H.-M.; Xu, X.-J.; Ju, X.-H. Detonation Velocities and Pressures, and Their Relationships with Electric Spark Sensitivities for Nitramines. Propell. Explos. Pyrot. 2006, 31, 102–109. [Google Scholar] [CrossRef]
- Qiu, L.; Xiao, H.-M.; Gong, X.-D.; Ju, X.-H.; Zhu, W.-H. Theoretical Studies on the Structures, Thermodynamic Properties, Detonation Properties, and Pyrolysis Mechanisms of Spiro Nitramines. J. Phys. Chem. A 2006, 110, 3797–3807. [Google Scholar] [CrossRef]
- Rice, B.M.; Hare, J.J.; Byrd, E.F.C. Accurate Predictions of Crystal Densities Using Quantum Mechanical Molecular Volumes. J. Phys. Chem. A 2007, 111, 10874–10879. [Google Scholar] [CrossRef]
- Politzer, P.; Martinez, J.; Murray, J.S.; Concha, M.C.; Alejandro, T.L. An Electrostatic Interaction Correction for Improved Crystal Density Prediction. Mol. Phys. 2009, 19, 2095–2101. [Google Scholar] [CrossRef]
- Keshavarz, M.H.; Soury, H.; Motamedoshariati, H.; Dashtizadeh, A. Improved Method for Prediction of Density of Energetic Compounds Using Their Molecular Structure. Struct. Chem. 2015, 26, 455–466. [Google Scholar] [CrossRef]
- Rice, B.M.; Byrd, E.F.C. Evaluation of Electrostatic Descriptors for Predicting Crystalline Density. J. Comput. Chem. 2013, 34, 2146–2151. [Google Scholar] [CrossRef] [PubMed]
- Ammon, H.L. Updated Atom/functional Group and Atom Code Volume Additivity Parameters for The Calculation of Crystal Densities of Single Molecules, Organic Salts, and Multi-fragment Materials Containing, H, C, B, N, O, F, S, P, Cl, Br and I. Propell. Explos. Pyrot. 2008, 33, 92–102. [Google Scholar] [CrossRef]
- Yang, J.; De, S.; Campell, J.E.; Li, S.; Ceriotti, M.; Day, G.M. Large-Scale Computational Screening of Molecular Organic Semiconductors Using Crystal Structure Prediction. Chem. Mater. 2018, 30, 4361–4371. [Google Scholar] [CrossRef]
- Curtis, F.; Li, X.-Y.; Rose, T.; Álvaro, V.M.; Bhattacharya, S.; Ghiringhelli, L.M.; Marom, N. GAtor: A First-Principles Genetic Algorithm for Molecular Crystal Structure Prediction. J. Chem. Theory Comput. 2018, 14, 2246–2264. [Google Scholar] [CrossRef]
- Semenok, D.V.; Kvashnin, A.G.I.; Kruglov, A.A.; Oganov, R. Actinium Hydrides AcH10, AcH12, and AcH16 as High-Temperature Conventional Superconductors. J. Phys. Chem. Lett. 2018, 9, 1920–1926. [Google Scholar] [CrossRef]
- Lepeshkin, S.V.; Baturin, V.S.; Uspenskii, Y.A.; Oganov, A.R. Simultaneous Prediction of Atomic Structure and Stability of Nanoclusters in A Wide Area of Compositions. J. Phys. Chem. Lett. 2019, 10, 102–106. [Google Scholar] [CrossRef]
- Monserrat, B.; Drummond, N.D.; Philip, D.S.; Howie, R.-S.; Ríos, P.L.; Gregoryanz, E.; Pickard, C.J.; Needs, R.J. Structure and Metallicity of Phase V of Hydrogen. Phys. Rev. Lett. 2018, 120, 255701. [Google Scholar] [CrossRef]
- Podryabinkin, E.V.; Tikhonov, E.V.; Shapeev, A.V.; Oganov, A.R. Accelerating Crystal Structure Prediction by Machine-learning Interatomic Potentials with Active Learning. Phys. Rev. B 2019, 99, 064114. [Google Scholar] [CrossRef]
- Oganov, A.R.; Pickard, C.J.; Zhu, Q.; Needs, R.J. Struture Prediction Drives Materials Discovery. Nat. Rev. Mater. 2019, 4, 331–348. [Google Scholar] [CrossRef]
- Nielsen, A.T. Calculation of Densities of Fuel and Explosives from Molar Volume Additive Increments, Naval Weapons Center Report: NWC TP 5452. February 1973. Available online: https://www.osti.gov/biblio/7225584 (accessed on 1 February 1973).
- Immirzi, A.; Perini, B. Prediction of Density in Organic Crystals. Acta. Cryst. A 1977, 33, 216–218. [Google Scholar] [CrossRef]
- Kitiagorodsky, A.I. Molecular Crystals and Molecules; Academic Press: New York, NY, USA, 1973; pp. 18–21. [Google Scholar]
- Ammon, H.L.; Mitchell, S. A new Atom/functional Group Volume Additivity Data Base for The Calculation of the Crystal Densities of C, H, N, O and F-containing Compounds. Propell. Explos. Pyrot. 1998, 23, 260–265. [Google Scholar] [CrossRef]
- Stine, J.R. Prediction of Crystal Densities of Organic Explosives by Group Aadditivity; Los Alamos National Laboratory’s Report: New Mexico, NM, USA, 1981. Available online: https://www.osti.gov/biblio/6254123 (accessed on 1 August 1981).
- Keshavarz, M.H. Predictions of Densities of Acyclic and Cyclic Nitramines, Nitrateesters and Nitroaliphatic Compounds for Evaluation of Their Detonation Performance. J. Hazard. Mater. 2007, 143, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Naef, R.; Acree, W. Calculation of the Surface Tension of Ordinary Organic and Ionic Liquids by Means of a Generally Applicable Computer Algorithm Based on the Group-Additivity Method. Molecules 2018, 23, 1224. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Meanwell, N.A.; Li, D.; Hageman, M.J. The Expanding Role of Prodrugs in Contemporary Drug Design and Fevelopment. Nat. Rev. Drug Discov. 2018, 17, 559–587. [Google Scholar] [CrossRef] [PubMed]
- Janbazi, H.; Hasemann, O.; Schulz, C.; Kempf, A.; Wlokas, I.; Peukert, S.I. Response Surface and Group Additivity Methodology for Estimation of Thermodynamic Properties of Organosilanes. J. Chem. Kinet. 2018, 50, 681–690. [Google Scholar] [CrossRef]
- Rudolf, N.; William, A. Application of a General Computer Algorithm Based on the Group-Additivity Method for the Calculation of Two Molecular Descriptors at Both Ends of Dilution: Liquid Viscosity and Activity Coefficient in Water at Infinite Dilution. Molecules 2018, 23, 5. [Google Scholar]
- Reynolds, C.H.; Reynolds, R.C. Group Additivity in Ligand Binding Affinity: An Alternative Approach to Ligand Efficiency. J. Chem. Inf. Model. 2017, 57, 3086–3093. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. Theoretical and Computational Chemistry; Politzer, P., Murray, J.S., Eds.; Elsevier Scientific: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Murray, J.S.; Brinck, T.; Politzer, P. Relationships of Molecular Surface Electrostatic Potentials to Some Macroscopic Properties. Chem. Phys. 1996, 204, 289–299. [Google Scholar] [CrossRef]
- Wang, G.-X.; Xiao, H.-M.; Ju, X.-H.; Gong, X.-D. Calculation of Detonation Velocity, Pressure, and Electric Sensitivity of Nitro Arenes Based on Quantum Chemistry. Propell. Explos. Pyrot. 2006, 31, 361–368. [Google Scholar] [CrossRef]
- Xu, X.-J.; Xiao, H.-M.; Ju, X.-H.; Gong, X.-D.; Zhu, W.-H. Computational Studies on Polynitrohexaazaadmantanes as Potential High Energy Density Materials. J. Phys. Chem. A 2006, 110, 5929–5993. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Xiao, H.-M.; Gong, X.-D.; Ju, X.-H.; Zhu, W.-H. Crystal Density Predictions for Nitramines Based on Quantum Chemistry. J. Hazard. Mater. 2007, 141, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Brinck, T.; Lane, P.; Paulsen, K.; Politzer, P. Statistically-based Interaction Indices Derived from Molecular Surface Electrostatic Potentials: A General Interaction Properties Function (GIPF). J. Mol. Struct. Theochem. 1994, 307, 55–64. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Perspectives on the Crystal Densities and Packing Coefficients of Explosive Compounds. Struct. Chem. 2016, 7, 401–408. [Google Scholar] [CrossRef]
- Zhang, Y.-Q.; Zhang, D.-C. Hydrogen Bond in the Crystals of Substituted Ntrrobenzenes. J. Suzhou. U-Nat. Sci. 1996, 12, 55–61. [Google Scholar]
- Ma, Y.; Zhang, A.-B.; Xue, X.-G.; Jiang, D.-J.; Zhu, Y.-Q.; Zhang, C.-Y. Crystal Packing of Impact-Sensitive High-Energy Explosives. Cryst. Growth. Des. 2014, 14, 6101–6114. [Google Scholar] [CrossRef]
- Boggs, P.T.; Byrd, R.H.; Schnabel, R.B. A stable and efficient algorithm for nonlinear orthogonal distance regression. SIAM J. Sci. Stat. Comp. 1987, 8, 1052–1078. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.-W. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.-W. Quantitative Analysis of Molecular Surface Based on Improved Marching Tetrahedra Algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef]
- Taherkhani, M.; Safabakhsh, R. A Novel Stability-Based Adaptive Inertia Weight for Particle Swarm Optimization. Appl. Soft Comput. 2016, 38, 281–295. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds refer to the computational models and results are available from the authors. |








{kind=link}
{kind=link}
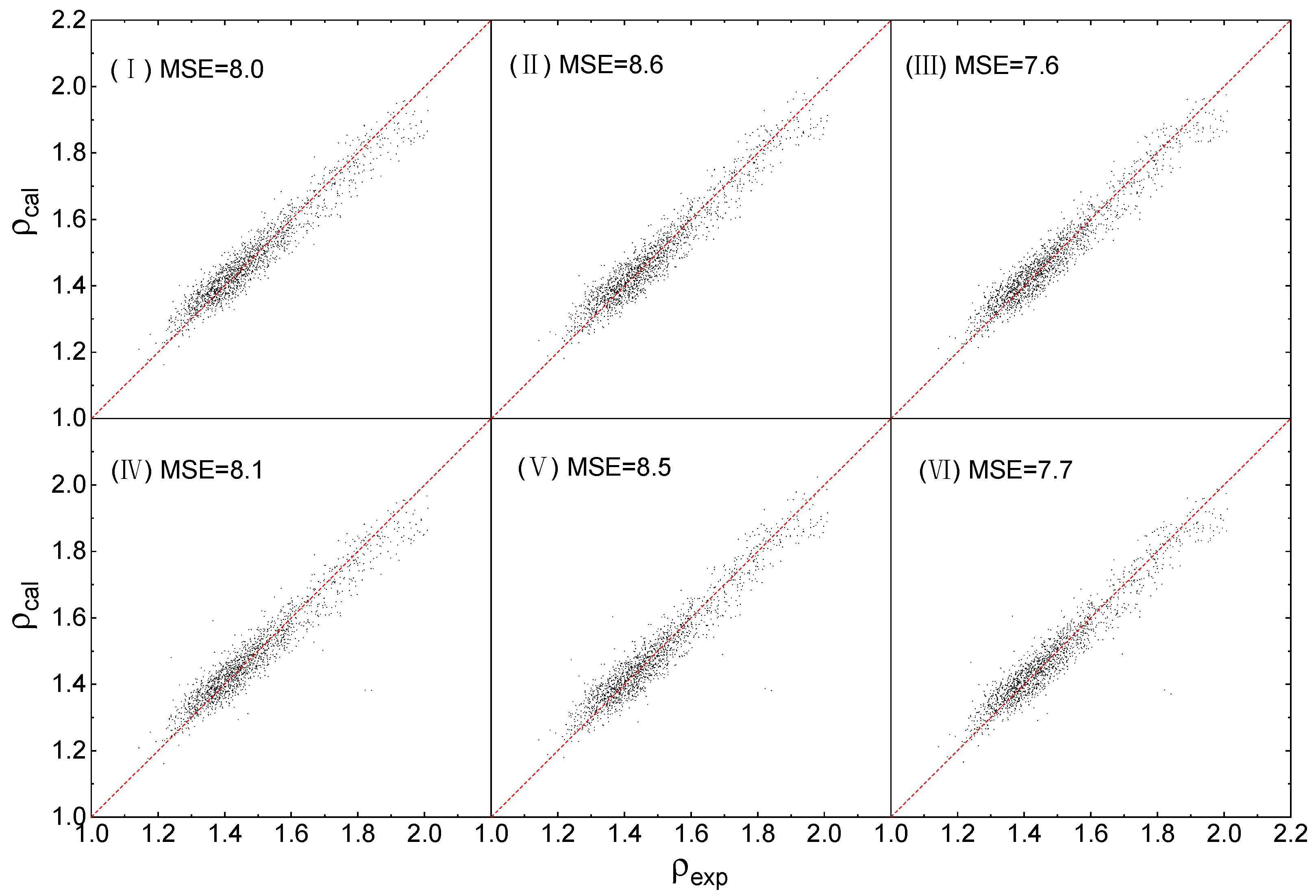{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Equation | DFT Level |
|---|---|---|
| I | (1) | B3PW91/6-31G(d,p) |
| II | (2) | B3PW91/6-31G(d,p) |
| III | (3) | B3PW91/6-31G(d,p) |
| IV | (1) | B3LYP/6-31G** |
| V | (2) | B3LYP/6-31G** |
| VI | (3) | B3LYP/6-31G** |
| Condition | α | β | β1 | β2 | γ | Δρmax (g/cm3) |
|---|---|---|---|---|---|---|
| I | 1 | 0.0010 | 0.1960 | - | −0.2549 | 0.15 |
| II | 1 | 0.0006 | - | 0.0036 | −0.0825 | 0.17 |
| III | 1 | 0.0005 | 0.1836 | 0.0033 | −0.2342 | 0.15 |
| IV | 1 | 0.0010 | 0.1802 | - | −0.2320 | 0.15 |
| V | 1 | 0.0006 | - | 0.0036 | −0.0721 | 0.16 |
| VI | 1 | 0.0005 | 0.1670 | 0.0033 | −0.2105 | 0.15 |
| Equation | B3PW91/6-31G(d,p) | B3LYP/6-31G** |
|---|---|---|
| (12) | 0.33 | 0.21 |
| (1) | 0.16 | 0.15 |
| (2) | 0.15 | 0.15 |
| (3) | 0.15 | 0.15 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Zhang, M.; Chen, J.; Su, L.; Zhao, S.; Zhang, C.; Liu, J.; Chen, C. Corrections of Molecular Morphology and Hydrogen Bond for Improved Crystal Density Prediction. Molecules 2020, 25, 161. https://doi.org/10.3390/molecules25010161
Wang L, Zhang M, Chen J, Su L, Zhao S, Zhang C, Liu J, Chen C. Corrections of Molecular Morphology and Hydrogen Bond for Improved Crystal Density Prediction. Molecules. 2020; 25(1):161. https://doi.org/10.3390/molecules25010161
Chicago/Turabian StyleWang, Linyuan, Miao Zhang, Jie Chen, Liang Su, Shicao Zhao, Chaoyang Zhang, Jian Liu, and Chunyan Chen. 2020. "Corrections of Molecular Morphology and Hydrogen Bond for Improved Crystal Density Prediction" Molecules 25, no. 1: 161. https://doi.org/10.3390/molecules25010161
APA StyleWang, L., Zhang, M., Chen, J., Su, L., Zhao, S., Zhang, C., Liu, J., & Chen, C. (2020). Corrections of Molecular Morphology and Hydrogen Bond for Improved Crystal Density Prediction. Molecules, 25(1), 161. https://doi.org/10.3390/molecules25010161