
Identifying Ortholog Selective Fragment Molecules for Bacterial Glutaredoxins by NMR and Affinity Enhancement by Modification with an Acrylamide Warhead

 ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. Identifying Orthologous GRX Candidates
2.2. Fragment Screening of the Three Orthologous Proteins
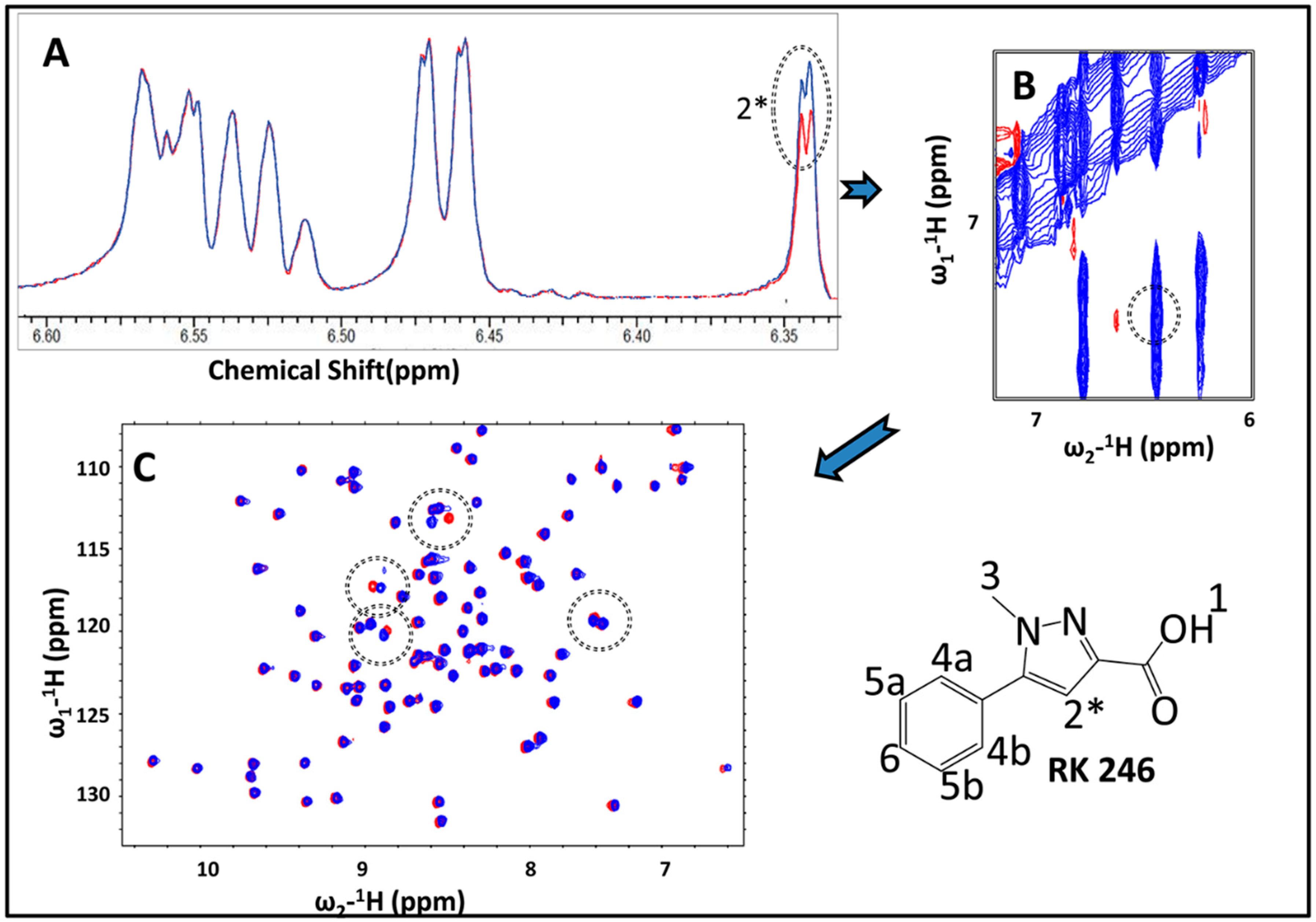
2.3. Hit Validation with trNOE and 15N HSQC
2.4. Dissociation Constant and Ligand Efficiency
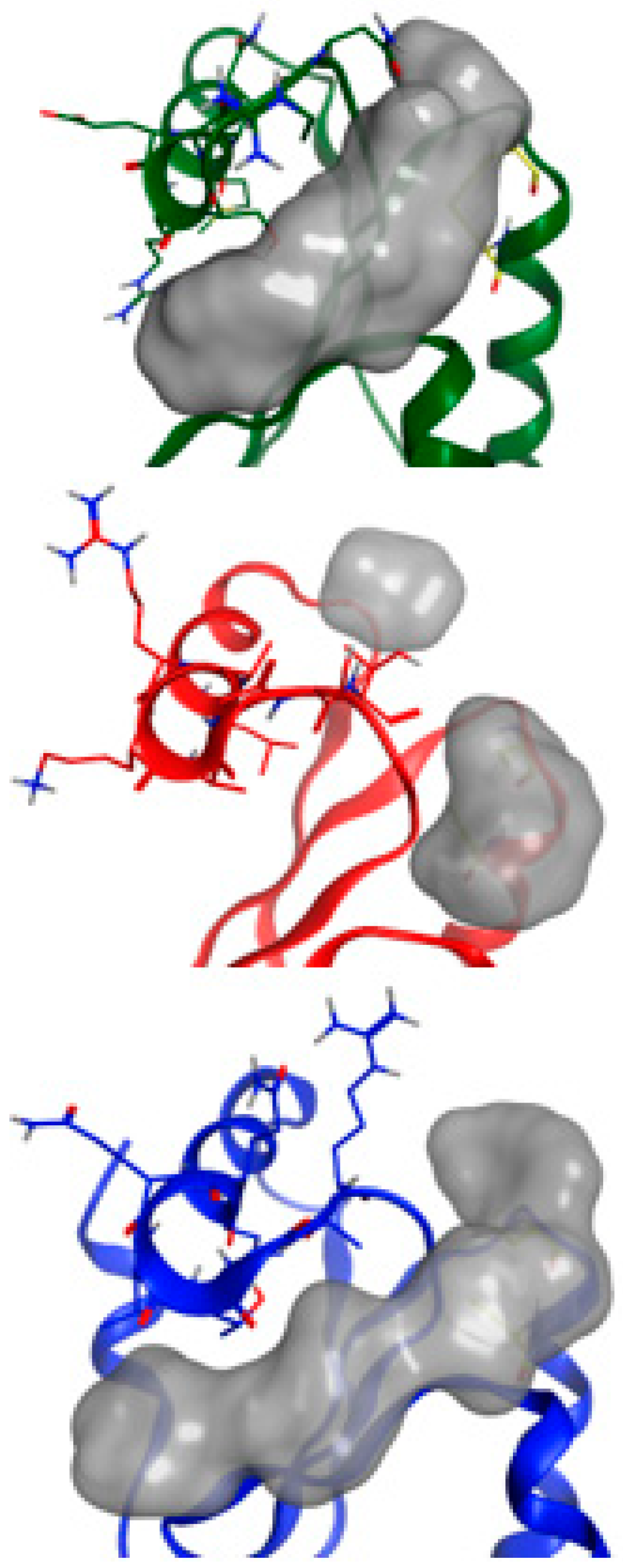
2.5. NMR-Based Modeling of Target Protein
2.6. Identification of the Binding Pose of the Winning RK395 Fragment with PaGRX
2.7. Acrylamide Warhead Development
2.8. Summary of Findings
3. Experimental
3.1. Library Design
3.2. Protein Expression and Purification
3.3. Ligand-Based NMR Experiments
3.4. NMR Backbone Resonance Assignments for Three Orthologous Proteins
3.5. NMR-Based Modeling of PaGRX
3.6. Protein-Observed NMR Experiments for Fragment Characterization
3.7. Dissociation Constant (Kd) Determination
3.8. Calculating Ligand Efficiency
3.9. Kinetic Studies of Warhead Reactivity
3.10. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FBDD | fragment-based drug discovery |
| STD | saturation transfer difference |
| rSTD% | relative saturation transfer difference |
| NMR | nuclear magnetic resonance |
| HSQC | heteronuclear single quantum correlation |
| SAR | structure activity relationship |
| BrmGRX | Brucella melitensis glutaredoxin |
| hGRX1 | human glutaredoxin 1 |
| PaGRX | Pseudomonas aeruginosa glutaredoxin |
| HTS | high throughput screening |
| logP | log of partition-coefficient |
| Kd | dissociation constant |
| L.E. | ligand efficiency |
| CSPs | chemical shift perturbations |
| trNOE | transferred nuclear overhauser effect |
References
- Hoelder, S.; Clarke, P.A.; Workman, P. Discovery of small molecule cancer drugs: Successes, challenges and opportunities. Mol. Oncol. 2012, 6, 155–176. [Google Scholar] [CrossRef]
- Van den Akker, F.; Bonomo, R.A. Exploring additional dimensions of complexity in inhibitor design for serine β-lactamases: Mechanistic and intra- and inter-molecular chemistry approaches. Front. Microbiol. 2018, 9, 622. [Google Scholar] [CrossRef]
- Lister, P.D.; Wolter, D.J.; Hanson, N.D. Antibacterial-resistant Pseudomonas aeruginosa: Clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610. [Google Scholar] [CrossRef]
- Leeper, T.; Zhang, S.; Van Voorhis, W.C.; Myler, P.J. Comparative analysis of glutaredoxin domains from bacterial opportunistic pathogens. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Carmi, C.; Galvani, E.; Vacondio, F.; Rivara, S.; Lodola, A.; Russo, S.; Aiello, S.; Bordi, F.; Costantino, G.; Cavazzoni, A.; et al. Irreversible inhibition of epidermal growth factor receptor activity by 3-aminopropanamides. J. Med. Chem. 2012, 55, 2251–2264. [Google Scholar] [CrossRef] [PubMed]
- Khattri, R.B.; Morris, D.L.; Davis, C.M.; Bilinovich, S.M.; Caras, A.J.; Panzner, M.J.; Debord, M.A.; Leeper, T.C. An NMR-guided screening method for selective fragment docking and synthesis of a warhead inhibitor. Molecules 2016, 21, 846. [Google Scholar] [CrossRef] [PubMed]
- Adeniyi, A.A.; Muthusamy, R.; Soliman, M.E. New drug design with covalent modifiers. Expert Opin. Drug Discov. 2016, 11, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Harner, M.J.; Frank, A.O.; Fesik, S.W. Fragment-based drug discovery using NMR spectroscopy. J. Biomol. NMR 2013, 56, 65–75. [Google Scholar] [CrossRef]
- Davies Thomas, G.; Hyvönen, M. Fragment-Based Drug Discovery and X-Ray Crystallography; Davies, T.G., Hyvönen, M., Eds.; Topics in Current Chemistry; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Navratilova, I.; Hopkins, A.L. Fragment screening by surface plasmon resonance. ACS Med. Chem. Lett. 2010, 1, 44–48. [Google Scholar] [CrossRef]
- Meiby, E.; Simmonite, H.; Le Strat, L.; Davis, B.; Matassova, N.; Moore, J.D.; Mrosek, M.; Murray, J.; Hubbard, R.E.; Ohlson, S. Fragment screening by weak affinity chromatography: Comparison with established techniques for screening against HSP90. Anal. Chem. 2013, 85, 6756–6766. [Google Scholar] [CrossRef]
- Pedro, L.; Quinn, R.J. Native mass spectrometry in fragment-based drug discovery. Molecules 2016, 21, 984. [Google Scholar] [CrossRef] [PubMed]
- Kobe, A.; Caaveiro, J.M.M.; Tashiro, S.; Kajihara, D.; Kikkawa, M.; Mitani, T.; Tsumoto, K. Incorporation of rapid thermodynamic data in fragment-based drug discovery. J. Med. Chem. 2013, 56, 2155–2159. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Wells, J.A.; Braisted, A.C. Tethering: Fragment-based drug discovery. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 199–223. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Greer, J. A decade of fragment-based drug design: Strategic advances and lessons learned. Nat. Rev. Drug Discov. 2007, 6, 211–219. [Google Scholar] [CrossRef]
- Rees, D.C.; Congreve, M.; Murray, C.W.; Carr, R. Fragment-based lead discovery. Nat. Rev. Drug Discov. 2004, 3, 660–672. [Google Scholar] [CrossRef]
- Jhoti, H.; Cleasby, A.; Verdonk, M.; Williams, G. Fragment-based screening using X-ray crystallography and NMR spectroscopy. Curr. Opin. Chem. Biol. 2007, 11, 485–493. [Google Scholar] [CrossRef]
- Amzel, L.M. Structure-based drug design. Curr. Opin. Biotechnol. 1998, 9, 366–369. [Google Scholar] [CrossRef]
- Meyer, B.; Peters, T. NMR Spectroscopy Techniques for Screening and Identifying Ligand Binding to Protein Receptors. Angew. Chem. Int. Ed. 2003, 42, 864–890. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001, 123, 6108–6117. [Google Scholar] [CrossRef]
- Venkitakrishnan, R.P.; Benard, O.; Max, M.; Markley, J.L.; Assadi-Porter, F.M. Use of NMR saturation transfer difference spectroscopy to study ligand binding to membrane proteins. Methods Mol. Biol. 2012, 914, 47–63. [Google Scholar]
- Wüthrich, K. NMR with proteins and nucleic Acids. Europhys. News 1986, 17, 11–13. [Google Scholar]
- Zhang, X.; Tang, H.; Ye, C.; Liu, M. Structure-based drug design: NMR-based approach for ligand–protein interactions. Drug Discov. Today Technol. 2006, 3, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Potenza, D.; Vasile, F.; Belvisi, L.; Civera, M.; Araldi, E.M. V STD and trNOESY NMR study of receptor-ligand interactions in living cancer cells. Chembiochem 2011, 12, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Ziarek, J.J.; Peterson, F.C.; Lytle, B.L.; Volkman, B.F. Binding Site Identification and Structure Determination of Protein–Ligand Complexes by NMR. Methods Enzymol. 2011, 493, 241–275. [Google Scholar]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef]
- Keserű, G.M.; Makara, G.M. Hit discovery and hit-to-lead approaches. Drug Discov. Today 2006, 11, 741–748. [Google Scholar] [CrossRef]
- Gao, G.; Liu, X.; Pavlovsky, A.; Viola, R.E. Identification of selective enzyme inhibitors by fragment library screening. J. Biomol. Screen. 2010, 15, 1042–1050. [Google Scholar] [CrossRef]
- Hesterkamp, T.; Whittaker, M. Fragment-based activity space: Smaller is better. Curr. Opin. Chem. Biol. 2008, 12, 260–268. [Google Scholar] [CrossRef]
- Alves, R.; Vilaprinyo, E.; Sorribas, A.; Herrero, E. Evolution based on domain combinations: The case of glutaredoxins. BMC Evol. Biol. 2009, 9, 66. [Google Scholar] [CrossRef]
- Fry, D.W.; Bridges, J.; Denny, W.; Doherty, A.; Greis, K.D.; Hicks, J.L.; Hook, K.E.; Keller, P.R.; Leopold, W.R.; Loo, J.A.; et al. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc. Natl. Acad. Sci. USA 1998, 95, 12022–12027. [Google Scholar] [CrossRef]
- Xu, Y.-Y.; Li, S.-N.; Yu, G.-J.; Hu, Q.-H.; Li, H.-Q. Discovery of novel 4-anilinoquinazoline derivatives as potent inhibitors of epidermal growth factor receptor with antitumor activity. Bioorg. Med. Chem. 2013, 21, 6084–6091. [Google Scholar] [CrossRef] [PubMed]
- Maresso, A.W.; Wu, R.; Kern, J.W.; Zhang, R.; Janik, D.; Missiakas, D.M.; Duban, M.E.; Joachimiak, A.; Schneewind, O. Activation of inhibitors by sortase triggers irreversible modification of the active site. J. Biol. Chem. 2007, 282, 23129–23139. [Google Scholar] [CrossRef] [PubMed]
- Barf, T.; Kaptein, A. Irreversible protein kinase inhibitors: Balancing the benefits and risks. J. Med. Chem. 2012, 55, 6243–6262. [Google Scholar] [CrossRef] [PubMed]
- Mather, B.D.; Viswanathan, K.; Miller, K.M.; Long, T.E. Michael addition reactions in macromolecular design for emerging technologies. Prog. Polym. Sci. 2006, 31, 487–531. [Google Scholar] [CrossRef]
- Siklos, M.; BenAissa, M.; Thatcher, G.R.J. Cysteine proteases as therapeutic targets: Does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B 2015, 5, 506–519. [Google Scholar] [CrossRef]
- Cocco, M.; Garella, D.; Di Stilo, A.; Borretto, E.; Stevanato, L.; Giorgis, M.; Marini, E.; Fantozzi, R.; Miglio, G.; Bertinaria, M. Electrophilic warhead-based design of compounds preventing NLRP3 inflammasome-dependent pyroptosis. J. Med. Chem. 2014, 57, 10366–10382. [Google Scholar] [CrossRef]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef]
- Madden, T. The BLAST Sequence Analysis Tool; National Center for Biotechnology Information: Bethesda, MD, USA, 2013; pp. 1–17. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Develop ment Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Cala, O.; Krimm, I. Ligand-Orientation Based Fragment Selection in STD NMR Screening. J. Med. Chem. 2015, 58, 8739–8742. [Google Scholar] [CrossRef]
- Wielens, J.; Headey, S.J.; Rhodes, D.I.; Mulder, R.J.; Dolezal, O.; Deadman, J.J.; Newman, J.; Chalmers, D.K.; Parker, M.W.; Peat, T.S.; et al. Parallel screening of low molecular weight fragment libraries: Do differences in methodology affect hit identification? J. Biomol. Screen. 2013, 18, 147–159. [Google Scholar] [CrossRef]
- Begley, D.W.; Moen, S.O.; Pierce, P.G.; Zartler, E.R. Saturation transfer difference NMR for fragment screening. Curr. Protoc. Chem. Biol. 2013, 5, 251–268. [Google Scholar] [PubMed]
- Pouliot, M.; Jeanmart, S. Pan Assay Interference Compounds (PAINS) and Other Promiscuous Compounds in Antifungal Research. J. Med. Chem. 2016, 59, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Barelier, S.; Pons, J.; Gehring, K.; Lancelin, J.-M.; Krimm, I. Ligand specificity in fragment-based drug design. J. Med. Chem. 2010, 53, 5256–5266. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.J.; Hubbard, R.E. Lessons for fragment library design: Analysis of output from multiple screening campaigns. J. Comput. Aided. Mol. Des. 2009, 23, 603–620. [Google Scholar] [CrossRef]
- Zartler, E.R.; Mo, H. Practical aspects of NMR-based fragment discovery. Curr. Top. Med. Chem. 2007, 7, 1592–1599. [Google Scholar] [CrossRef]
- Shimotakahara, S.; Furihata, K.; Tashiro, M. Application of NMR screening techniques for observing ligand binding with a protein receptor. Magn. Reson. Chem 2005, 43, 69–72. [Google Scholar] [CrossRef]
- Dalvit, C. NMR methods in fragment screening: Theory and a comparison with other biophysical techniques. Drug Discov. Today 2009, 14, 1051–1057. [Google Scholar] [CrossRef]
- London, R.E. Theoretical analysis of the inter-ligand overhauser effect: A new approach for mapping structural relationships of macromolecular ligands. J. Magn. Reson. 1999, 141, 301–311. [Google Scholar] [CrossRef]
- Breukels, V.; Konijnenberg, A.; Nabuurs, S.M.; Doreleijers, J.F.; Kovalevskaya, N.V.; Vuister, G.W. Overview of the Use of NMR to Examine Protein Structure. In Current Protocols in Protein Science; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Herrmann, T.; Gntert, P.; Whrich, K.; Güntert, P.; Wüthrich, K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 2002, 319, 209–227. [Google Scholar] [CrossRef]
- Güntert, P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004, 278, 353–378. [Google Scholar]
- Xia, T.H.; Bushweller, J.H.; Sodano, P.; Billeter, M.; Björnberg, O.; Holmgren, A.; Wüthrich, K. NMR structure of oxidized Escherichia coli glutaredoxin: Comparison with reduced E. coli glutaredoxin and functionally related proteins. Protein Sci. 1992, 1, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Berardi, M.J.; Bushweller, J.H. The NMR solution structure of human glutaredoxin in the fully reduced form. J. Mol. Biol. 1998, 280, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Rajarathnam, K. 13C NMR chemical shifts can predict disulfide bond formation. J. Biomol. NMR 2000, 18, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [PubMed]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Huth, J.R.; Fesik, S.W. Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 2005, 48, 2518–2525. [Google Scholar] [CrossRef]
- Fielding, L. NMR methods for the determination of protein–ligand dissociation constants. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 219–242. [Google Scholar] [CrossRef]
- Batas, B.; Schiraldi, C.; Chaudhuri, J. Inclusion body purification and protein refolding using microfiltration and size exclusion chromatography. J. Biotechnol. 1999, 68, 149–158. [Google Scholar] [CrossRef]
- Advanced Chemistry Development, I. ACD/Structure Elucidator. Available online: https://books.google.com.br/books?id=voxsDwAAQBAJ&pg=PT380&lpg=PT380&dq=Advanced+Chemistry+Development,+I.+ACD/Structure+Elucidator,+version+15.01+2015.&source=bl&ots=3Wnfrd16oq&sig=ACfU3U2yLOBsejUrsPMeetW7uFvn8ZXozw&hl=en&sa=X&redir_esc=y#v=onepage&q=Advanced%20Chemistry%20Development%2C%20I.%20ACD%2FStructure%20Elucidator%2C%20version%2015.01%202015.&f=false (accessed on 15 January 2015).
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef]
- Ulrich, E.L.; Akutsu, H.; Doreleijers, J.F.; Harano, Y.; Ioannidis, Y.E.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z.; et al. BioMagResBank. Nucleic Acids Res. 2008, 36, D402–D408. [Google Scholar] [CrossRef] [PubMed]
- Stockman, B.J.; Dalvit, C. NMR screening techniques in drug discovery and drug design. Prog. Nucl. Magn. Reson. Spectrosc. 2002, 41, 187–231. [Google Scholar] [CrossRef]
- Shen, Y.; Bax, A. Protein structural information derived from NMR chemical shift with the neural network program TALOS-N. In Methods in Molecular Biology (Clifton, N.J.); Springer: New York, NY, USA, 2015; Volume 1260, pp. 17–32. ISBN 9781493922390. [Google Scholar]
- Laskowski, R.A.; Rullmann, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B.; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System. Schrödinger LLC 2002, Version 1. Available online: http://www.pymol.org (accessed on 28 December 2019).
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.B.; Meyer, E.F.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The protein data bank: A computer-based archival file for macromolecular structures. Arch. Biochem. Biophys. 1978, 185, 584–591. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- WaveMetrics Inc. IGOR Pro, 6.36; WaveMetrics Inc.: Tigard, OR, USA, 2014. [Google Scholar]
- Arai, M.; Ferreon, J.C.; Wright, P.E. Quantitative analysis of multisite protein-ligand interactions by NMR: Binding of intrinsically disordered p53 transactivation subdomains with the TAZ2 domain of CBP. J. Am. Chem. Soc. 2012, 134, 3792–3803. [Google Scholar] [CrossRef]
- Tanaka, D.; Tsuda, Y.; Shiyama, T.; Nishimura, T.; Chiyo, N.; Tominaga, Y.; Sawada, N.; Mimoto, T.; Kusunose, N. A practical use of ligand efficiency indices out of the fragment-based approach: Ligand efficiency-guided lead identification of soluble epoxide hydrolase inhibitors. J. Med. Chem. 2011, 54, 851–857. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Abadzapatero, C.; Metz, J. Ligand efficiency indices as guideposts for drug discovery. Drug Discov. Today 2005, 10, 464–469. [Google Scholar] [CrossRef]
- Johnson, S.; Barile, E.; Farina, B.; Purves, A.; Wei, J.; Chen, L.H.; Shiryaev, S.; Zhang, Z.; Rodionova, I.; Agrawal, A.; et al. Targeting Metalloproteins by Fragment-Based Lead Discovery. Chem. Biol. Drug Des. 2011, 78, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, N.-N.; Yin, P.-D.; Cui, P.-X.; Zhou, C.-Z. Glutathionylation-triggered conformational changes of glutaredoxin Grx1 from the yeast Saccharomyces cerevisiae. Proteins 2008, 72, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Yin, S.; Dokholyan, N.V. Rapid flexible docking using a stochastic rotamer library of ligands. J. Chem. Inf. Model. 2010, 50, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided. Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE). Available online: https://www.cell.com/heliyon/pdf/S2405-8440(18)34930-2.pdf (accessed on 1 August 2013).
Sample Availability: Samples of the compounds are not available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Fragment | Selectivity | BrmGRX Kd ± S.D. (mM) | hGRX1 Kd ± S.D. (mM) | PaGRX Kd ± S.D. (mM) | BrmGRX L.E. | hGRX1 L.E. | PaGRX L.E. |
|---|---|---|---|---|---|---|---|---|
 | RK207 | BrmGRX | 0.98 ± 0.24 | 2.45 ± 0.52 | 1.90 ± 0.34 | 0.29 | 0.25 | 0.27 |
 | RK144 | BrmGRX, PaGRX | 5.24 ± 0.92 | 5.42 ± 0.9 | 1.92 ± 0.42 | 0.22 | 0.22 | 0.26 |
 | RK246 | BrmGRX, PaGRX | 10.63 ± 5.18 | 5.22 ± 1.16 | 8.48 ± 2.06 | 0.18 | 0.21 | 0.19 |
 | RK395 | PaGRX | 5.49 ± 3.22 | 1.86 ± 1.15 | 0.51 ± 0.37 | 0.22 | 0.27 | 0.32 |
 | RK104 | hGRX1 | 0.90 ± 0.15 | 2.89 ± 0.47 | 0.72 ± 0.16 | 0.32 | 0.27 | 0.33 |
 | RK208 | hGRX1, PaGRX | 2.28 ± 0.30 | 1.61 ± 0.19 | 0.56 ± 0.08 | 0.26 | 0.27 | 0.32 |
 | RK192 | hGRX1, PaGRX | 5.56 ± 0.51 | 5.05 ± 1.27 | 5.26 ± 1.12 | 0.22 | 0.22 | 0.22 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khattri, R.B.; Morris, D.L.; Bilinovich, S.M.; Manandhar, E.; Napper, K.R.; Sweet, J.W.; Modarelli, D.A.; Leeper, T.C. Identifying Ortholog Selective Fragment Molecules for Bacterial Glutaredoxins by NMR and Affinity Enhancement by Modification with an Acrylamide Warhead. Molecules 2020, 25, 147. https://doi.org/10.3390/molecules25010147
Khattri RB, Morris DL, Bilinovich SM, Manandhar E, Napper KR, Sweet JW, Modarelli DA, Leeper TC. Identifying Ortholog Selective Fragment Molecules for Bacterial Glutaredoxins by NMR and Affinity Enhancement by Modification with an Acrylamide Warhead. Molecules. 2020; 25(1):147. https://doi.org/10.3390/molecules25010147
Chicago/Turabian StyleKhattri, Ram B., Daniel L. Morris, Stephanie M. Bilinovich, Erendra Manandhar, Kahlilah R. Napper, Jacob W. Sweet, David A. Modarelli, and Thomas C. Leeper. 2020. "Identifying Ortholog Selective Fragment Molecules for Bacterial Glutaredoxins by NMR and Affinity Enhancement by Modification with an Acrylamide Warhead" Molecules 25, no. 1: 147. https://doi.org/10.3390/molecules25010147
APA StyleKhattri, R. B., Morris, D. L., Bilinovich, S. M., Manandhar, E., Napper, K. R., Sweet, J. W., Modarelli, D. A., & Leeper, T. C. (2020). Identifying Ortholog Selective Fragment Molecules for Bacterial Glutaredoxins by NMR and Affinity Enhancement by Modification with an Acrylamide Warhead. Molecules, 25(1), 147. https://doi.org/10.3390/molecules25010147