
Substituted N-(Pyrazin-2-yl)benzenesulfonamides; Synthesis, Anti-Infective Evaluation, Cytotoxicity, and In Silico Studies

 , , , , , ,
, , , , , ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Lipophilicity
2.3. Acidobasic Properties
2.4. Biological Activity
2.4.1. Antimycobacterial Activity Evaluation against Mycobacterium tuberculosis, Mycobacterium kansasii, and Mycobacterium avium
2.4.2. Antimycobacterial Activity Evaluation against Mycobacterium smegmatis and Mycobacterium aurum
2.4.3. Antibacterial and Antifungal Activity Evaluation
2.4.4. In Vitro Cytotoxicity Assays
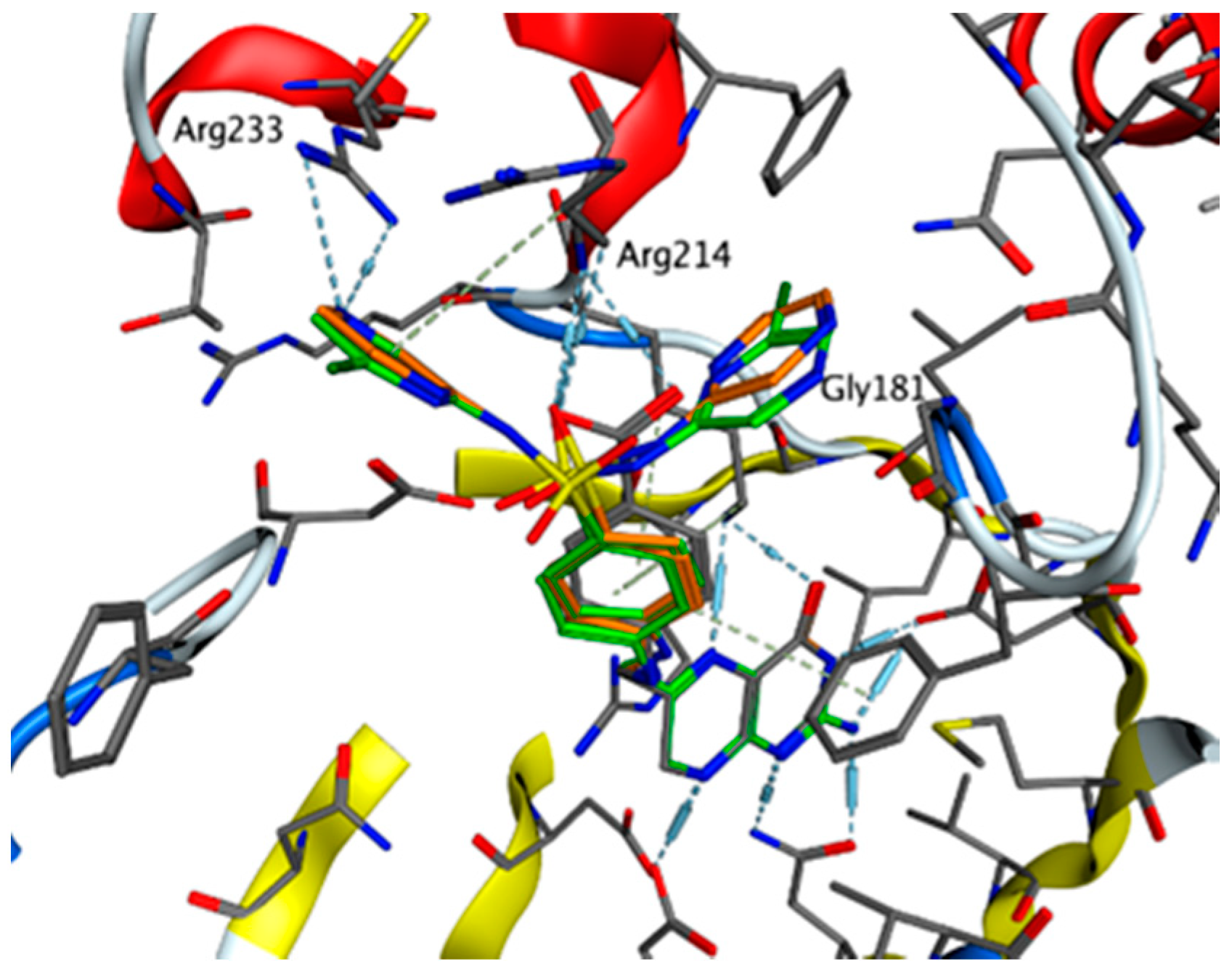
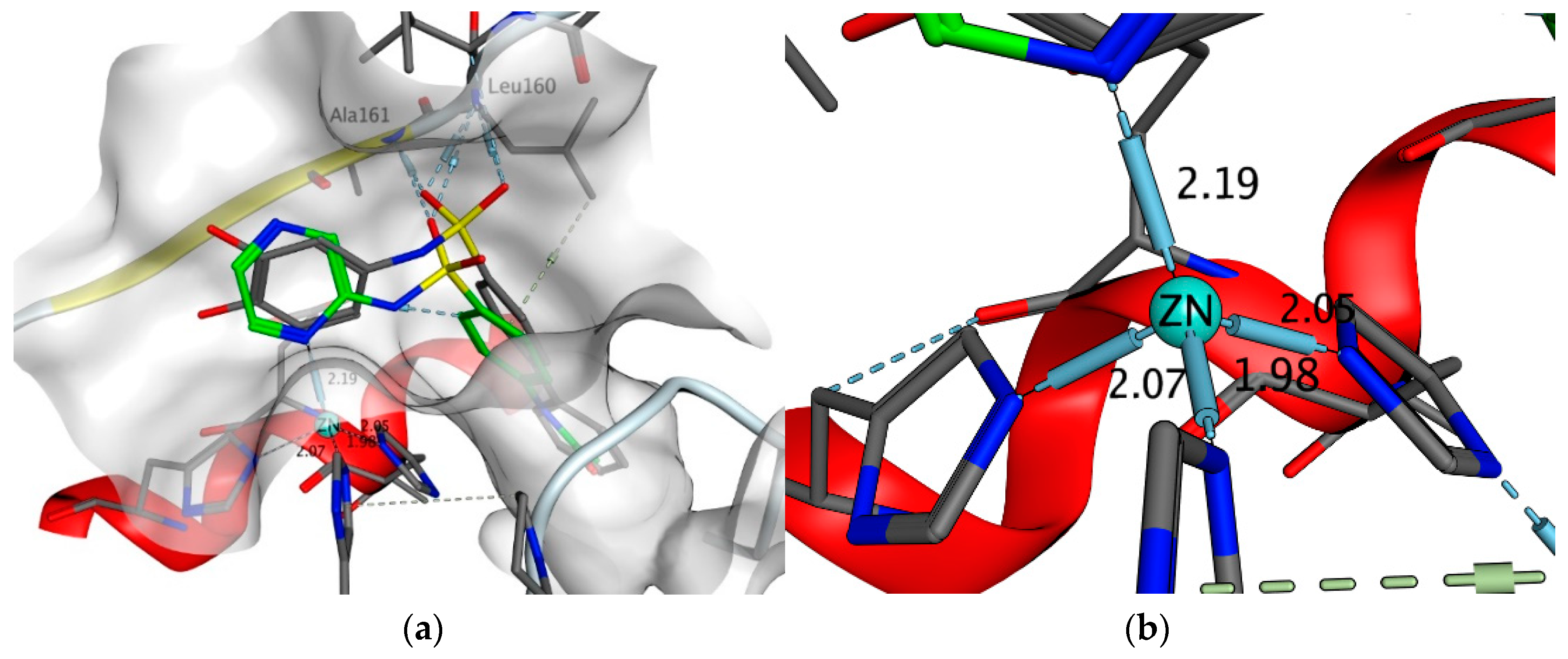
2.4.5. Exploring Potential Targets and Activities using Docking Studies
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. Synthesis of Final Compounds, General Procedure
3.2.2. Compounds 4a and 4b
3.3. Analytical Data of the Prepared Compounds
3.4. Log k Determination
3.5. pKa Determination
3.6. Biological Assays
3.7. In Silico Studies
3.7.1. DHPS Docking
3.7.2. Target Fishing
3.7.3. Other Targets Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2018. WHO/CDC/TB/2018.20. Available online: http://www.who.int/tb/publications/global_report/en/ (accessed on 20 June 2019).
- Zitko, J.; Servusová, B.; Paterová, P.; Mandíková, J.; Kubíček, V.; Kučera, R.; Hrabcová, V.; Kuneš, J.; Soukup, O.; Doležal, M. Synthesis, antimycobacterial activity and in vitro cytotoxicity of 5-chloro-N-phenylpyrazine-2-carboxamides. Molecules 2013, 18, 14807–14825. [Google Scholar] [CrossRef] [PubMed]
- Zitko, J.; Barbora, S.V.; Paterová, P.; Navrátilová, L.; Trejtnar, F.; Kuneš, J.; Doležal, M. Design, synthesis and anti-mycobacterial evaluation of some new N-phenylpyrazine-2-carboxamides. Chem. Pap. 2016, 70, 649–657. [Google Scholar] [CrossRef]
- Zitko, J.; Mindlová, A.; Valášek, O.; Jand’ourek, O.; Paterová, P.; Janoušek, J.; Konečná, K.; Doležal, M. Design, Synthesis and Evaluation of N-pyrazinylbenzamides as Potential Antimycobacterial Agents. Molecules 2018, 23, 2390. [Google Scholar] [CrossRef] [PubMed]
- Alafeefy, A.; Alqasoumi, S.; Ashour, A.; Alshebly, M. Quinazoline-sulfonamides as potential antitumor agents: Synthesis and biological testing. J. Enzym. Inhib. Med. Chem. 2013, 28, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Kumar, R. Novel inhibitors of cyclooxygenase-2: The sulfones and sulfonamides of 1,2-diaryl-4,5-difluorobenzene. Analysis of quantitative structure-activity relationship. J. Enzym. Inhib. 1998, 13, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Focken, T.; Burford, K.; Grimwood, M.E.; Zenova, A.; Andrez, J.-C.; Gong, W.; Wilson, M.; Taron, M.; Decker, S.; Lofstrand, V.; et al. Identification of CNS-Penetrant Aryl Sulfonamides as Isoform-Selective NaV1.6 Inhibitors with Efficacy in Mouse Models of Epilepsy. J. Med. Chem. 2019, 62, 9618–9641. [Google Scholar] [CrossRef]
- Chohan, Z.; Shad, H. Metal-based new sulfonamides: Design, synthesis, antibacterial, antifungal, and cytotoxic properties. J. Enzym. Inhib. Med. Chem. 2012, 27, 403–412. [Google Scholar] [CrossRef]
- Doležal, M.; Zitko, J. Pyrazine derivatives: A patent review (June 2012—present). Expert Opin. Ther. Pat. 2015, 25, 33–47. [Google Scholar] [CrossRef]
- Shepard, D.; Dreicer, R. Zibotentan for the treatment of castrate-resistant prostate cancer. Expert Opin. Investig. Drugs 2010, 19, 899–908. [Google Scholar] [CrossRef]
- Baxter, A.; Johnson, T.; Kindon, N.; Roberts, B.; Stocks, M. N-Pyrazinyl-Phenyl-Phenylsulphonamides and Their Use in the Treatment of Chemokine Mediated Diseases. WO Patent 2003/059893, 24 July 2003. [Google Scholar]
- Aleksa, V.; Prozorova, V. Pharmacokinetics and the clinical use of sulfalene. Sov. Meditsina 1985, 10, 106–108. [Google Scholar]
- Forgacs, P.; Wengenack, N.; Hall, L.; Zimmerman, S.; Silverman, M.; Roberts, G. Tuberculosis and Trimethoprim-Sulfamethoxazole. Antimicrob. Agents Chemother. 2009, 53, 4789–4793. [Google Scholar] [CrossRef] [PubMed]
- Palomino, J.; Martin, A. The potential role of trimethoprim-sulfamethoxazole in the treatment of drug-resistant tuberculosis. Future Microbiol. 2016, 11, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Burchall, J. Mechanism of action of trimethoprim-sulfamethoxazole. II. J. Infect. Dis. 1973, 128 (Suppl. S3), S437–S441. [Google Scholar] [CrossRef]
- Hitchings, G. Mechanism of action of trimethoprim-sulfamethoxazole. I. J. Infect. Dis. 1973, 128 (Suppl. S3), S433–S436. [Google Scholar] [CrossRef]
- Alderwick, L.; Harrison, J.; Lloyd, G.; Birch, H. The Mycobacterial Cell Wall--Peptidoglycan and Arabinogalactan. Cold Spring Harb. Perspect. Med. 2015, 5, a021113. [Google Scholar] [CrossRef] [PubMed]
- Doležal, M.; Palek, L.; Vinsova, J.; Buchta, V.; Jampilek, J.; Kralova, K. Substituted pyrazinecarboxamides: Synthesis and biological evaluation. Molecules 2006, 11, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Servusova-Vanaskova, B.; Paterová, P.; Garaj, V.; Mandíková, J.; Kunes, J.; Naesens, L.; Jílek, P.; Doležal, M.; Zitko, J. Synthesis and Antimicrobial Evaluation of 6-Alkylamino-N-phenylpyrazine-2-carboxamides. Chem. Boil. Drug Des. 2015, 86, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Tostmann, A.; Boeree, M.J.; Peters, W.H.; Roelofs, H.M.; Aarnoutse, R.E.; Van Der Ven, A.J.; Dekhuijzen, P.R. Isoniazid and its toxic metabolite hydrazine induce in vitro pyrazinamide toxicity. Int. J. Antimicrob. Agents 2008, 31, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE); 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2019.
- Bell, P.H.; Richard, O.R., Jr. Studies in Chemotherapy. VII. A Theory of the Relation of Structure to Activity of Sulfanilamide Type Compounds1. J. Am. Chem. Soc. 1942, 64, 2905–2917. [Google Scholar] [CrossRef]
- Bamford, M.; Dean, D.; Naylor, A.; Takle, A.; Wilson, D. Nitrogen-Containing Heterocyclic Compounds as Inhibitors of B-Raf Kinase. U.S. Patent 7297693b2, 20 November 2007. [Google Scholar]
- Bengtsson, M.; Larsson, J.; Nikitidis, G.; Storm, P.; Bailey, J.P.; Griffen, E.J.; Arnould, J.-C.; Bird, T.G.C. Preparation of 5-Heteroaryl Thiazoles and Their Use as Phosphoinositide 3-Kinase (PI3K) Inhibitors. WO Patent 2006051270A1, 18 May 2006. [Google Scholar]
- Dea-Ayuela, M.A.; Castillo, E.; González-Álvarez, M.; Vega, C.; Rolon, M.; Bolás-Fernández, F.; Borras, J.; González-Rosende, M.E. In vivo and in vitro anti-leishmanial activities of 4-nitro-N-pyrimidin- and N-pyrazin-2-ylbenzenesulfonamides, and N2-(4-nitrophenyl)-N1-propylglycinamide. Bioorganic Med. Chem. 2009, 17, 7449–7456. [Google Scholar] [CrossRef]
- Coatney, G.R.; Cooper, W.C.; Young, M.D.; Burgess, R.W.; Smarr, R.G. Studies in human malaria: II. the suppressive action of sulfadiazine and sulfapyrazine against sporozoite-induced vivax malaria (St. Elizabeth strain). Am. J. Hyg. 1947, 46, 105–118. [Google Scholar] [PubMed]
- Coatney, G.R.; Cooper, W.C.; Young, M.D.; Mclendon, S.B. Studies in Human Malaria: I. The Protective Action of Sulfadiazine and Sulfapyrazine against Sporozoite—Induced Falciparum Malaria. Am. J. Hyg. 1947, 46, 84–104. [Google Scholar]
- Kuenzel, W.; Kirby, J. Method for the Stimulation of Sperm Production and Gonadal Development in Animals. WO Patent 1999038376A1, 5 August 1999. [Google Scholar]
- Modir, S.; Mansouri, B.; Kiaeis, S. The effect of sulfaclozine 30% (Esb3) on experimental coccidiosis in broiler cockerels. Iran. J. Vet. Res. 2009, 5, 62–69. [Google Scholar]
- Sinha, S. Sulfonamide derivatives as Mycobacterium tuberculosis inhibitors: In silico approach. In Silico Pharmacol. 2018, 6, 4. [Google Scholar]
- Ayers, K. Structural and Kinetics Studies of the Enzyme Dihydropteroate Synthase and the Implications for Antibiotic Resistance. Master’s Thesis, University of Tennessee Health Science Center, Memphis, TN, USA, 2009. Theses and Dissertations (ETD). Paper 333. [Google Scholar] [CrossRef]
- Nopponpunth, V.; Sirawaraporn, W.; Greene, P.; Santi, D. Cloning and expression of Mycobacterium tuberculosis and Mycobacterium leprae dihydropteroate synthase in Escherichia coli. J. Bacteriol. 1999, 181, 6814–6821. [Google Scholar]
- Yun, M.-K.; Wu, Y.; Li, Z.; Zhao, Y.; Waddell, M.B.; Ferreira, A.M.; Lee, R.E.; Bashford, N.; White, S.W. Catalysis and Sulfa Drug Resistance in Dihydropteroate Synthase. Science 2012, 335, 1110–1114. [Google Scholar] [CrossRef]
- Hammoudeh, D.; Zhao, Y.; White, S.; Lee, R. Replacing sulfa drugs with novel DHPS inhibitors. Future Med. Chem. 2013, 5, 1331–1340. [Google Scholar] [CrossRef]
- Chotpatiwetchkul, W.; Boonyarattanakalin, K.; Gleeson, D.; Gleeson, M. Exploring the catalytic mechanism of dihydropteroate synthase: Elucidating the differences between the substrate and inhibitor. Org. Biomol. Chem. 2017, 15, 5593–5601. [Google Scholar] [CrossRef]
- Anand, N. Mechanism of Action of Antimicrobial and Antitumor Agents; Corcoran, J.W., Hahn, F., Snell, J., Arora, K., Eds.; Springer: Berlin/Heidelberg, Germany, 1975; pp. 668–698. [Google Scholar]
- Akopian, T.; Kandror, O.; Tsu, C.; Lai, J.H.; Wu, W.; Liu, Y.; Zhao, P.; Park, A.; Wolf, L.; Dick, L.R.; et al. Cleavage Specificity of Mycobacterium tuberculosis ClpP1P2 Protease and Identification of Novel Peptide Substrates and Boronate Inhibitors with Anti-bacterial Activity. J. Boil. Chem. 2015, 290, 11008–11020. [Google Scholar] [CrossRef]
- Draper, P. The aliphatic acylamide amidohydrolase of Mycobacterium smegmatis: Its inducible nature and relation to acyl-transfer to hydroxylamine. J. Gen. Microbiol. 1967, 46, 111–123. [Google Scholar] [CrossRef]
- Hamburger, M., Jr.; Ruegsegger, J.; Brookens, N.; Eakin, E. The Absorption, Excretion, and Distribution of 2-Sulfanilamidopyrazine(Sulfapyrazine)in Man. Am. J. Med Sci. 1942, 204, 186–193. [Google Scholar] [CrossRef]
- Liu, C.; Wang, S.; Zhang, Q.; Shao, Y. Influence of three coccidiostats on the pharmacokinetics of florfenicol in rabbits. Exp. Anim. 2015, 64, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.; De Simone, G. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [PubMed]
- EUCAST DISCUSSION DOCUMENT E.Dis 5.1. Determination of Minimum Inhibitory Concentrations (MICs) of Antibacterial Agents by Broth Dilution. Clin. Microbiol. Infect. 2003, 9, 1–7. [CrossRef]
- Tauro, M.; Laghezza, A.; Loiodice, F.; Piemontese, L.; CaraDonna, A.; Capelli, D.; Montanari, R.; Pochetti, G.; Di Pizio, A.; Agamennone, M.; et al. Catechol-based matrix metalloproteinase inhibitors with additional antioxidative activity. J. Enzym. Inhib. Med. Chem. 2016, 31, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Ogryzek, M.; Chylewska, A.; Turecka, K.; Lesiak, D.; Królicka, A.; Banasiuk, R.; Nidzworski, D.; Makowski, M. Coordination chemistry of pyrazine derivatives analogues of PZA: Design, synthesis, characterization and biological activity. RSC Adv. 2016, 6, 52009–52025. [Google Scholar] [CrossRef]
- Pedersen, K.S.; Perlepe, P.; Aubrey, M.L.; Woodruff, D.N.; Reyes-Lillo, S.E.; Reinholdt, A.; Voigt, L.; Li, Z.; Borup, K.; Rouzières, M.; et al. Formation of the layered conductive magnet CrCl2(pyrazine)(2) through redox-active coordination chemistry. Nat. Chem. 2018, 10, 1056–1061. [Google Scholar] [CrossRef]
- McDonald, R. Pyrazine Chemistry. I. Derivatives of 3-Aminopyrazinoic Acid. J. Am. Chem. Soc. 1945, 67, 1711–1713. [Google Scholar]
Sample Availability: Samples of compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a | b | IC50 (µM) | |||||
|---|---|---|---|---|---|---|---|
 |  | ||||||
| No. | R (the Cut Line Represents the Attachment Point) | Antimycobacterial Activity MIC (µg/mL) | |||||
| Mtb H37Rv | M. kansasii | M. avium | M. smeg | M. aurum | |||
| 1a |  | >100 | 100 | >100 | ≥250 | ≥500 | >1000 |
| 1b | 50 | 100 | >100 | 250 | ≥500 | 731 | |
| 2a |  | >100 | 25 | >100 | ≥500 | ≥500 | >500 * |
| 2b | >100 | 25 | >100 | 250 | ≥500 | >500 * | |
| 3a |  | >100 | >100 | >100 | >500 | >500 | >1000 |
| 3b | >100 | >100 | >100 | 250 | 125 | >1000 | |
| 4a |  | 6.25 | 1.56 | 100 | ≥500 | 250 | 775.6 * |
| 4b *** | 6.25 | 12.5 | 25 | ≥500 | 250 | >500 | |
| 5a |  | >100 | 50 | >100 | 250 | 250 | >1000 |
| 5b | 100 | >100 | >100 | 250 | 250 | 364 | |
| 6a |  | >100 | >100 | >100 | ≥500 | ≥500 | >1000 |
| 6b | >100 | 100 | >100 | ≥500 | ≥500 | >1000 | |
| 7b |  | 100 | 50 | >100 | 125 | 125 | 308 |
| 8a |  | >100 | 50 | >100 | ≥500 | 250 | >500 * |
| 8b | 50 | >100 | 50 | ≥500 | ≥500 | 523.3 | |
| 9a |  | >100 | >100 | >100 | 125 | ≥500 | >250 * |
| 9b | >100 | 50 | >100 | ≥500 | ≥500 | 179.5 | |
| 10a |  | >100 | 50 | >100 | ≥500 | ≥500 | >250 * |
| 11a |  | >100 | >100 | >100 | ≥500 | 250 | >1000 |
| 11b | >100 | 25 | >100 | ≥500 | ≥500 | 778.5 | |
| 12a |  | 100 | 100 | >100 | ≥500 | ≥500 | >250 * |
| 12b | 100 | 12.5 | >100 | ≥500 | ≥500 | 302 | |
| 13a |  | >100 | 50 | >100 | 62.5 | ≥500 | >500 * |
| 13b | 50 | 50 | >100 | 125 | 250 | >1000 | |
| 14a |  | >100 | 100 | >100 | ≥500 | ≥500 | >250 * |
| Sulfamethoxazole | 3.13 | 3.13 | 12.5 | 250 | ≥500 | n.t. | |
| Sulfaclozine | 6.25 | 12.5 | 25 | 250 | 250 | n.t. | |
| Sulfaquinoxaline | 3.13 | 12.5 | 50 | 125 | 250 | n.t. | |
| Benzenesulfonamide | >100 | >100 | >100 | 31.25 | 125 | n.t. | |
| PZA ** | >100 | >100 | >100 | ≥500 | ≥500 | >104 [20] | |
| INH | 0.2 | 25 | 12.5 | 15.625 | 3.91 | 79 × 103 [20] | |
| RFM | n.t. | n.t. | n.t. | 25 | 1.56 | n.t. | |
| CPX | n.t. | n.t. | n.t. | 0.125 | 0.008 | n.t. | |
| Experimental | Calculated | ||||
|---|---|---|---|---|---|
| No. | Absorbance Method | Potentiometric Method | ChemAxon | MOE | ChemDraw |
| 6b | 6.95 | 7.21 | 6.21 | 8.30 | NA |
| 9b | 6.32 | 6.38 | 6.29 | 8.30 | NA |
| 13b | 5.63 | 5.75 | 6.01 | 8.30 | NA |
| No. | pKa | No. | pKa | No. | pKa | No. | pKa | No. | pKa |
|---|---|---|---|---|---|---|---|---|---|
| 1a | 6.11 | 3b | 5.76 | 6a | 6.30 | 9a | 6.39 | 12a | 6.11 |
| 1b | 6.05 | 4a | 6.61 | 6b | 6.21 | 9b | 6.29 | 12b | 6.05 |
| 2a | 6.16 | 4b | 6.49 | 7b | 6.03 | 10a | 6.38 | 13a | 6.07 |
| 2b | 6.09 | 5a | 6.10 | 8a | 6.12 | 11a | 6.16 | 13b | 6.01 |
| 3a | 5.80 | 5b | 6.03 | 8b | 6.05 | 11b | 6.09 | 14a | 6.07 |
| Substituents | Antimycobacterial Activity MIC (µg/mL) against Mtb H37Rv | |||
|---|---|---|---|---|
| R1 | R2 | Amide | Retro-amide | Sulfonamide |
| H | H | 100 | >100 | >100 (1a) |
| H | 4-OCH3 | 25 | >100 | >100 (6a) |
| 6-Cl | H | >100 | 25 | 50 (1b) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouz, G.; Juhás, M.; Pausas Otero, L.; Paredes de la Red, C.; Janďourek, O.; Konečná, K.; Paterová, P.; Kubíček, V.; Janoušek, J.; Doležal, M.; et al. Substituted N-(Pyrazin-2-yl)benzenesulfonamides; Synthesis, Anti-Infective Evaluation, Cytotoxicity, and In Silico Studies. Molecules 2020, 25, 138. https://doi.org/10.3390/molecules25010138
Bouz G, Juhás M, Pausas Otero L, Paredes de la Red C, Janďourek O, Konečná K, Paterová P, Kubíček V, Janoušek J, Doležal M, et al. Substituted N-(Pyrazin-2-yl)benzenesulfonamides; Synthesis, Anti-Infective Evaluation, Cytotoxicity, and In Silico Studies. Molecules. 2020; 25(1):138. https://doi.org/10.3390/molecules25010138
Chicago/Turabian StyleBouz, Ghada, Martin Juhás, Lluis Pausas Otero, Cristina Paredes de la Red, Ondřej Janďourek, Klára Konečná, Pavla Paterová, Vladimír Kubíček, Jiří Janoušek, Martin Doležal, and et al. 2020. "Substituted N-(Pyrazin-2-yl)benzenesulfonamides; Synthesis, Anti-Infective Evaluation, Cytotoxicity, and In Silico Studies" Molecules 25, no. 1: 138. https://doi.org/10.3390/molecules25010138
APA StyleBouz, G., Juhás, M., Pausas Otero, L., Paredes de la Red, C., Janďourek, O., Konečná, K., Paterová, P., Kubíček, V., Janoušek, J., Doležal, M., & Zitko, J. (2020). Substituted N-(Pyrazin-2-yl)benzenesulfonamides; Synthesis, Anti-Infective Evaluation, Cytotoxicity, and In Silico Studies. Molecules, 25(1), 138. https://doi.org/10.3390/molecules25010138