
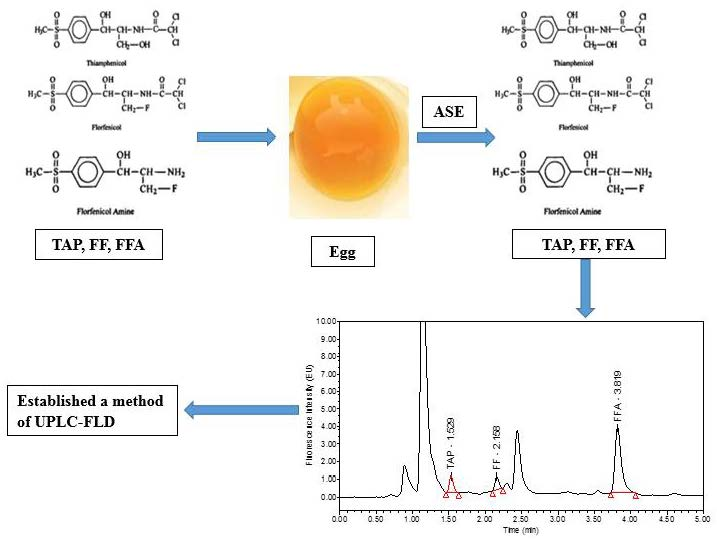
Development of an Accelerated Solvent Extraction-Ultra-Performance Liquid Chromatography-Fluorescence Detection Method for Quantitative Analysis of Thiamphenicol, Florfenicol and Florfenicol Amine in Poultry Eggs


Abstract
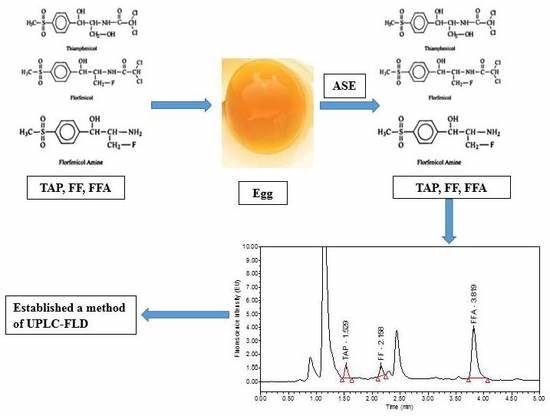
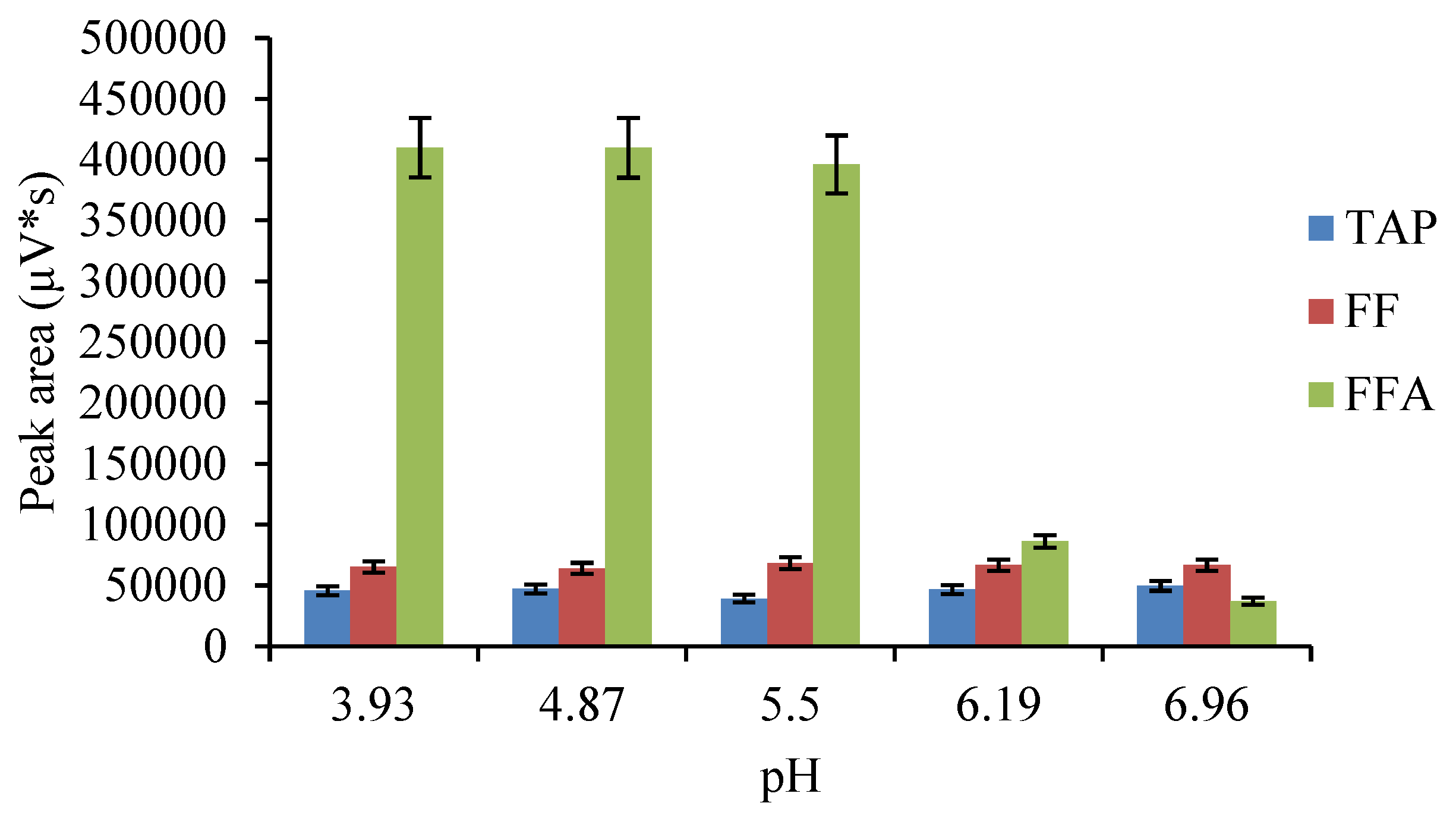
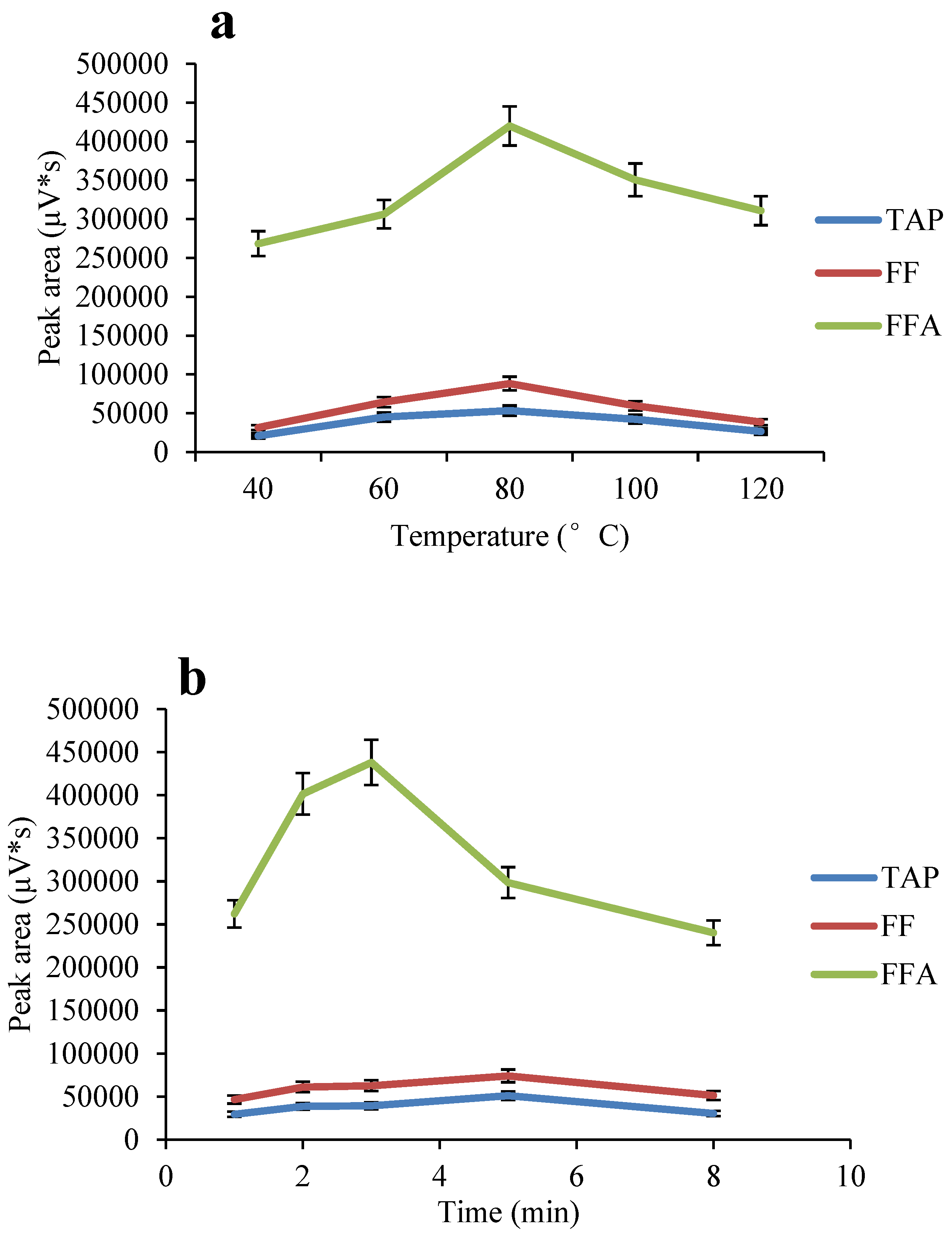
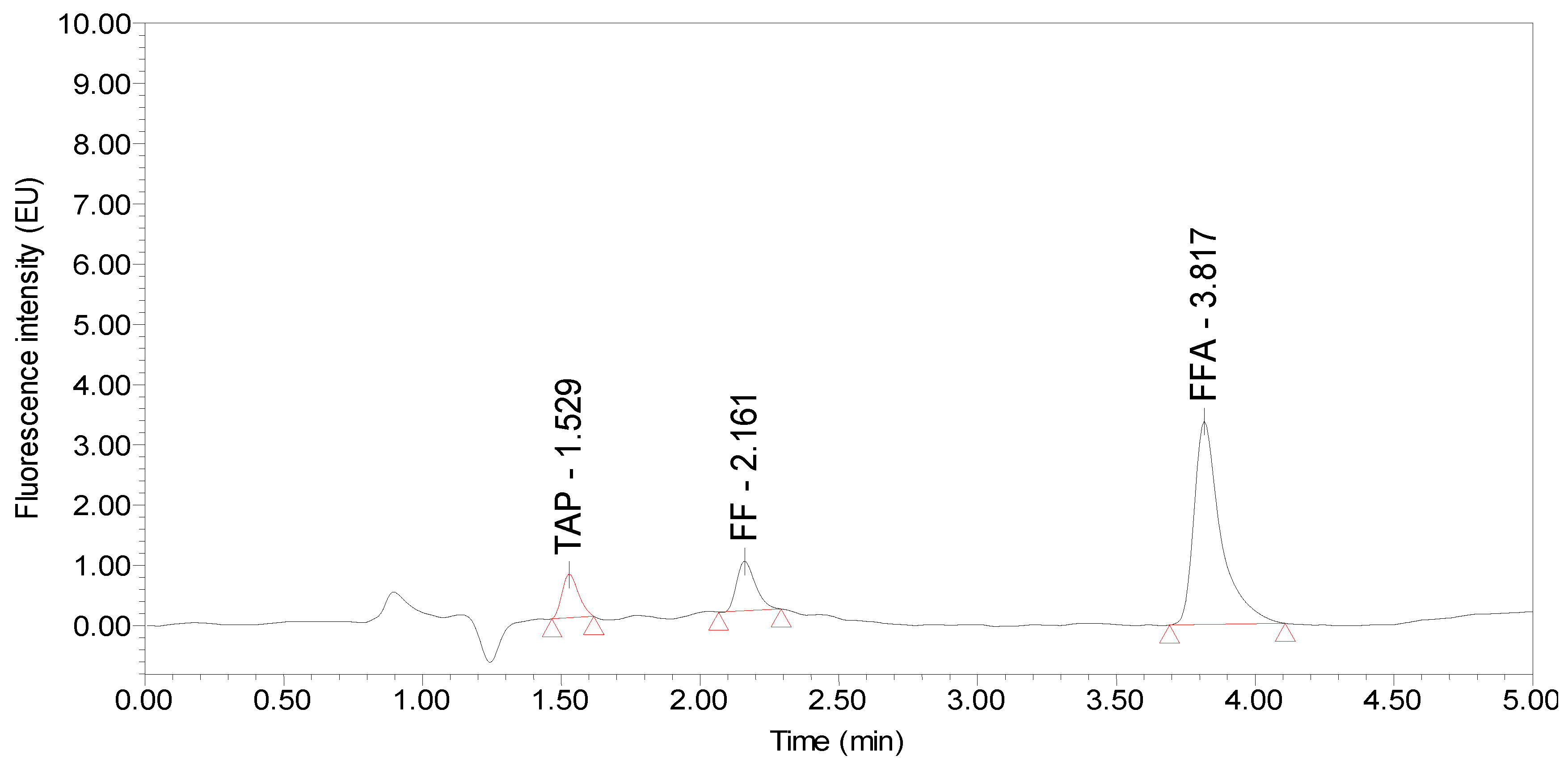
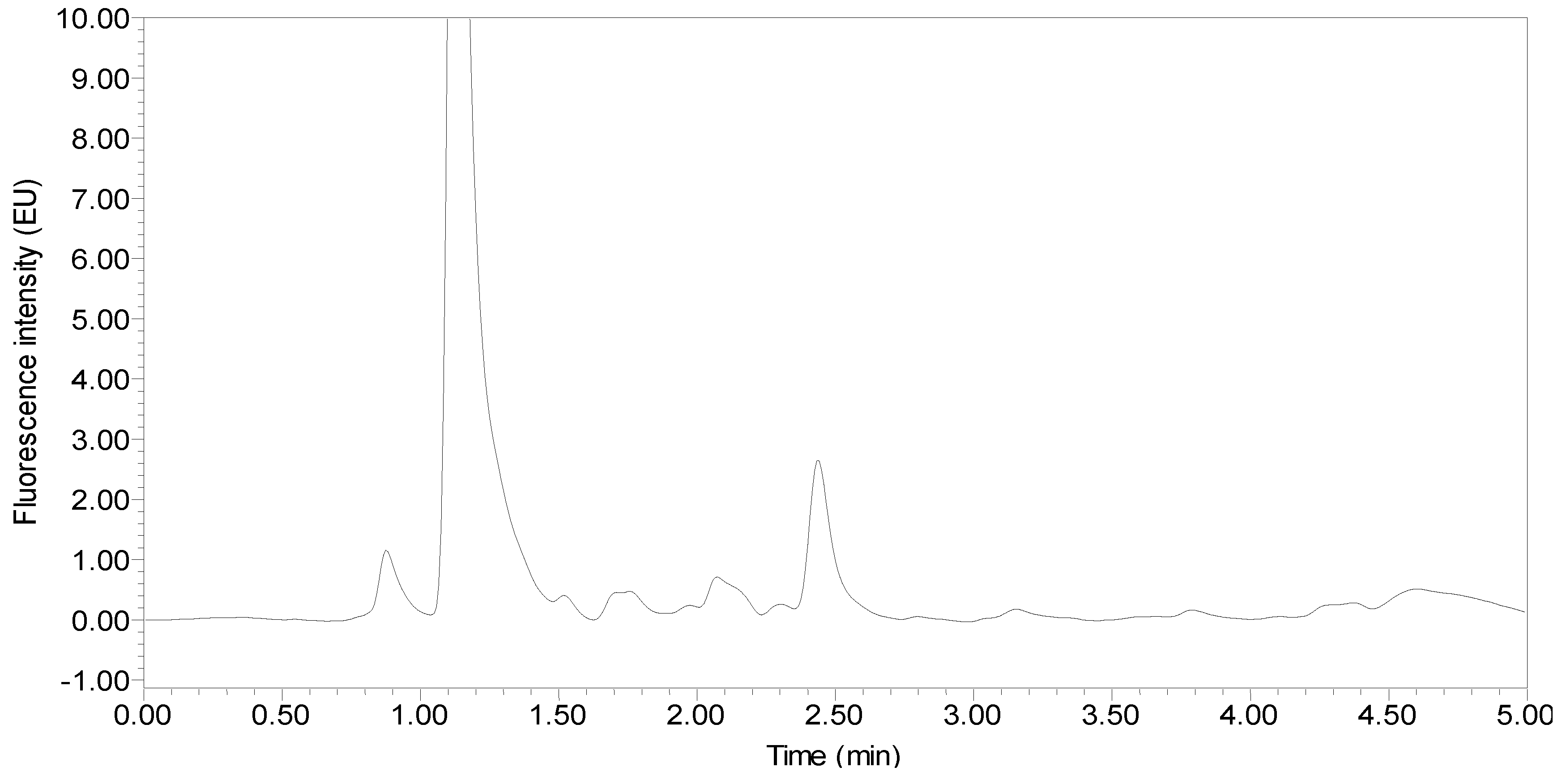
:
1. Introduction
2. Results and Discussion
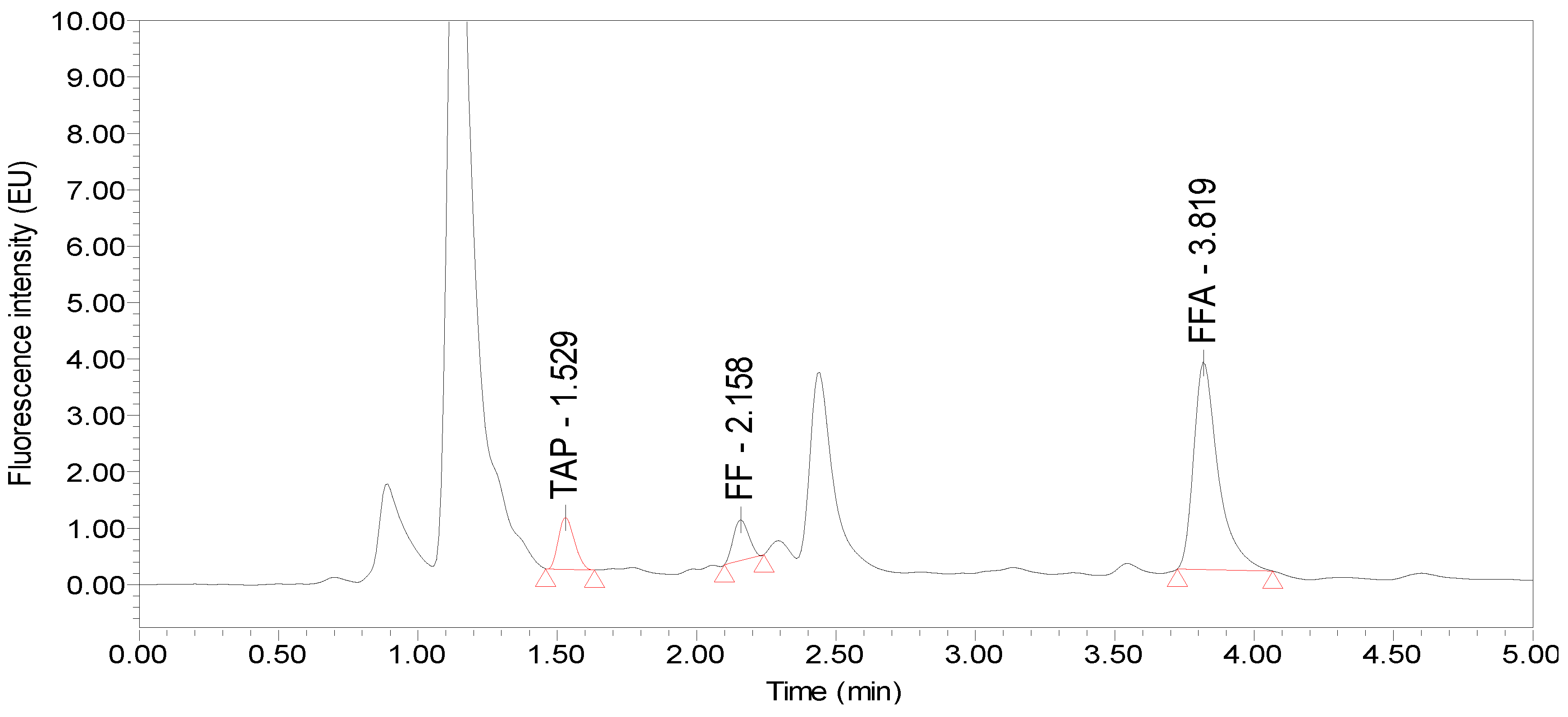
2.1. Selection of the Chromatographic Column and Mobile Phase
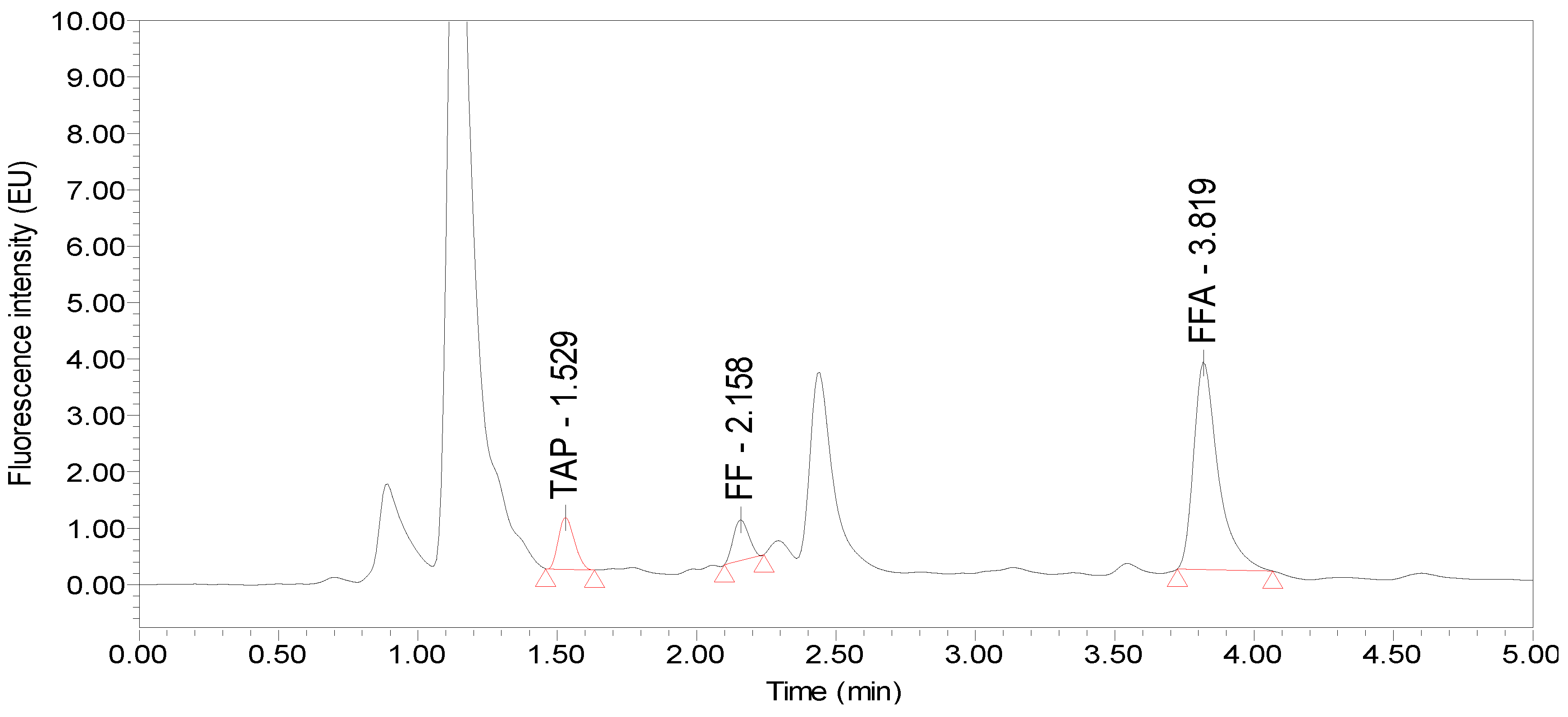
2.2. Determination of the Detection Wavelength
2.3. Selection and Optimization of the Extraction Solvent and Extraction Method
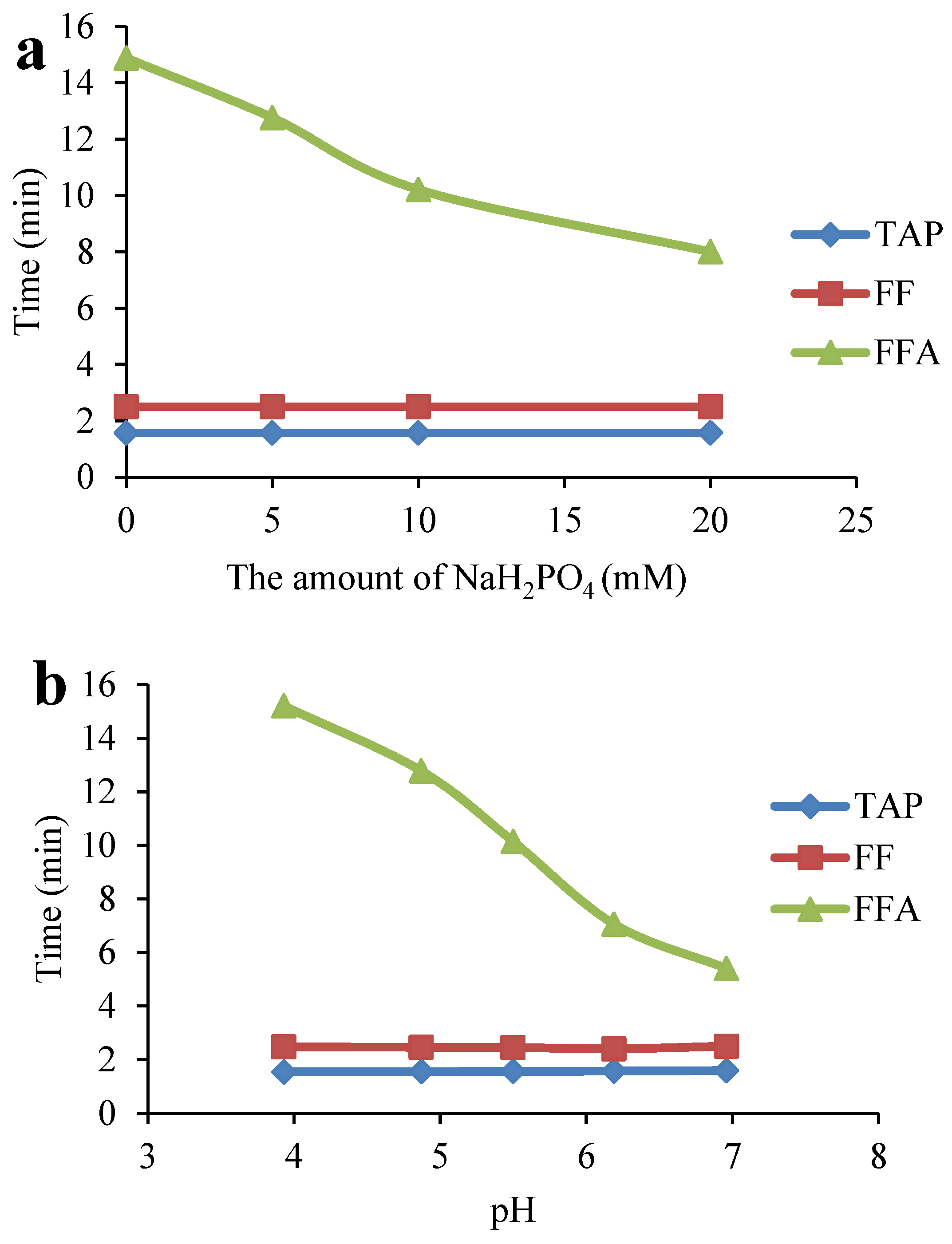
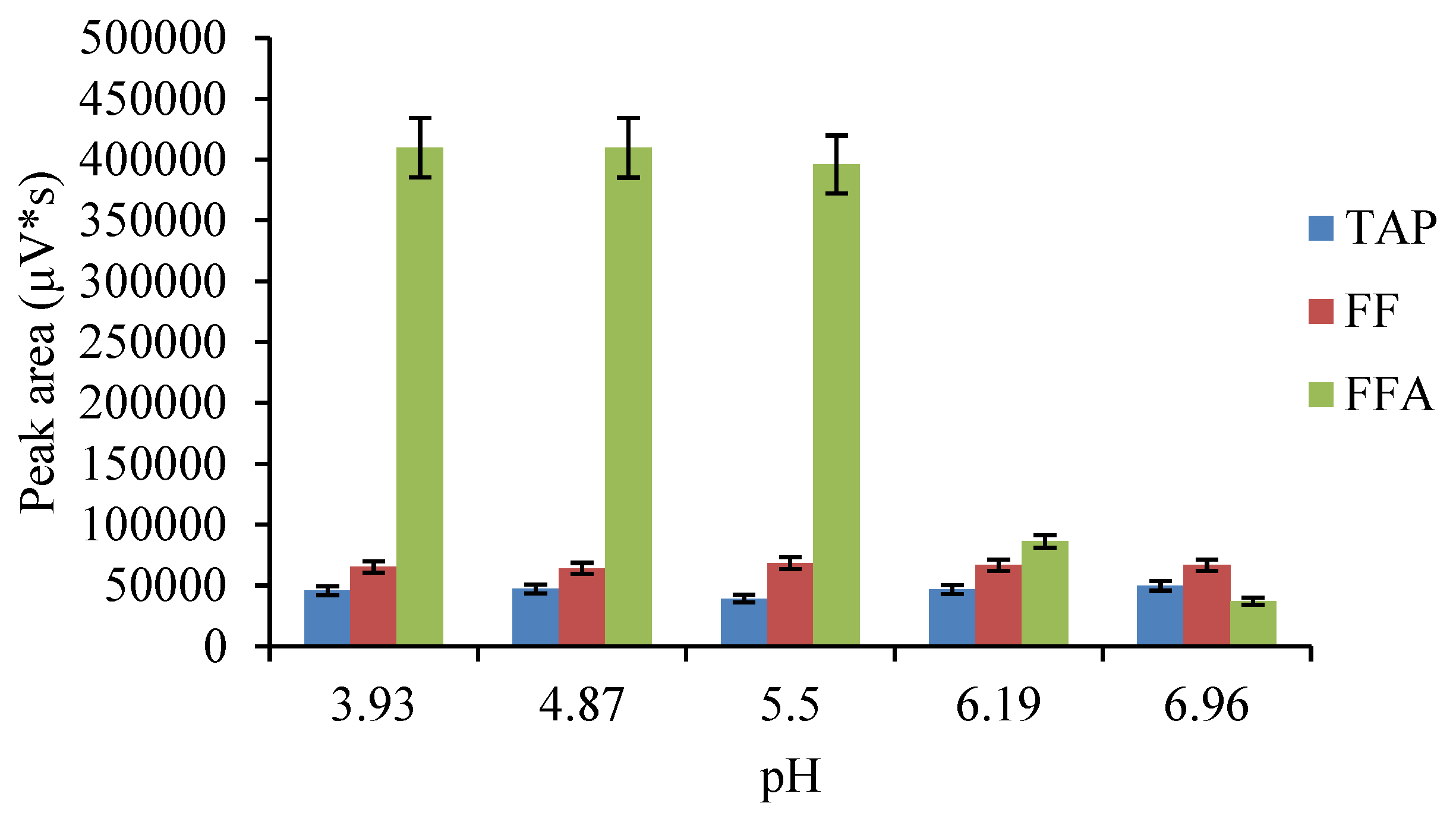
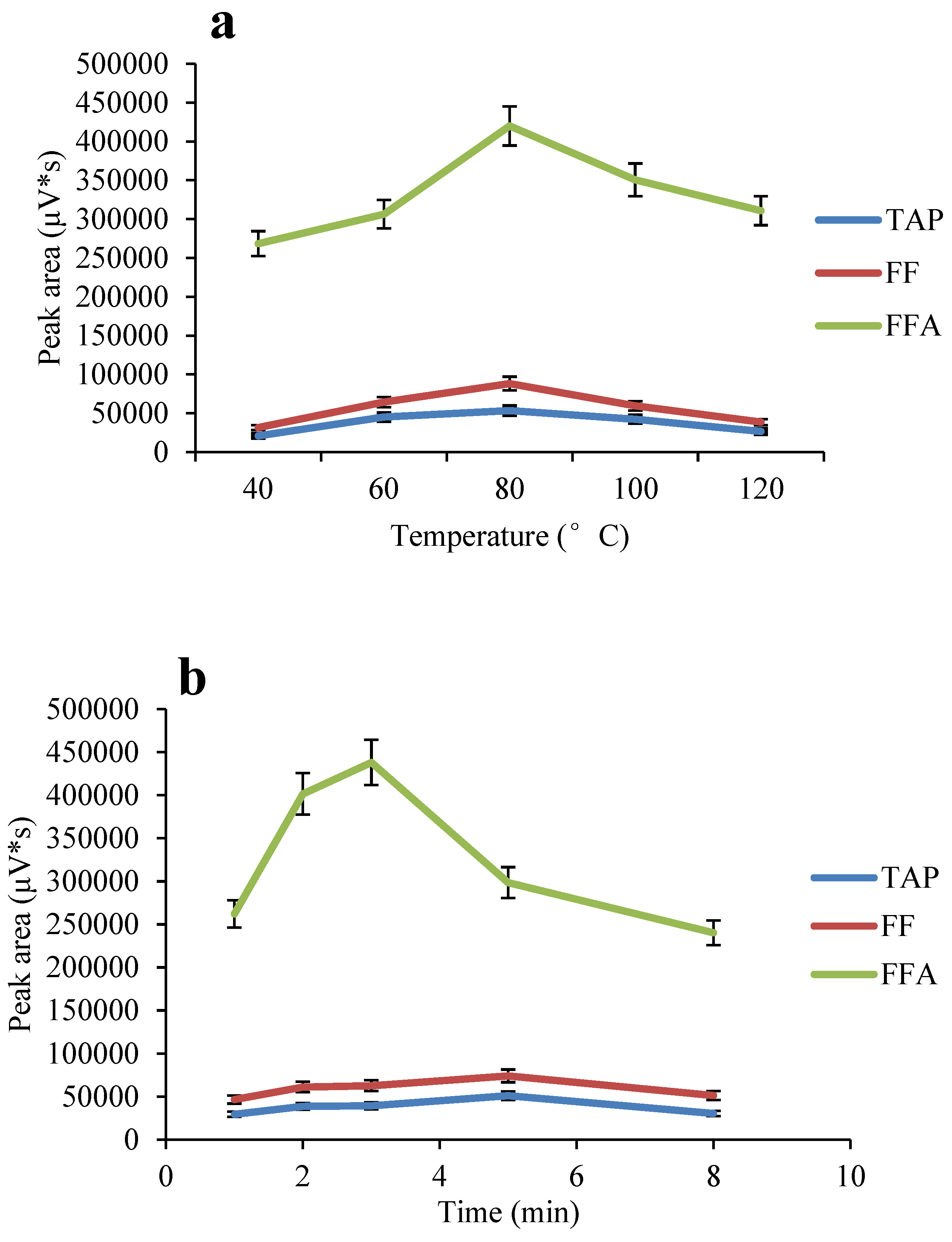
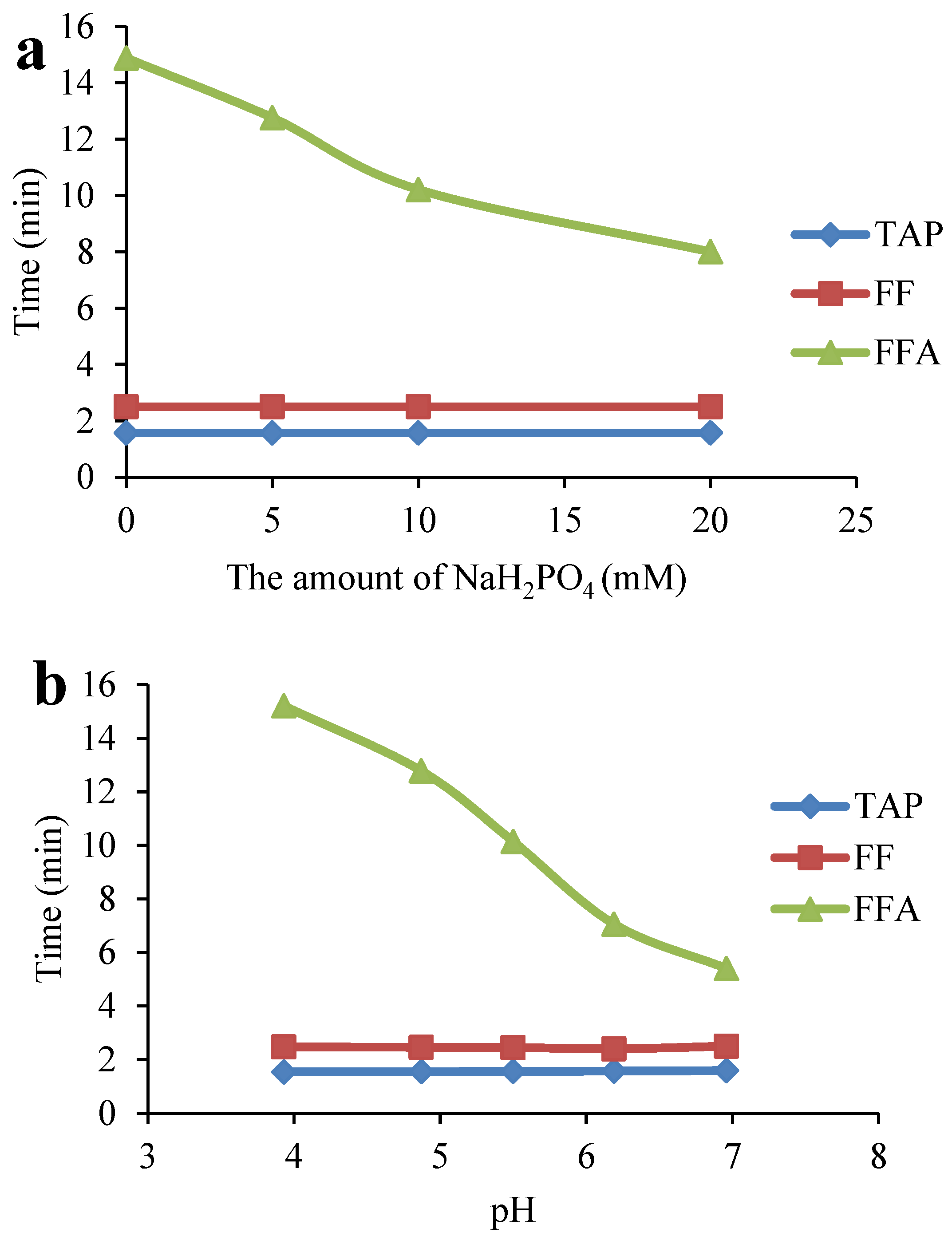
2.4. Optimization of the ASE Method
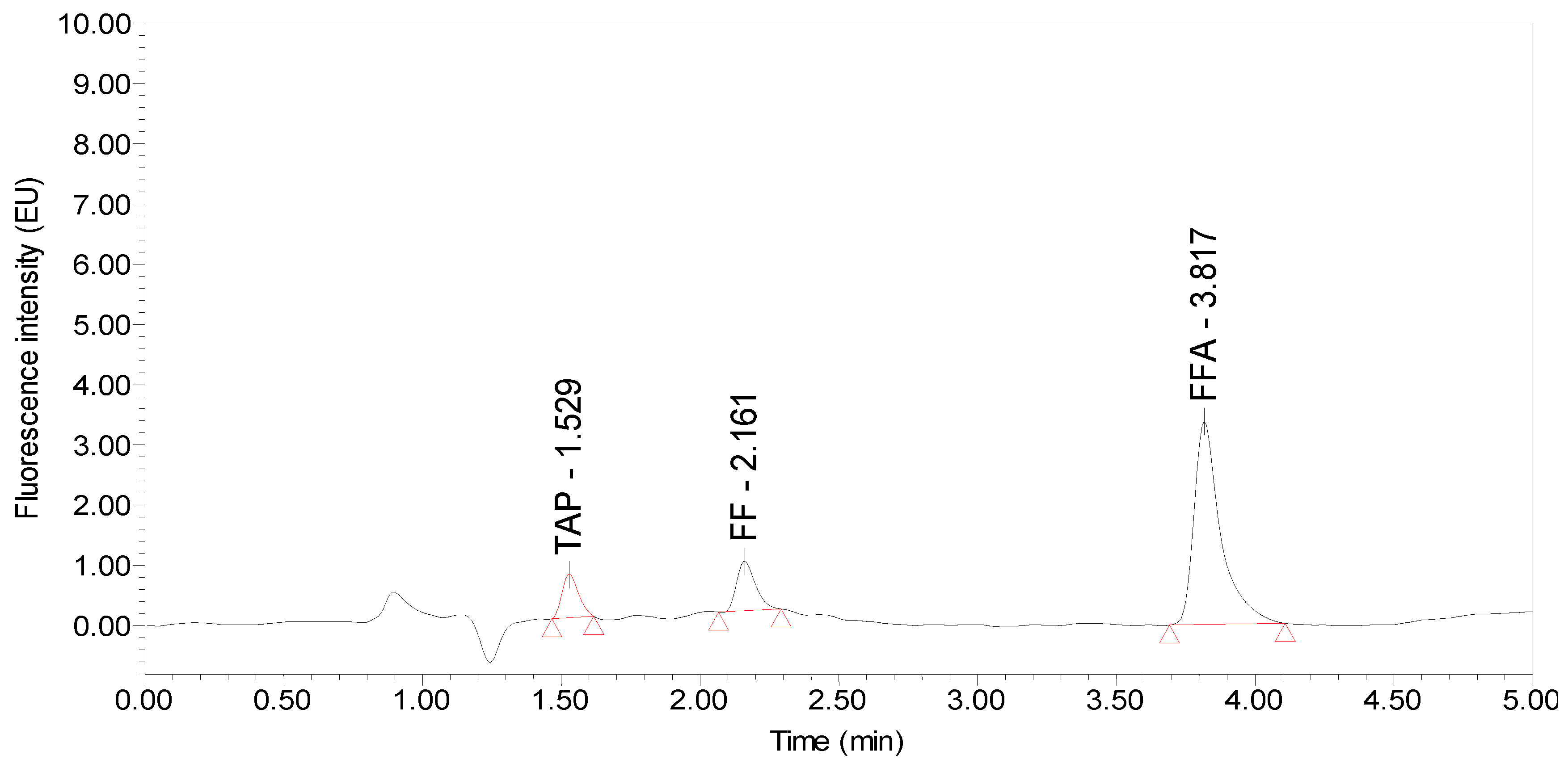
2.5. Bioanalytical Method Validation
2.6. Real Sample Analysis
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Standard and Working Solutions
3.3. UPLC-FLD Instrumentation and Conditions
3.4. Sample Preparation
3.4.1. ASE Extraction
3.4.2. Ultrasonic Extraction
3.4.3. Vortex Shock Extraction
3.4.4. Vortex Oscillating + Ultrasonic Extraction
3.4.5. Sample Purification
3.5. Method Validation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lesnierowski, G.; Stangierski, J. What’s new in chicken egg research and technology for human health promotion? - a review. Trends Food Sci. Technol. 2018, 71, 46–51. [Google Scholar] [CrossRef]
- Ashraf, A.; Rahman, F.A.; Abdullah, N. Poultry feed in Malaysia: an insight into the Halalan Toyyiban issues. In Proceedings of the 3rd International Halal Conference (INHAC 2016), Selangor, Malaysia, 21–22 November 2016; Hashim, N.M., Shariff, N.N.M., Mahamood, S.F., Harun, H.M.F., Shahruddin, M.S., Bhari, A., Eds.; Springer: Singapore, 2018; pp. 511–531. [Google Scholar]
- Tao, X.; He, Z.; Cao, X.; Jiang, H.; Li, H. Approaches for the determination of florfenicol and thiamphenicol in pork using a chemiluminescent ELISA. Anal. Methods 2015, 7, 8386–8392. [Google Scholar] [CrossRef]
- Festing, M.F.; Diamanti, P.; Turton, J.A. Strain differences in haematological response to chloramphenicol succinate in mice: implications for toxicological research. Food Chem. Toxicol. 2001, 39, 375–383. [Google Scholar] [CrossRef]
- Hanekamp, J.C.; Bast, A. Antibiotics exposure and health risks: chloramphenicol. Environ. Toxicol. Pharmacol. 2015, 39, 213–220. [Google Scholar] [CrossRef]
- Hu, D.; Han, Z.; Li, C.; Lv, L.; Cheng, Z.; Liu, S. Florfenicol induces more severe hemotoxicity and immunotoxicity than equal doses of chloramphenicol and thiamphenicol in Kunming mice. Immunopharmacol. Immunotoxicol. 2016, 38, 472–485. [Google Scholar] [CrossRef]
- Sadeghi, A.S.; Mohsenzadeh, M.; Abnous, K.; Taghdisi, S.M.; Ramezani, M. Development and characterization of DNA aptamers against florfenicol: fabrication of a sensitive fluorescent aptasensor for specific detection of florfenicol in milk. Talanta 2018, 182, 193–201. [Google Scholar] [CrossRef]
- Sams, R.A. Florfenicol: chemistry and metabolism of a novel-broad-spectrum antibiotic. Tieraerztliche Umschau 1995, 50, 703–707. [Google Scholar]
- Cornejo, J.; Pokrant, E.; Riquelme, R.; Briceno, C.; Maddaleno, A.; Araya-Jordan, C.; Martin, B.S. Single-laboratory validation of an LC-MS/MS method for determining florfenicol (FF) and florfenicol amine (FFA) residues in chicken feathers and application to a residue-depletion study. Food Addit. Contam. Part A 2017, 34, 469–476. [Google Scholar] [CrossRef]
- Yang, F.; Yang, F.; Kong, T.; Wang, G.; Bai, D.; Liu, B. Pharmacokinetics of florfenicol and its metabolite florfenicol amine in crucian carp (Carassius auratus) at three temperatures after one single intramuscular injection. J. Vet. Pharmacol. Ther. 2018, 41, 739–745. [Google Scholar] [CrossRef]
- The European Medicines Agency. Commission Regulation (EU) No. 37/2010 of 22 December 2009 on Pharmacologically Active Substances and Their Classification Regarding Maximum Residue Limits in Foodstuffs of Animal Origin; The European Commission: Brussels, Belgium, 2010. [Google Scholar]
- U.S. Food and Drug Administration. CFR-Code of Federal Regulations Title 21 Part 556 Tolerances for Residue of New Animal Drugs in Food; U.S. Food and Drug Administration: Rockville, MD, USA, 2014. [Google Scholar]
- Ministry of Agriculture of the People ‘s Republic of China. Maxium Residue Level of Veterinary Drugs in Food of Animal Origin. Notice No. 235; Ministry of Agriculture: Beijing, China, 2002.
- Xie, K.; Jia, L.; Yao, Y.; Xu, D.; Chen, S.; Xie, X.; Pei, Y.; Bao, W.; Dai, G.; Wang, J.; et al. Simultaneous determination of thiamphenicol, florfenicol and florfenicol amine in eggs by reversed-phase high-performance liquid chromatography with fluorescence detection. J. Chromatogr. B 2011, 879, 2351–2354. [Google Scholar] [CrossRef]
- Huang, S.; Gan, N.; Liu, H.; Zhou, Y.; Chen, Y.; Cao, Y. Simultaneous and specific enrichment of several amphenicol antibiotics residues in food based on novel aptamer functionalized magnetic adsorbents using HPLC-DAD. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2017, 1060, 247–254. [Google Scholar] [CrossRef]
- Yang, J.; Sun, G.; Qian, M.; Huang, L.; Ke, X.; Yang, B. Development of a high-performance liquid chromatography method for the determination of florfenicol in animal feedstuffs. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2017, 1068–1069, 9–14. [Google Scholar] [CrossRef]
- Yang, Q.H.; Xiao-Hui, A.I.; Rong, L.I. Simultaneous determination of chloramphenicol, thiamphnicol, florfenicol and forfenicol-amine in aquatic products by gas chromatographic method with solid phase extraction. Chin. J. Anal. Lab. 2015, 34, 533–537. [Google Scholar]
- Peng, S.W.; Xin, L.J.; Mei, S.S. Determination of 3 chloramphenicols medicine residuesin animal-origin Chinese medicinal materials pheretima and hirudoin the retailer by gas chromatography. Chin. J. Vet. Drug 2017, 51, 65–69. [Google Scholar]
- Chen, D.; Yu, J.; Tao, Y.; Pan, Y.; Xie, S.; Huang, L.; Peng, D.; Wang, X.; Wang, Y.; Liu, Z.; et al. Qualitative screening of veterinary anti-microbial agents in tissues, milk, and eggs of food-producing animals using liquid chromatography coupled with tandem mass spectrometry. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2016, 1017–1018, 82–88. [Google Scholar] [CrossRef]
- Imran, M.; Habib, F.E.; Majeed, S.; Tawab, A.; Rauf, W.; Rahman, M.; Umer, M.; Iqbal, M. LC-MS/MS-based determination of chloramphenicol, thiamphenicol, florfenicol and florfenicol amine in poultry meat from the Punjab-Pakistan. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2018, 35, 1530–1542. [Google Scholar] [CrossRef]
- Marques, T.V.; Paschoal, J.A.R.; Barone, R.S.C.; Cyrino, J.E.P.; Rath, S. Depletion study and estimation of withdrawal periods for florfenicol and florfenicol amine in pacu (Piaractus mesopotamicus). Aquac. Res. 2018, 49, 111–119. [Google Scholar] [CrossRef]
- Xie, X.; Wang, B.; Pang, M.; Zhao, X.; Xie, K.; Zhang, Y.; Wang, Y.; Guo, Y.; Liu, C.; Bu, X.; et al. Quantitative analysis of chloramphenicol, thiamphenicol, florfenicol and florfenicol amine in eggs via liquid chromatography-electrospray ionization tandem mass spectrometry. Food Chem. 2018, 269, 542–548. [Google Scholar] [CrossRef]
- Shen, J.; Xia, X.; Jiang, H.; Li, C.; Li, J.; Li, X.; Ding, S. Determination of chloramphenicol, thiamphenicol, florfenicol, and florfenicol amine in poultry and porcine muscle and liver by gas chromatography-negative chemical ionization mass spectrometry. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2009, 877, 1523–1529. [Google Scholar] [CrossRef]
- Azzouz, A.; Ballesteros, E. Multiresidue method for the determination of pharmacologically active substances in egg and honey using a continuous solid-phase extraction system and gas chromatography-mass spectrometry. Food Chem. 2015, 178, 63–69. [Google Scholar] [CrossRef]
- Amorello, D.; Barreca, S.; Gambacurta, S.; Gulotta, M.G.; Orecchio, S.; Pace, A. An analytical method for monitoring micro-traces of landfill leachate in groundwater using fluorescence excitation–emission matrix spectroscopy. Anal. Methods 2016, 8, 3475–3480. [Google Scholar] [CrossRef]
- Fedeniuk, R.W.; Mizuno, M.; Neiser, C.; O’Byrne, C. Development of LC-MS/MS methodology for the detection/determination and confirmation of chloramphenicol, chloramphenicol 3-O-beta-d-glucuronide, florfenicol, florfenicol amine and thiamphenicol residues in bovine, equine and porcine liver. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2015, 991, 68–78. [Google Scholar] [CrossRef]
- Shin, D.; Kang, H.-S.; Jeong, J.; Kim, J.; Choe, W.J.; Lee, K.S.; Rhee, G.-S. Multi-residue determination of veterinary drugs in fishery products using liquid chromatography-tandem mass spectrometry. Food Anal. Methods 2018, 11, 1815–1831. [Google Scholar] [CrossRef]
- Moretti, S.; Dusi, G.; Giusepponi, D.; Pellicciotti, S.; Rossi, R.; Saluti, G.; Cruciani, G.; Galarini, R. Screening and confirmatory method for multiclass determination of 62 antibiotics in meat. J. Chromatogr. A 2016, 1429, 175–188. [Google Scholar] [CrossRef]
- Wang, K.; Lin, K.; Huang, X.; Chen, M. A simple and fast extraction method for the determination of multiclass antibiotics in eggs using LC-MS/MS. J. Agric. Food Chem. 2017, 65, 5064–5073. [Google Scholar] [CrossRef]
- Orlando, E.A.; Costa Roque, A.G.; Losekann, M.E.; Simionato, A.V.C. UPLC-MS/MS determination of florfenicol and florfenicol amine antimicrobial residues in tilapia muscle. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1035, 8–15. [Google Scholar] [CrossRef]
- Barreto, F.; Ribeiro, C.; Barcellos Hoff, R.; Costa, T.D. Determination of chloramphenicol, thiamphenicol, florfenicol and florfenicol amine in poultry, swine, bovine and fish by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A. 2016, 1449, 48–53. [Google Scholar] [CrossRef]
- Feng, J.B.; Huang, D.R.; Zhong, M.; Liu, P.; Dong, J.D. Pharmacokinetics of florfenicol and behaviour of its metabolite florfenicol amine in orange-spotted grouper (Epinephelus coioides) after oral administration. J. Fish. Dis. 2016, 39, 833–843. [Google Scholar] [CrossRef]
- Yang, F.; Fang, Y.; Liu, Z.C. Determination of thiamphenicol, florfenicol and florfenicol amine residues in aquatic products by HPLC with fluorescence detection. Chin. J. Vet. Drug 2008, 42, 14–16. [Google Scholar]
- Karami-Osboo, R.; Miri, R.; Javidnia, K.; Kobarfard, F. Simultaneous chloramphenicol and florfenicol determination by a validated DLLME-HPLC-UV method in pasteurized milk. Iran. J. Pharm. Res. 2016, 15, 361–368. [Google Scholar]
- Zhai, Z.Y.; Xu, Y. Determination of thiamphenicol, florfenicol and florfenicol amine in lyophilized royal jelly powder by HPLC. J. Prev. Med. Inf. 2010, 26, 82–84. [Google Scholar]
- Xie, K.Z.; Xu, D.; Chen, S.Q.; Xie, X.; Jia, L.F.; Huang, Y.P.; Guo, H.S.; Wang, J.Y.; Liu, Z.P. Simultaneous determination of residues of thiamphenicol forfenicol and forfenicol amine in chicken muscle by HPLC with fluorescence detection. Chin. J. Anal. Lab. 2011, 30, 31–35. [Google Scholar]
- Alechaga, E.; Moyano, E.; Galceran, M.T. Ultra-high performance liquid chromatography-tandem mass spectrometry for the analysis of phenicol drugs and florfenicol-amine in foods. Analyst 2012, 137, 2486–2494. [Google Scholar] [CrossRef]
- Lehotay, S.J.; Lightfield, A.R. Simultaneous analysis of aminoglycosides with many other classes of drug residues in bovine tissues by ultrahigh-performance liquid chromatography-tandem mass spectrometry using an ion-pairing reagent added to final extracts. Anal. Bioanal. Chem. 2018, 410, 1095–1109. [Google Scholar] [CrossRef]
- Yang, H.S.; Meng, Y.Z.; Meiqin, W.G. Simultaneous UHPLC-MS/MS determination of chloramphenicol and florfenicol in aquatic products with accelerated solvent extraction. Phy. Chem. Test. Chem. 2012, 48, 1353–1356. [Google Scholar]
- Xiao, Z.; Song, R.; Rao, Z.; Wei, S.; Jia, Z.; Suo, D.; Fan, X. Development of a subcritical water extraction approach for trace analysis of chloramphenicol, thiamphenicol, florfenicol, and florfenicol amine in poultry tissues. J. Chromatogr. A 2015, 1418, 29–35. [Google Scholar] [CrossRef] [PubMed]
- US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Veterinary Medicine. Guidance for Industry: Bioanalytical Method Validation; US Department of Health and Human Services: Washington, DC, USA, 2001.
- The European Communities. Commission decision 2002/657/EC of 12 august 2002 implementing council directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Commun. 2002, 221, 8–36. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix | Analyte | Extraction Reagents (acetonitrile:ammonia, v/v) | ||||
|---|---|---|---|---|---|---|
| 99:1 | 98:2 | 97:3 | 96:4 | 95:5 | ||
| Hen eggs | TAP | 93.4 ± 1.8 | 92.8 ± 1.1 | 89.4 ± 2.3 | 81.6 ± 2.2 | 73.9 ± 2.0 |
| FF | 92.6 ± 2.7 | 93.2 ± 3.0 | 84.2 ± 2.2 | 74.3 ± 1.9 | 69.6 ± 2.0 | |
| FFA | 86.9 ± 2.1 | 92.4 ± 2.2 | 88.4 ± 3.1 | 78.8 ± 2.9 | 69.3 ± 1.9 | |
| Duck eggs | TAP | 90.2 ± 2.3 | 91.3 ± 2.3 | 85.3 ± 2.4 | 80.8 ± 2.4 | 70.3 ± 2.2 |
| FF | 93.0 ± 1.8 | 91.2 ± 2.5 | 84.2 ± 2.0 | 74.6 ± 1.9 | 68.7 ± 2.3 | |
| FFA | 85.0 ± 2.5 | 93.2 ± 1.8 | 84.3 ± 2.3 | 73.6 ± 1.9 | 61.5 ± 1.9 | |
| Goose eggs | TAP | 90.7 ± 2.2 | 93.4 ± 2.6 | 86.7 ± 2.4 | 75.8 ± 2.7 | 67.9 ± 2.0 |
| FF | 93.1 ± 1.8 | 91.3 ± 1.9 | 87.3 ± 2.3 | 71.4 ± 2.2 | 60.4 ± 2.4 | |
| FFA | 85.0 ± 1.8 | 92.3 ± 2.0 | 84.1 ± 1.8 | 73.0 ± 1.9 | 63.8 ± 2.2 | |
| Pigeon eggs | TAP | 92.4 ± 2.6 | 92.6 ± 2.1 | 82.3 ± 3.1 | 75.5 ± 2.4 | 63.2 ± 2.3 |
| FF | 92.3 ± 2.4 | 93.1 ± 2.0 | 83.8 ± 1.9 | 76.1 ± 3.0 | 61.0 ± 2.2 | |
| FFA | 88.2 ± 2.2 | 96.3 ± 2.1 | 86.5 ± 2.7 | 73.1 ± 2.1 | 64.8 ± 2.3 | |
| Quail eggs | TAP | 91.3 ± 2.3 | 92.8 ± 2.3 | 87.3 ± 2.0 | 70.8 ± 1.9 | 65.7 ± 2.1 |
| FF | 92.7 ± 2.2 | 94.2 ± 2.5 | 86.2 ± 2.5 | 74.5 ± 2.4 | 68.5 ± 2.5 | |
| FFA | 86.5 ± 1.8 | 93.0 ± 1.9 | 84.7 ± 2.5 | 76.2 ± 1.9 | 70.4 ± 1.8 | |
| Matrix | Analyte | Extraction Method | |||
|---|---|---|---|---|---|
| Ultrasonic | Vortex Oscillation | Vortex Oscillation + Ultrasonic | ASE | ||
| Hen egg | TAP | 34.4 ± 2.5 | 78.6 ± 2.6 | 88.3 ± 2.2 | 92.8 ± 2.4 |
| FF | 37.1 ± 2.3 | 81.5 ± 3.1 | 92.0 ± 2.7 | 96.0 ± 2.3 | |
| FFA | 54.4 ± 2.2 | 84.0 ± 2.9 | 91.9 ± 2.8 | 93.3 ± 2.0 | |
| Duck eggs | TAP | 30.9 ± 2.0 | 70.0 ± 2.7 | 84.9 ± 2.2 | 90.2 ± 1.9 |
| FF | 33.4 ± 2.5 | 79.5 ± 2.7 | 89.7 ± 2.4 | 92.5 ± 2.1 | |
| FFA | 49.0 ± 2.4 | 74.4 ± 3.2 | 86.0 ± 2.9 | 91.7 ± 2.2 | |
| Goose eggs | TAP | 31.3 ± 2.9 | 71.9 ± 3.0 | 82.7 ± 2.6 | 93.3 ± 1.9 |
| FF | 35.6 ± 2.7 | 72.0 ± 2.6 | 88.4 ± 3.0 | 92.4 ± 2.5 | |
| FFA | 40.5 ± 3.4 | 77.0 ± 2.8 | 85.0 ± 2.4 | 90.4 ± 2.1 | |
| Pigeon eggs | TAP | 34.4 ± 3.0 | 69.9 ± 2.7 | 85.3 ± 2.9 | 86.9 ± 2.2 |
| FF | 34.7 ± 2.6 | 70.0 ± 3.2 | 87.0 ± 2.6 | 89.0 ± 2.4 | |
| FFA | 40.7 ± 2.8 | 72.6 ± 2.9 | 80.3 ± 2.3 | 95.1 ± 1.6 | |
| Quail eggs | TAP | 36.1 ± 2.9 | 72.1 ± 2.8 | 83.5 ± 2.7 | 90.1 ± 2.0 |
| FF | 30.1 ± 3.0 | 73.4 ± 2.3 | 88.5 ± 3.3 | 92.6 ± 2.4 | |
| FFA | 45.5 ± 2.8 | 69.7 ± 3.1 | 83.7 ± 2.5 | 92.7 ± 2.7 | |
| Matrix | Analyte | Linear Range (µg/kg) | Linear Regression Equation | Determination Coefficient (R2) | LOD (µg/kg) | LOQ (µg/kg) |
|---|---|---|---|---|---|---|
| Hen eggs | TAP | 9.7–250.0 | y = 1030.2x + 295.52 | 0.9996 | 3.3 | 9.7 |
| FF | 10.5–400.0 | y = 737.43x + 714.06 | 0.9998 | 4.7 | 10.5 | |
| FFA | 4.3–400.0 | y = 3020.3x + 164.01 | 0.9998 | 1.8 | 4.3 | |
| Duck eggs | TAP | 9.9–250.0 | y = 562.84x + 634.05 | 0.9997 | 3.4 | 9.9 |
| FF | 11.7–400.0 | y = 844.1x + 616.34 | 0.9997 | 4.9 | 11.7 | |
| FFA | 4.7–400.0 | y = 4495.8x + 520.7 | 0.9996 | 1.9 | 4.7 | |
| Goose eggs | TAP | 9.8–250.0 | y = 618.73x + 139.95 | 0.9996 | 3.4 | 9.8 |
| FF | 11.2–400.0 | y = 713.68x + 738.78 | 0.9996 | 4.8 | 11.2 | |
| FFA | 4.7–400.0 | y = 3081.1x – 258.32 | 0.9997 | 1.9 | 4.7 | |
| Pigeon eggs | TAP | 9.9–250.0 | y = 683.44x + 543.48 | 0.9998 | 3.4 | 9.9 |
| FF | 11.2–400.0 | y = 762.9x + 761.11 | 0.9993 | 4.8 | 11.2 | |
| FFA | 4.8–400.0 | y = 4019.1x + 490.45 | 0.9998 | 1.9 | 4.8 | |
| Quail eggs | TAP | 9.7–250.0 | y = 667.44x + 483.91 | 0.9993 | 3.3 | 9.7 |
| FF | 10.6–400.0 | y = 753.98x – 193.97 | 0.9999 | 4.7 | 10.6 | |
| FFA | 4.6–400.0 | y = 4824.6x – 229.06 | 0.9994 | 1.8 | 4.6 |
| Matrix | Analyte | Spiking Level (µg/kg) | Recovery (%) (n = 6) | RSD (%) (n = 6) | Intraday RSD (%) (n = 6) | Interday RSD (%) (n = 18) |
|---|---|---|---|---|---|---|
| Hen eggs | TAP | 9.7 | 85.6 ± 1.8 | 2.1 | 2.6 | 3.6 |
| 25 | 90.5 ± 2.7 | 3.0 | 3.7 | 5.0 | ||
| 50 α | 92.7 ± 1.5 | 1.6 | 2.5 | 3.1 | ||
| 100 | 91.5 ± 2.0 | 2.2 | 2.5 | 2.9 | ||
| FF | 10.5 | 84.9 ± 3.4 | 4.0 | 4.7 | 5.1 | |
| 50 | 90.2 ± 2.5 | 2.8 | 3.8 | 4.8 | ||
| 100 α | 93.5 ± 2.6 | 2.8 | 4.0 | 4.3 | ||
| 200 | 94.9 ± 2.7 | 2.8 | 2.3 | 3.4 | ||
| FFA | 4.3 | 86.7 ± 3.6 | 4.2 | 3.9 | 5.3 | |
| 50 | 91.5 ± 1.8 | 2.0 | 3.1 | 3.6 | ||
| 100 α | 96.7 ± 3.5 | 3.6 | 3.2 | 2.8 | ||
| 200 | 98.0 ± 1.8 | 1.8 | 2.4 | 2.7 | ||
| Duck eggs | TAP | 9.9 | 84.8 ± 2.0 | 2.4 | 3.9 | 4.2 |
| 25 | 93.8 ± 1.8 | 1.9 | 3.6 | 4.6 | ||
| 50 α | 94.5 ± 2.4 | 2.5 | 1.9 | 2.6 | ||
| 100 | 92.8 ± 1.1 | 1.2 | 2.4 | 2.4 | ||
| FF | 11.7 | 85.1 ± 1.6 | 1.9 | 1.2 | 2.4 | |
| 50 | 89.4 ± 2.0 | 2.2 | 1.8 | 1.8 | ||
| 100 α | 94.8 ± 1.5 | 1.6 | 2.5 | 3.6 | ||
| 200 | 94.5 ± 3.3 | 3.5 | 2.6 | 4.1 | ||
| FFA | 4.7 | 87.5 ± 1.9 | 2.2 | 3.1 | 3.7 | |
| 50 | 96.5 ± 2.2 | 2.3 | 3.1 | 3.4 | ||
| 100 α | 96.9 ± 1.7 | 1.8 | 2.7 | 3.6 | ||
| 200 | 96.1 ± 1.7 | 1.8 | 3.1 | 3.3 | ||
| Goose eggs | TAP | 9.8 | 85.5 ± 1.7 | 2.0 | 2.5 | 2.9 |
| 25 | 93.3 ± 2.1 | 2.3 | 3.5 | 4.0 | ||
| 50 α | 93.1 ± 2.9 | 3.1 | 4.3 | 5.4 | ||
| 100 | 94.5 ± 4.0 | 4.2 | 3.9 | 4.0 | ||
| FF | 11.2 | 80.7 ± 3.5 | 4.3 | 5.2 | 6.2 | |
| 50 | 93.9 ± 2.2 | 2.3 | 2.7 | 3.1 | ||
| 100 α | 95.2 ± 1.5 | 1.6 | 2.2 | 3.0 | ||
| 200 | 95.2 ± 2.5 | 2.6 | 3.1 | 3.4 | ||
| FFA | 4.7 | 83.9 ± 2.8 | 3.3 | 3.5 | 4.1 | |
| 50 | 93.7 ± 2.5 | 2.7 | 3.2 | 3.8 | ||
| 100 α | 94.5 ± 2.0 | 2.1 | 3.2 | 3.5 | ||
| 200 | 96.1 ± 2.6 | 2.7 | 3.3 | 4.4 |
| Matrix | Analyte | Spiking Level (µg/kg) | Recovery (%) (n = 6) | RSD (%) (n = 6) | Intraday RSD (%) (n = 6) | Interday RSD (%) (n = 18) |
|---|---|---|---|---|---|---|
| Pigeon eggs | TAP | 9.9 | 80.1 ± 2.5 | 3.1 | 2.5 | 3.6 |
| 25 | 93.6 ± 3.3 | 3.5 | 4.7 | 5.4 | ||
| 50 α | 92.3 ± 2.4 | 2.6 | 2.7 | 3.2 | ||
| 100 | 94.4 ± 3.3 | 3.5 | 2.2 | 4.3 | ||
| FF | 11.2 | 84.4 ± 2.2 | 2.6 | 3.2 | 3.3 | |
| 50 | 95.9 ± 2.5 | 2.6 | 4.4 | 4.2 | ||
| 100 α | 97.9 ± 4.1 | 4.2 | 3.3 | 5.0 | ||
| 200 | 98.6 ± 2.8 | 2.8 | 3.1 | 3.1 | ||
| FFA | 4.8 | 85.7 ± 2.0 | 2.3 | 3.8 | 4.1 | |
| 50 | 94.0 ± 2.3 | 2.4 | 2.9 | 3.2 | ||
| 100 α | 95.7 ± 3.2 | 3.3 | 4.2 | 5.4 | ||
| 200 | 97.7 ± 1.8 | 1.8 | 2.3 | 3.7 | ||
| Quail eggs | TAP | 9.7 | 84.1 ± 3.0 | 3.6 | 3.9 | 4.0 |
| 25 | 95.4 ± 2.0 | 2.1 | 3.3 | 3.9 | ||
| 50 α | 93.4 ± 2.8 | 3.0 | 3.9 | 4.4 | ||
| 100 | 96.3 ± 2.2 | 2.3 | 3.7 | 5.4 | ||
| FF | 10.6 | 86.5 ± 3.5 | 4.0 | 5.5 | 5.5 | |
| 50 | 94.9 ± 2.1 | 2.2 | 2.7 | 3.8 | ||
| 100 α | 95.9 ± 3.1 | 3.2 | 4.0 | 4.6 | ||
| 200 | 96.7 ± 2.3 | 2.4 | 3.2 | 3.4 | ||
| FFA | 4.6 | 87.5 ± 2.7 | 3.1 | 3.7 | 4.1 | |
| 50 | 96.0 ± 3.0 | 3.1 | 4.7 | 5.6 | ||
| 100 α | 95.5 ± 3.4 | 3.6 | 3.5 | 5.5 | ||
| 200 | 96.2 ± 3.6 | 3.7 | 5.4 | 6.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Xie, X.; Zhao, X.; Xie, K.; Diao, Z.; Zhang, G.; Zhang, T.; Dai, G. Development of an Accelerated Solvent Extraction-Ultra-Performance Liquid Chromatography-Fluorescence Detection Method for Quantitative Analysis of Thiamphenicol, Florfenicol and Florfenicol Amine in Poultry Eggs. Molecules 2019, 24, 1830. https://doi.org/10.3390/molecules24091830
Wang B, Xie X, Zhao X, Xie K, Diao Z, Zhang G, Zhang T, Dai G. Development of an Accelerated Solvent Extraction-Ultra-Performance Liquid Chromatography-Fluorescence Detection Method for Quantitative Analysis of Thiamphenicol, Florfenicol and Florfenicol Amine in Poultry Eggs. Molecules. 2019; 24(9):1830. https://doi.org/10.3390/molecules24091830
Chicago/Turabian StyleWang, Bo, Xing Xie, Xia Zhao, Kaizhou Xie, Zhixiang Diao, Genxi Zhang, Tao Zhang, and Guojun Dai. 2019. "Development of an Accelerated Solvent Extraction-Ultra-Performance Liquid Chromatography-Fluorescence Detection Method for Quantitative Analysis of Thiamphenicol, Florfenicol and Florfenicol Amine in Poultry Eggs" Molecules 24, no. 9: 1830. https://doi.org/10.3390/molecules24091830
APA StyleWang, B., Xie, X., Zhao, X., Xie, K., Diao, Z., Zhang, G., Zhang, T., & Dai, G. (2019). Development of an Accelerated Solvent Extraction-Ultra-Performance Liquid Chromatography-Fluorescence Detection Method for Quantitative Analysis of Thiamphenicol, Florfenicol and Florfenicol Amine in Poultry Eggs. Molecules, 24(9), 1830. https://doi.org/10.3390/molecules24091830