
ICAM3-Fc Outperforms Receptor-Specific Antibodies Targeted Nanoparticles to Dendritic Cells for Cross-Presentation

 ,
,
Abstract
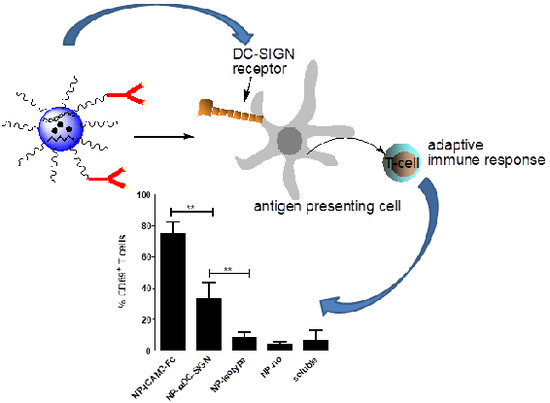
:
1. Introduction
2. Results
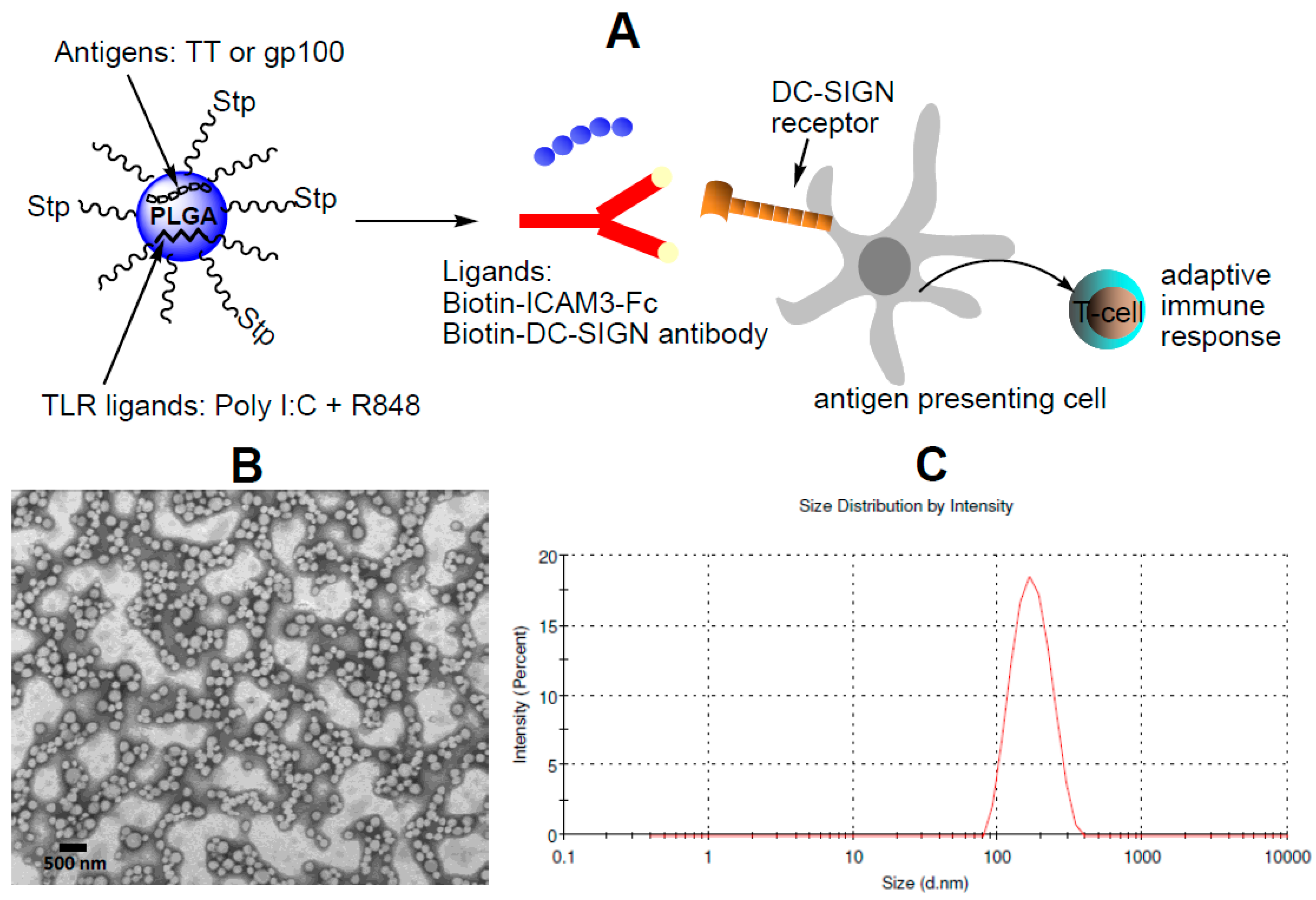
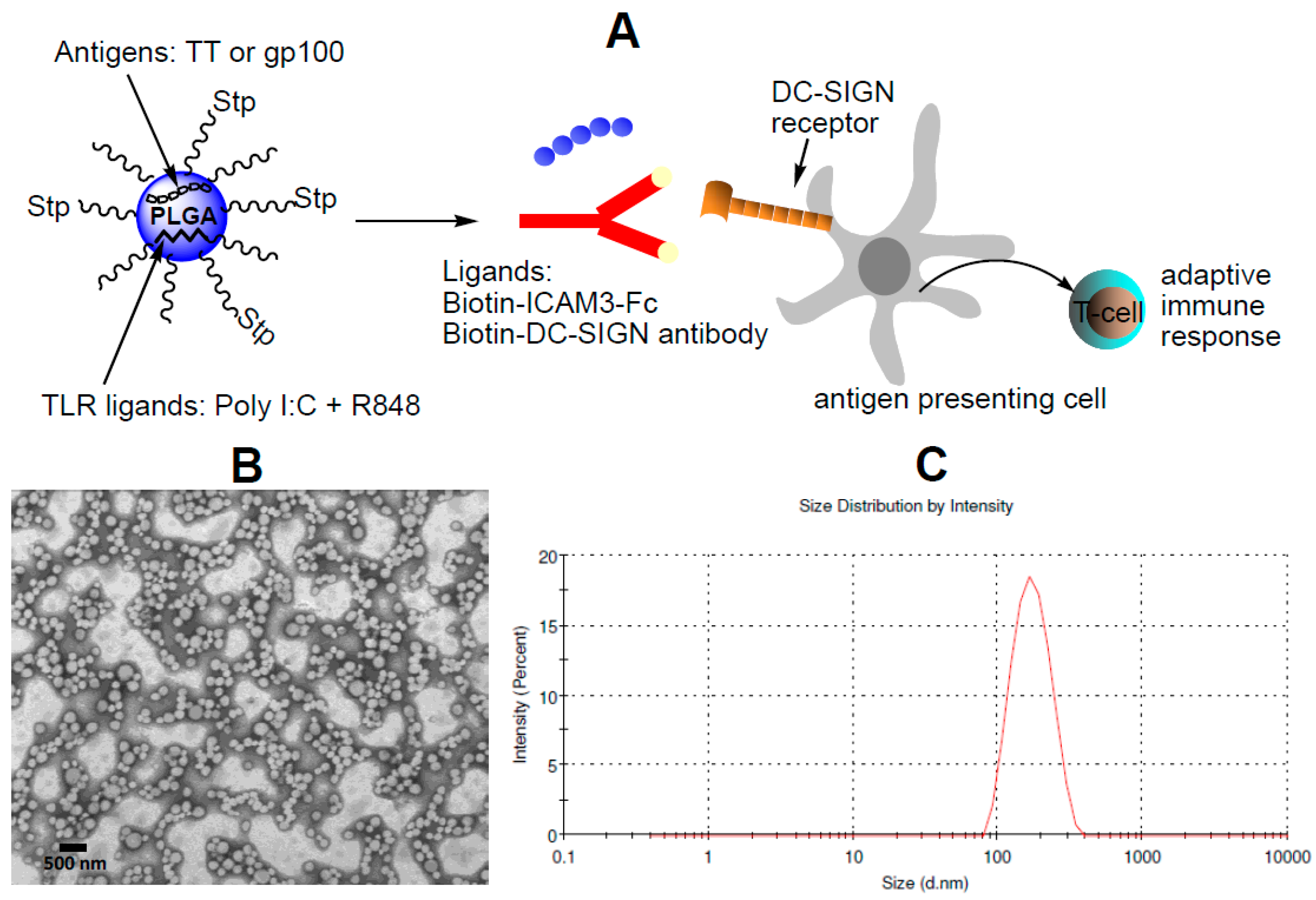
2.1. Characterization of NP Vaccine
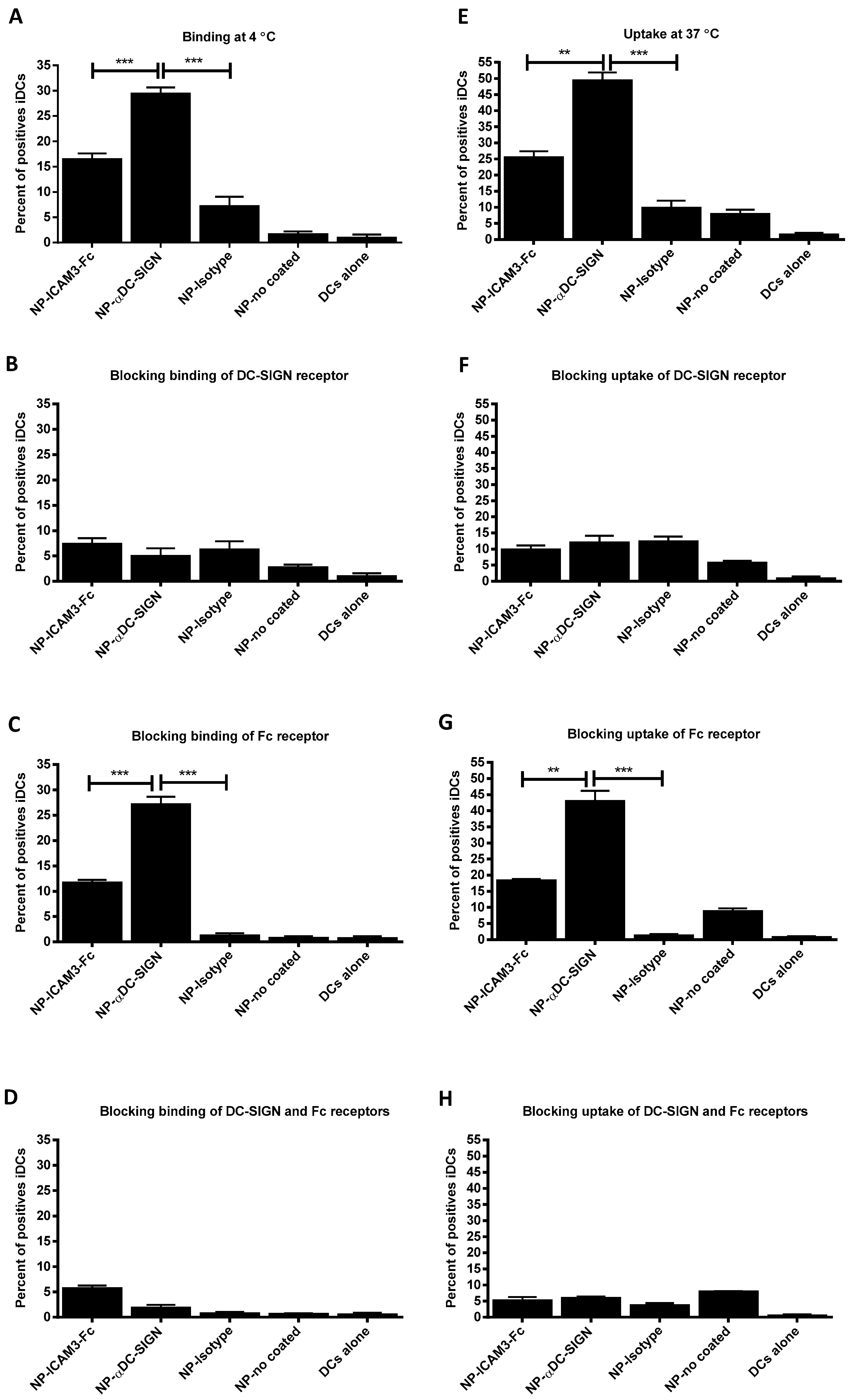
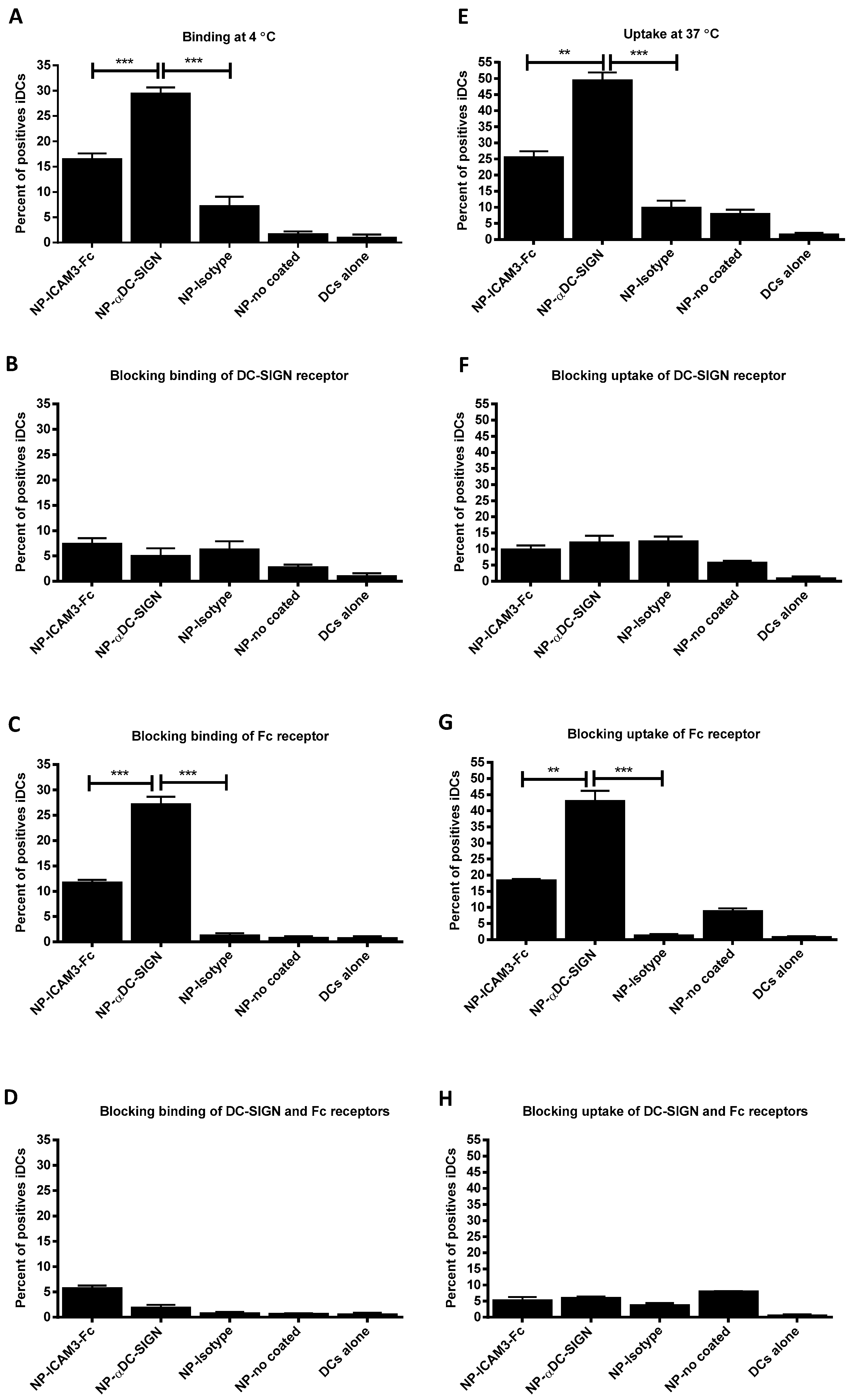
2.2. Binding and Uptake on DCs of NP Vaccine Coated with DC-SIGN Ligands
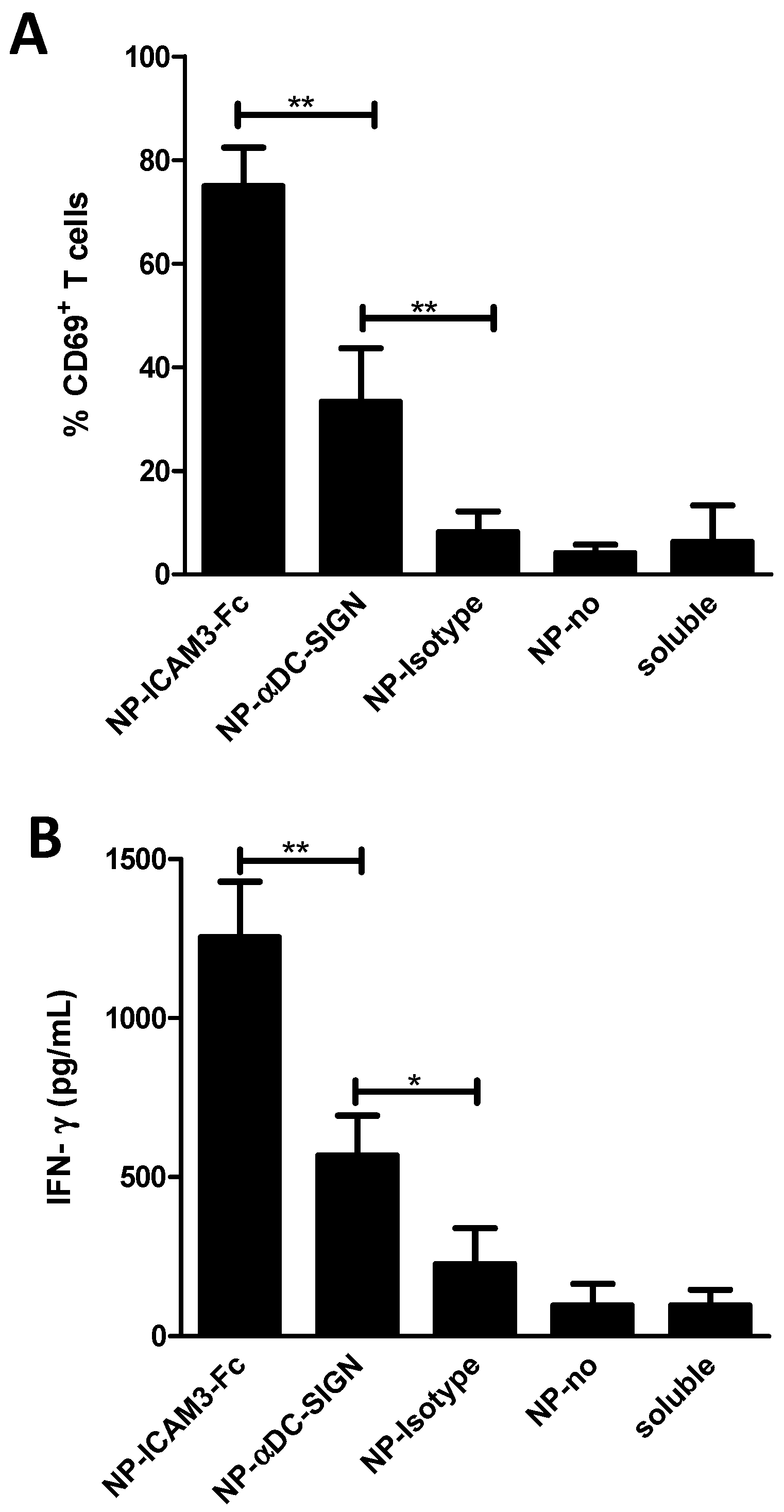
2.3. NP Vaccines Targeted by Anti-DC-SIGN and ICAM3-Fc Activate DCs
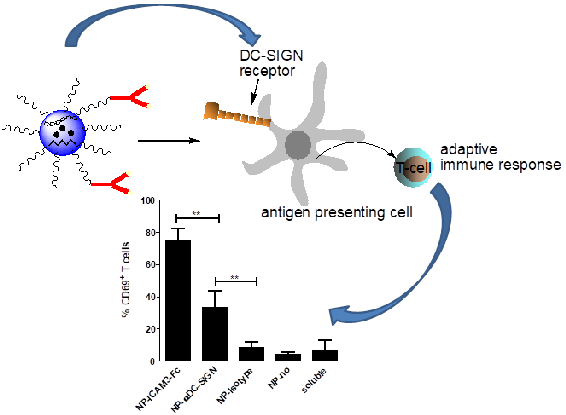
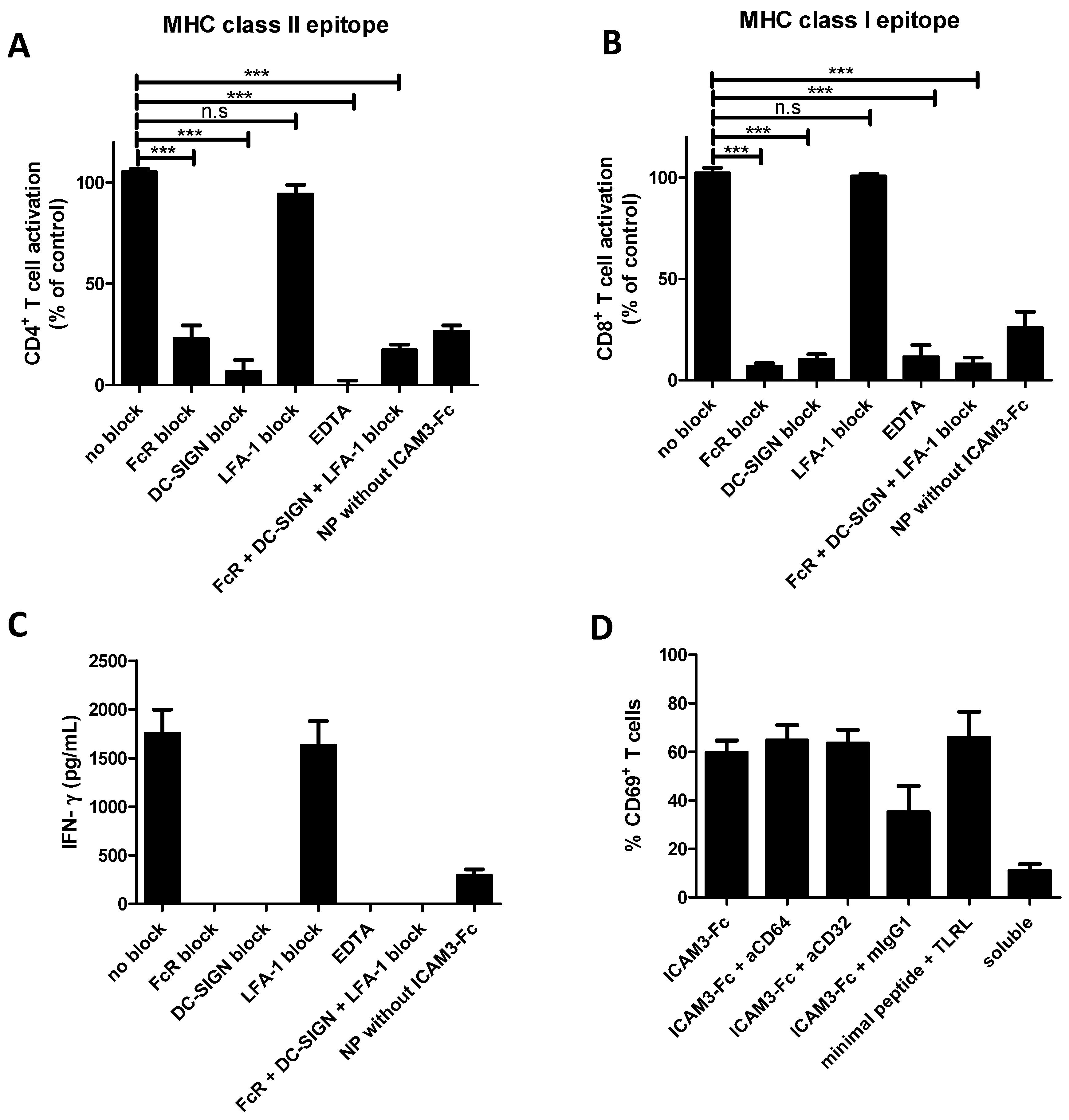
2.4. ICAM3-Fc Coating NP Vaccine Induces the Highest Levels of Cross-Presentation
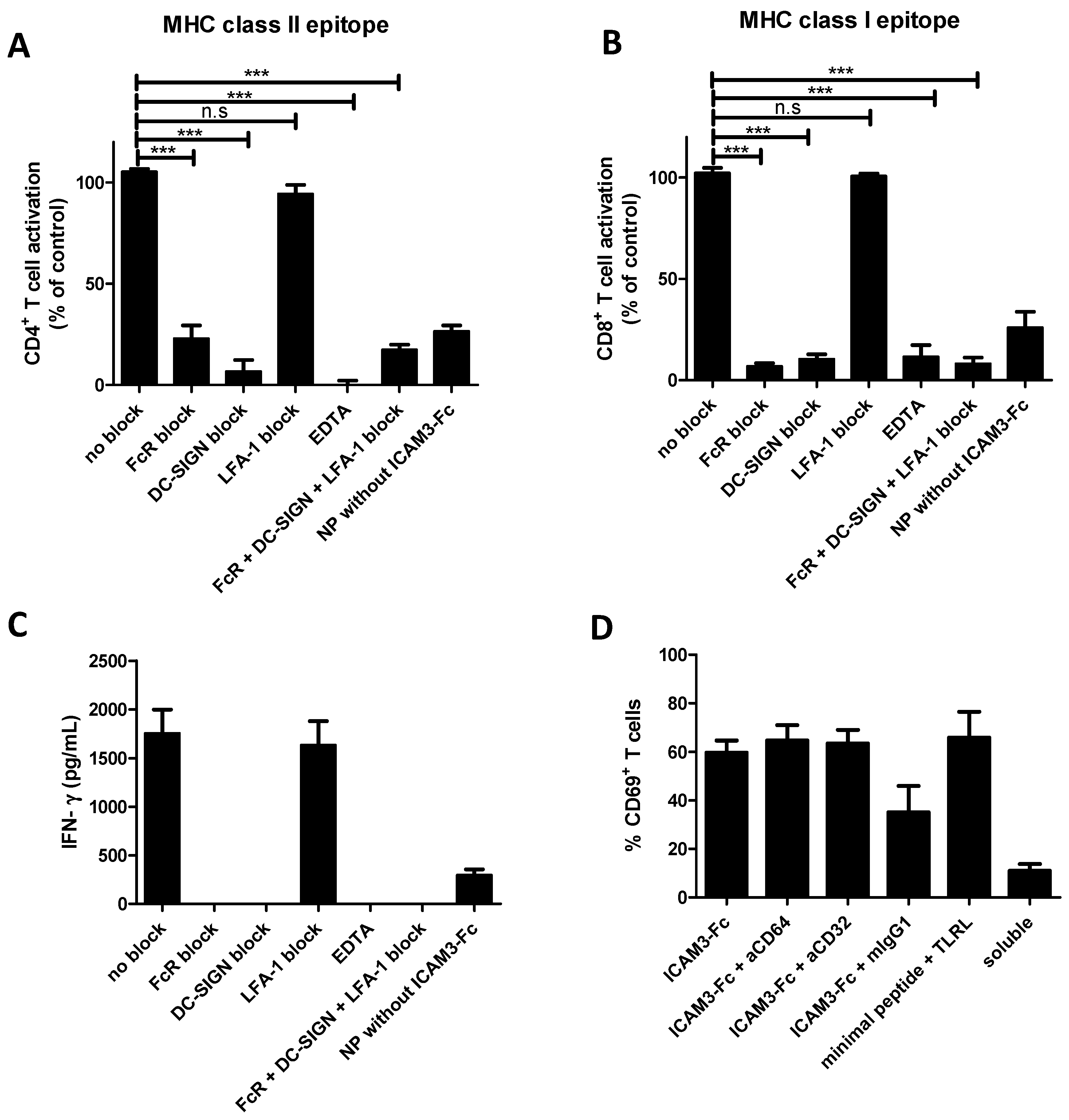
2.5. NP Vaccines Coated with ICAM3-Fc Induce Cross-Presentation via DC-SIGN but does not Require FcRs CD32 or CD64
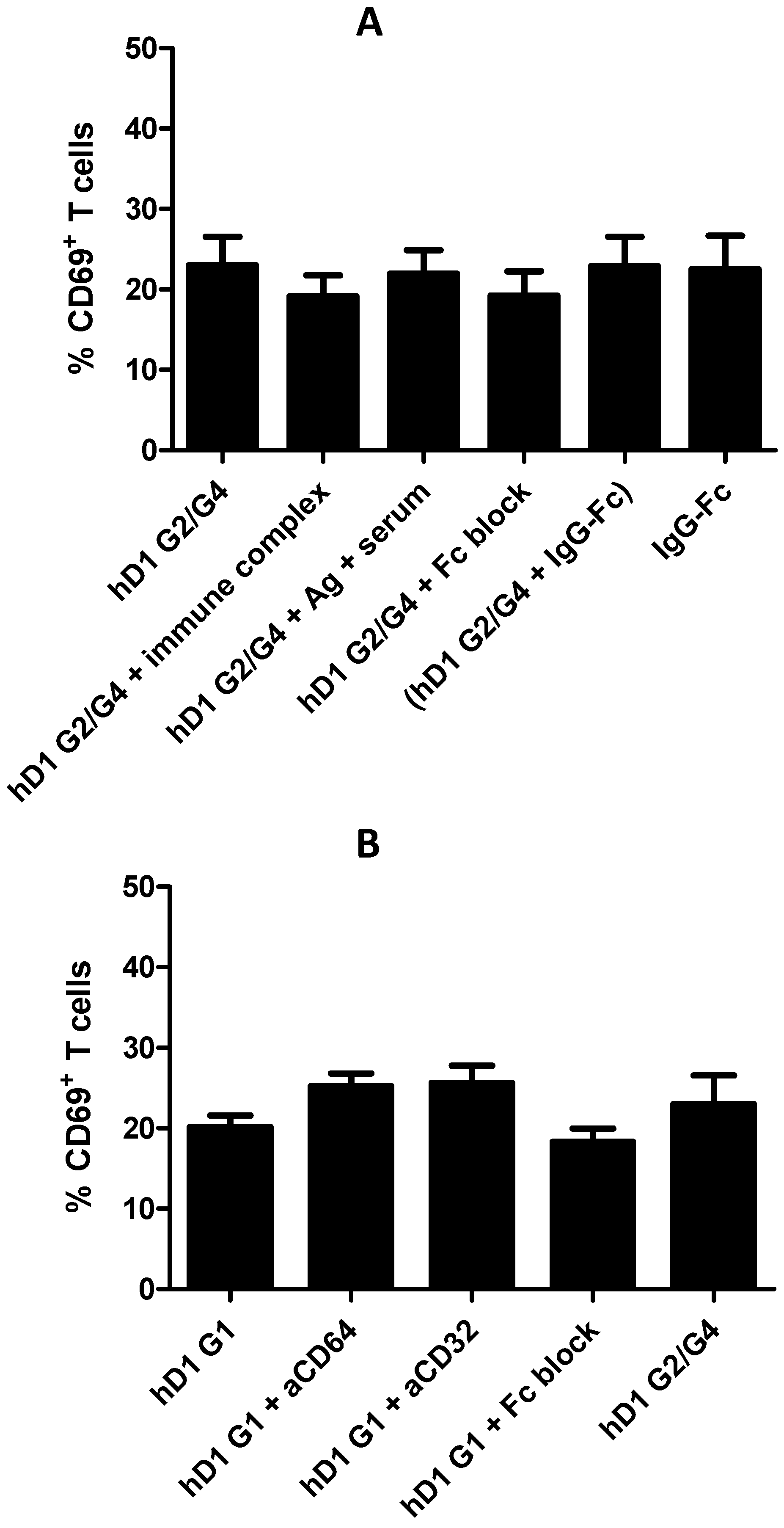
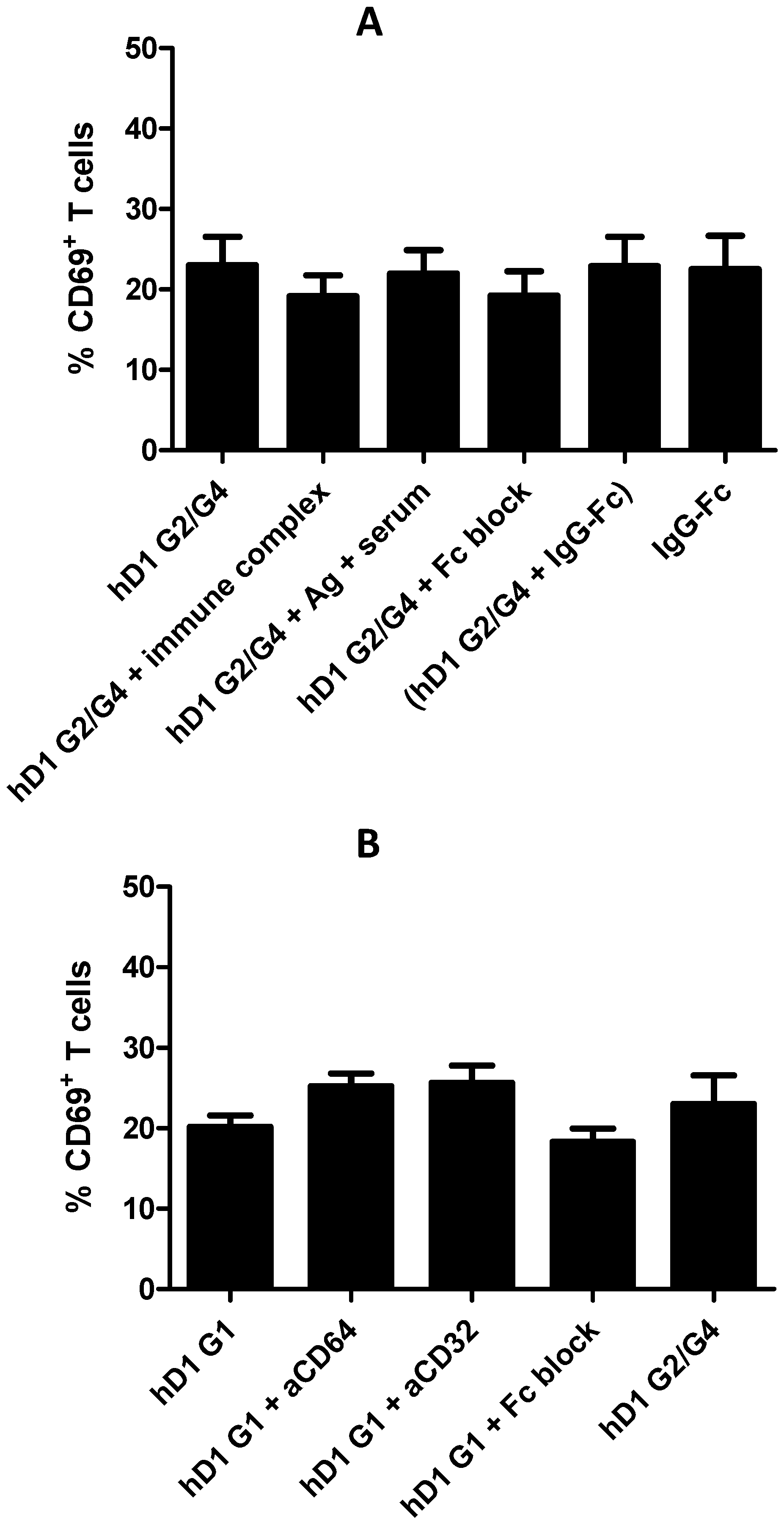
2.6. Adding FcR Binding Capacity to Antibody Coated NP Vaccine does not Improve Cross-Presentation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cells
4.3. Peptide Synthesis
4.4. Generation of Targeted NP Vaccine
4.5. Dynamic Light Scattering, Zeta-Potential and Transmission Electron Microscopy
4.6. Flow Cytometry Analysis of Binding and Uptake of the Various NP Vaccines by DCs
4.7. DC Maturation and Activation
4.8. Human CD8+ T Cell Activation
4.9. Human CD4+ T Cell Activation
4.10. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CRD | carbohydrate-recognition domain |
| DC | dendritic cell |
| DC-SIGN | dendritic cell specific intercellular adhesion molecule-3-grabbing non-integrin |
| DIEA | N,N-diisopropylethylamine |
| DLS | dynamic light scattering |
| DMF | N,N-dimethylformamide |
| DCM | dichloromethane |
| FITC | fluorescein isothiocyanate |
| FDA | food and drug administration |
| HOBt | 1-hydroxybenzotriazole |
| HPLC | high pressure liquid chromatography |
| NP | nanoparticle |
| PBL | peripheral blood lymphocyte |
| PBMC | peripheral blood mononuclear cell |
| PEG | polyethylene glycol |
| PVA | polyvinyl alcohol |
| TT | tetanus toxoid |
| EDAC | (1-ethyl-3-[3-dimethylaminopropyl] carbodiimide) |
| NHS | (Nhydroxysuccinimide) |
| TLR | Toll-like receptor |
| TLRL | Toll-like receptor ligand |
| Ag | Antigen |
| Abs | Antibodies |
References
- Hutchings, C.L.; Gilbert, S.C.; Hill, A.V.; Moore, A.C. Novel protein and poxvirus-based vaccine combinations for simultaneous induction of humoral and cell-mediated immunity. J. Immunol. 2005, 175, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Paschen, A.; Eichmuller, S.; Schadendorf, D. Identification of tumor antigens and t-cell epitopes, and its clinical application. Cancer Immunol. Immunother. 2004, 53, 196–203. [Google Scholar] [CrossRef]
- Spagnoli, G.C.; Adamina, M.; Bolli, M.; Weber, W.P.; Zajac, P.; Marti, W.; Oertli, D.; Heberer, M.; Harder, F. Active antigen-specific immunotherapy of melanoma: From basic science to clinical investigation. World J. Surg. 2005, 29, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; de Vries, I.J.; Torensma, R.; Figdor, C.G. Dendritic-cell immunotherapy: From ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 2007, 7, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Swider, E.; Koshkina, O.; Tel, J.; Cruz, L.J.; de Vries, I.J.M.; Srinivas, M. Customizing poly(lactic-co-glycolic acid) particles for biomedical applications. Acta Biomater. 2018, 73, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (plga) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Tacken, P.J.; Eich, C.; Rueda, F.; Torensma, R.; Figdor, C.G. Controlled release of antigen and toll-like receptor ligands from plga nanoparticles enhances immunogenicity. Nanomedicine 2017, 12, 491–510. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Tacken, P.J.; Fokkink, R.; Figdor, C.G. The influence of peg chain length and targeting moiety on antibody-mediated delivery of nanoparticle vaccines to human dendritic cells. Biomaterials 2011, 32, 6791–6803. [Google Scholar] [CrossRef] [PubMed]
- Shan, M.; Klasse, P.J.; Banerjee, K.; Dey, A.K.; Iyer, S.P.; Dionisio, R.; Charles, D.; Campbell-Gardener, L.; Olson, W.C.; Sanders, R.W.; et al. Hiv-1 gp120 mannoses induce immunosuppressive responses from dendritic cells. PLoS Pathog. 2007, 3, e169. [Google Scholar] [CrossRef]
- Sarkar, R.; Mitra, D.; Chakrabarti, S. Hiv-1 gp120 protein downregulates nef induced il-6 release in immature dentritic cells through interplay of dc-sign. PLoS ONE 2013, 8, e59073. [Google Scholar] [CrossRef]
- Wille-Reece, U.; Flynn, B.J.; Lore, K.; Koup, R.A.; Kedl, R.M.; Mattapallil, J.J.; Weiss, W.R.; Roederer, M.; Seder, R.A. Hiv gag protein conjugated to a toll-like receptor 7/8 agonist improves the magnitude and quality of th1 and cd8+ t cell responses in nonhuman primates. Proc. Natl. Acad. Sci. USA 2005, 102, 15190–15194. [Google Scholar] [CrossRef] [PubMed]
- Reis e Sousa, C. Harnessing dendritic cells. Semin. Immunol. 2011, 23, 1. [Google Scholar] [CrossRef]
- Cruz, L.J.; Tacken, P.J.; Fokkink, R.; Joosten, B.; Stuart, M.C.; Albericio, F.; Torensma, R.; Figdor, C.G. Targeted plga nano- but not microparticles specifically deliver antigen to human dendritic cells via dc-sign in vitro. J. Control. Release 2010, 144, 118–126. [Google Scholar] [CrossRef]
- Cruz, L.J.; Rosalia, R.A.; Kleinovink, J.W.; Rueda, F.; Lowik, C.W.; Ossendorp, F. Targeting nanoparticles to cd40, dec-205 or cd11c molecules on dendritic cells for efficient CD8(+) t cell response: A comparative study. J. Control. Release 2014, 192, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; Zeelenberg, I.S.; Cruz, L.J.; van Hout-Kuijer, M.A.; van de Glind, G.; Fokkink, R.G.; Lambeck, A.J.; Figdor, C.G. Targeted delivery of tlr ligands to human and mouse dendritic cells strongly enhances adjuvanticity. Blood 2011, 118, 6836–6844. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Tacken, P.J.; Bonetto, F.; Buschow, S.I.; Croes, H.J.; Wijers, M.; de Vries, I.J.; Figdor, C.G. Multimodal imaging of nanovaccine carriers targeted to human dendritic cells. Mol. Pharm. 2011, 8, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Schreibelt, G.; Klinkenberg, L.J.; Cruz, L.J.; Tacken, P.J.; Tel, J.; Kreutz, M.; Adema, G.J.; Brown, G.D.; Figdor, C.G.; de Vries, I.J. The c-type lectin receptor clec9a mediates antigen uptake and (cross-)presentation by human blood bdca3+ myeloid dendritic cells. Blood 2012, 119, 2284–2292. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Tacken, P.J.; Pots, J.M.; Torensma, R.; Buschow, S.I.; Figdor, C.G. Comparison of antibodies and carbohydrates to target vaccines to human dendritic cells via dc-sign. Biomaterials 2012, 33, 4229–4239. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Sittig, S.P.; Blom, R.A.; Cruz, L.J.; Schreibelt, G.; Figdor, C.G.; de Vries, I.J. Targeting uptake receptors on human plasmacytoid dendritic cells triggers antigen cross-presentation and robust type i ifn secretion. J. Immunol. 2013, 191, 5005–5012. [Google Scholar] [CrossRef]
- Ghinnagow, R.; Cruz, L.J.; Macho-Fernandez, E.; Faveeuw, C.; Trottein, F. Enhancement of adjuvant functions of natural killer t cells using nanovector delivery systems: Application in anticancer immune therapy. Front. Immunol. 2017, 8, 879. [Google Scholar] [CrossRef]
- Rosalia, R.A.; Cruz, L.J.; van Duikeren, S.; Tromp, A.T.; Silva, A.L.; Jiskoot, W.; de Gruijl, T.; Lowik, C.; Oostendorp, J.; van der Burg, S.H.; et al. Cd40-targeted dendritic cell delivery of plga-nanoparticle vaccines induce potent anti-tumor responses. Biomaterials 2015, 40, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Bonifaz, L.C.; Bonnyay, D.P.; Charalambous, A.; Darguste, D.I.; Fujii, S.; Soares, H.; Brimnes, M.K.; Moltedo, B.; Moran, T.M.; Steinman, R.M. In vivo targeting of antigens to maturing dendritic cells via the dec-205 receptor improves t cell vaccination. J. Exp. Med. 2004, 199, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Cambi, A.; Figdor, C.G. Levels of complexity in pathogen recognition by c-type lectins. Curr. Opin. Immunol. 2005, 17, 345–351. [Google Scholar] [CrossRef]
- Figdor, C.G.; van Kooyk, Y.; Adema, G.J. C-type lectin receptors on dendritic cells and langerhans cells. Nat. Rev. Immunol. 2002, 2, 77–84. [Google Scholar] [CrossRef]
- Moris, A.; Nobile, C.; Buseyne, F.; Porrot, F.; Abastado, J.P.; Schwartz, O. Dc-sign promotes exogenous mhc-i-restricted HIV-1 antigen presentation. Blood 2004, 103, 2648–2654. [Google Scholar] [CrossRef]
- Moris, A.; Pajot, A.; Blanchet, F.; Guivel-Benhassine, F.; Salcedo, M.; Schwartz, O. Dendritic cells and hiv-specific CD4+ t cells: Hiv antigen presentation, t-cell activation, and viral transfer. Blood 2006, 108, 1643–1651. [Google Scholar] [CrossRef]
- Engering, A.; Geijtenbeek, T.B.; van Vliet, S.J.; Wijers, M.; van Liempt, E.; Demaurex, N.; Lanzavecchia, A.; Fransen, J.; Figdor, C.G.; Piguet, V.; et al. The dendritic cell-specific adhesion receptor dc-sign internalizes antigen for presentation to t cells. J. Immunol. 2002, 168, 2118–2126. [Google Scholar] [CrossRef]
- Kretz-Rommel, A.; Qin, F.; Dakappagari, N.; Torensma, R.; Faas, S.; Wu, D.; Bowdish, K.S. In vivo targeting of antigens to human dendritic cells through dc-sign elicits stimulatory immune responses and inhibits tumor growth in grafted mouse models. J. Immunother 2007, 30, 715–726. [Google Scholar] [CrossRef]
- Dakappagari, N.; Maruyama, T.; Renshaw, M.; Tacken, P.; Figdor, C.; Torensma, R.; Wild, M.A.; Wu, D.; Bowdish, K.; Kretz-Rommel, A. Internalizing antibodies to the c-type lectins, l-sign and dc-sign, inhibit viral glycoprotein binding and deliver antigen to human dendritic cells for the induction of t cell responses. J. Immunol. 2006, 176, 426–440. [Google Scholar] [CrossRef]
- Cambi, A.; Lidke, D.S.; Arndt-Jovin, D.J.; Figdor, C.G.; Jovin, T.M. Ligand-conjugated quantum dots monitor antigen uptake and processing by dendritic cells. Nano Lett. 2007, 7, 970–977. [Google Scholar] [CrossRef]
- Cambi, A.; Beeren, I.; Joosten, B.; Fransen, J.A.; Figdor, C.G. The c-type lectin dc-sign internalizes soluble antigens and hiv-1 virions via a clathrin-dependent mechanism. Eur. J. Immunol. 2009, 39, 1923–1928. [Google Scholar] [CrossRef]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Singh, S.K.; Stephani, J.; Schaefer, M.; Kalay, H.; García-Vallejo, J.J.; den Haan, J.; Saeland, E.; Sparwasser, T.; van Kooyk, Y. Targeting glycan modified ova to murine dc-sign transgenic dendritic cells enhances mhc class i and ii presentation. Mol. Immunol. 2009, 47, 164–174. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Adema, G.J.; van Kooyk, Y.; Figdor, C.G. Identification of dc-sign, a novel dendritic cell-specific icam-3 receptor that supports primary immune responses. Cell 2000, 100, 575–585. [Google Scholar] [CrossRef]
- Tacken, P.J.; de Vries, I.J.; Gijzen, K.; Joosten, B.; Wu, D.; Rother, R.P.; Faas, S.J.; Punt, C.J.; Torensma, R.; Adema, G.J.; et al. Effective induction of naive and recall t-cell responses by targeting antigen to human dendritic cells via a humanized anti-dc-sign antibody. Blood 2005, 106, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, J.S.; Kilgannon, P.; Gallatin, W.M. The intercellular adhesion molecule (icam) family of proteins. New members and novel functions. Immunol. Res. 1998, 17, 313–327. [Google Scholar] [CrossRef]
- Anthony, R.M.; Kobayashi, T.; Wermeling, F.; Ravetch, J.V. Intravenous gammaglobulin suppresses inflammation through a novel t(h)2 pathway. Nature 2011, 475, 110–113. [Google Scholar] [CrossRef]
- Anthony, R.M.; Wermeling, F.; Ravetch, J.V. Novel roles for the igg fc glycan. Ann. N. Y. Acad. Sci. 2012, 1253, 170–180. [Google Scholar]
- Sondermann, P.; Pincetic, A.; Maamary, J.; Lammens, K.; Ravetch, J.V. General mechanism for modulating immunoglobulin effector function. Proc. Natl. Acad. Sci. USA 2013, 110, 9868–9872. [Google Scholar] [CrossRef]
- Bruhns, P. Properties of mouse and human igg receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Rueda, F.; Cordobilla, B.; Simon, L.; Hosta, L.; Albericio, F.; Domingo, J.C. Targeting nanosystems to human dcs via fc receptor as an effective strategy to deliver antigen for immunotherapy. Mol. Pharm. 2011, 8, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Regnault, A.; Lankar, D.; Lacabanne, V.; Rodriguez, A.; Thery, C.; Rescigno, M.; Saito, T.; Verbeek, S.; Bonnerot, C.; Ricciardi-Castagnoli, P.; et al. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class i-restricted antigen presentation after immune complex internalization. J. Exp. Med. 1999, 189, 371–380. [Google Scholar] [CrossRef]
- Schuurhuis, D.H.; Ioan-Facsinay, A.; Nagelkerken, B.; van Schip, J.J.; Sedlik, C.; Melief, C.J.; Verbeek, J.S.; Ossendorp, F. Antigen-antibody immune complexes empower dendritic cells to efficiently prime specific cd8+ ctl responses in vivo. J. Immunol. 2002, 168, 2240–2246. [Google Scholar] [CrossRef]
- Kalergis, A.M.; Ravetch, J.V. Inducing tumor immunity through the selective engagement of activating fcgamma receptors on dendritic cells. J. Exp. Med. 2002, 195, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Amigorena, S.; Bonnerot, C. Fc receptors for igg and antigen presentation on mhc class i and class ii molecules. Semin. Immunol. 1999, 11, 385–390. [Google Scholar] [CrossRef]
- Houghten, R.A.; Bray, M.K.; Degraw, S.T.; Kirby, C.J. Simplified procedure for carrying out simultaneous multiple hydrogen fluoride cleavages of protected peptide resins. Int. J. Pept. Protein Res. 1986, 27, 673–678. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the first author. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NP Vaccines | R848 (mg/mg NP) (% w/w) | Poly I:C (mg/mg NP) (% w/w) | Ags (mg/mg NP) (% w/w) |
|---|---|---|---|
| NP-(FITC-TT) | ------ | ------ | 25.9 ± 4.8 (64.8%) |
| NP-(gp100, R848, Poly I:C) | 6.4 ± 2.3 (40%) | 20.1 ± 3.1 (14.4%) | 30.0 ± 5.2 (75%) |
| NP-(FITC-TT, R848, Poly I:C) | 4.0 ± 1.2 (25%) | 18.9 ± 4.2 (23.3%) | 20.1 ± 2.9 (50.3%) |
| NP Vaccines with Distinct Targeting Moiety | Nanosphere Size ± S.D. (nm) | PDI | Width (nm) | Zeta Potential ± S.D. (mV) | Ligands (# Ligands/NP) (Experimental Values) |
|---|---|---|---|---|---|
| NP-none | 205.1 ± 12.3 | 0.160 | 75.6 | −15.76 ± 1.60 | ---- |
| NP-ICAM3-Fc | 234.1 ± 17.2 | 0.217 | 89.4 | −16.12 ± 6.46 | 520 |
| NP-αDC-SIGN | 237.1 ± 31.9 | 0.213 | 112.7 | −12.96 ± 1.57 | 390 |
| NP-isotype (h5G1.1) | 236.6 ± 27.2 | 0.187 | 92.5 | −11.66 ± 2.34 | 401 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz, L.J.; Tacken, P.J.; van der Schoot, J.M.S.; Rueda, F.; Torensma, R.; Figdor, C.G. ICAM3-Fc Outperforms Receptor-Specific Antibodies Targeted Nanoparticles to Dendritic Cells for Cross-Presentation. Molecules 2019, 24, 1825. https://doi.org/10.3390/molecules24091825
Cruz LJ, Tacken PJ, van der Schoot JMS, Rueda F, Torensma R, Figdor CG. ICAM3-Fc Outperforms Receptor-Specific Antibodies Targeted Nanoparticles to Dendritic Cells for Cross-Presentation. Molecules. 2019; 24(9):1825. https://doi.org/10.3390/molecules24091825
Chicago/Turabian StyleCruz, Luis J., Paul J. Tacken, Johan M.S. van der Schoot, Felix Rueda, Ruurd Torensma, and Carl G. Figdor. 2019. "ICAM3-Fc Outperforms Receptor-Specific Antibodies Targeted Nanoparticles to Dendritic Cells for Cross-Presentation" Molecules 24, no. 9: 1825. https://doi.org/10.3390/molecules24091825
APA StyleCruz, L. J., Tacken, P. J., van der Schoot, J. M. S., Rueda, F., Torensma, R., & Figdor, C. G. (2019). ICAM3-Fc Outperforms Receptor-Specific Antibodies Targeted Nanoparticles to Dendritic Cells for Cross-Presentation. Molecules, 24(9), 1825. https://doi.org/10.3390/molecules24091825