
Sodium-Hydrogen Exchanger Isoform-1 Inhibition: A Promising Pharmacological Intervention for Resuscitation from Cardiac Arrest


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
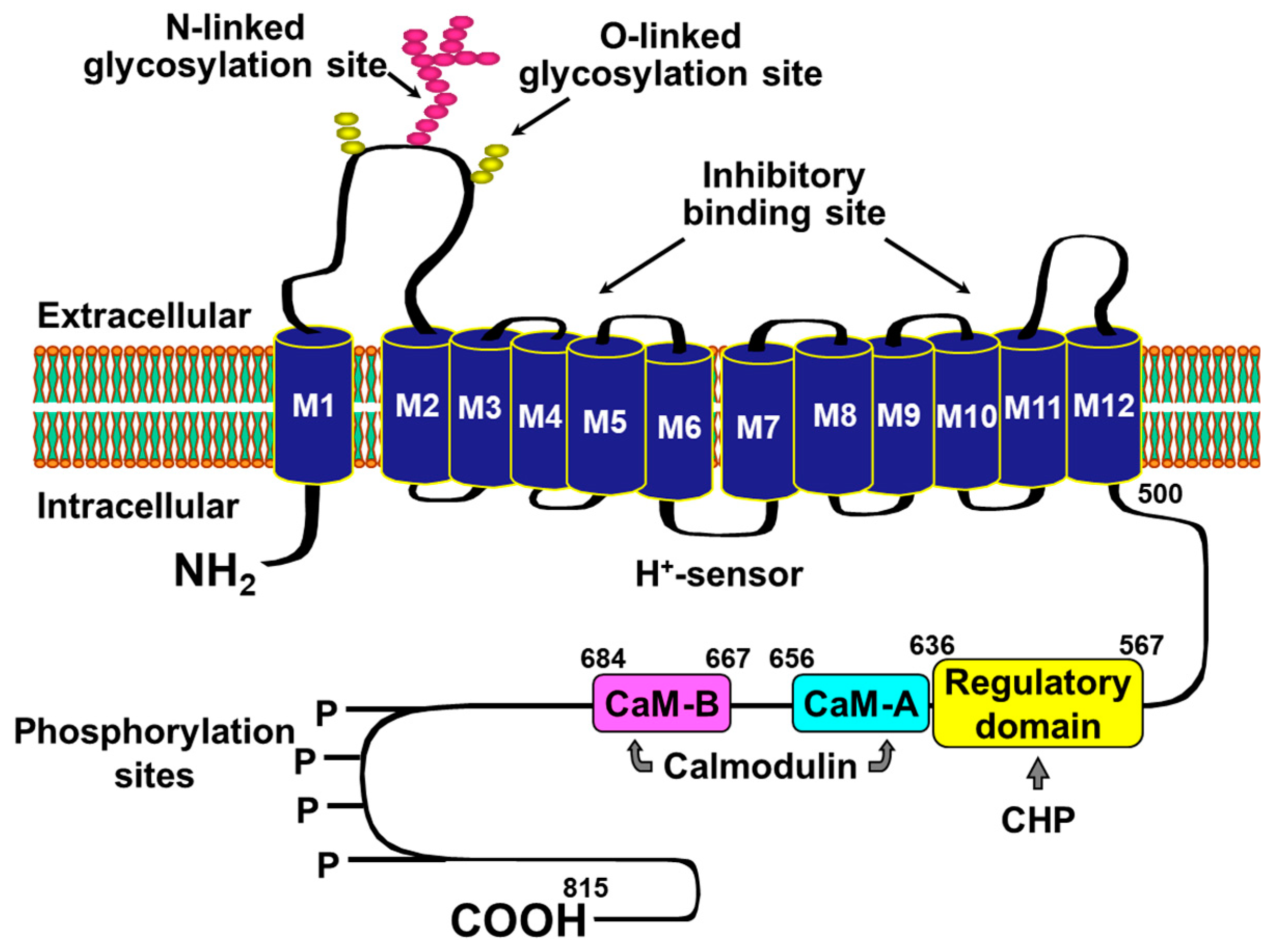
2. Rationale for Targeting NHE-1 during Cardiac Resuscitation
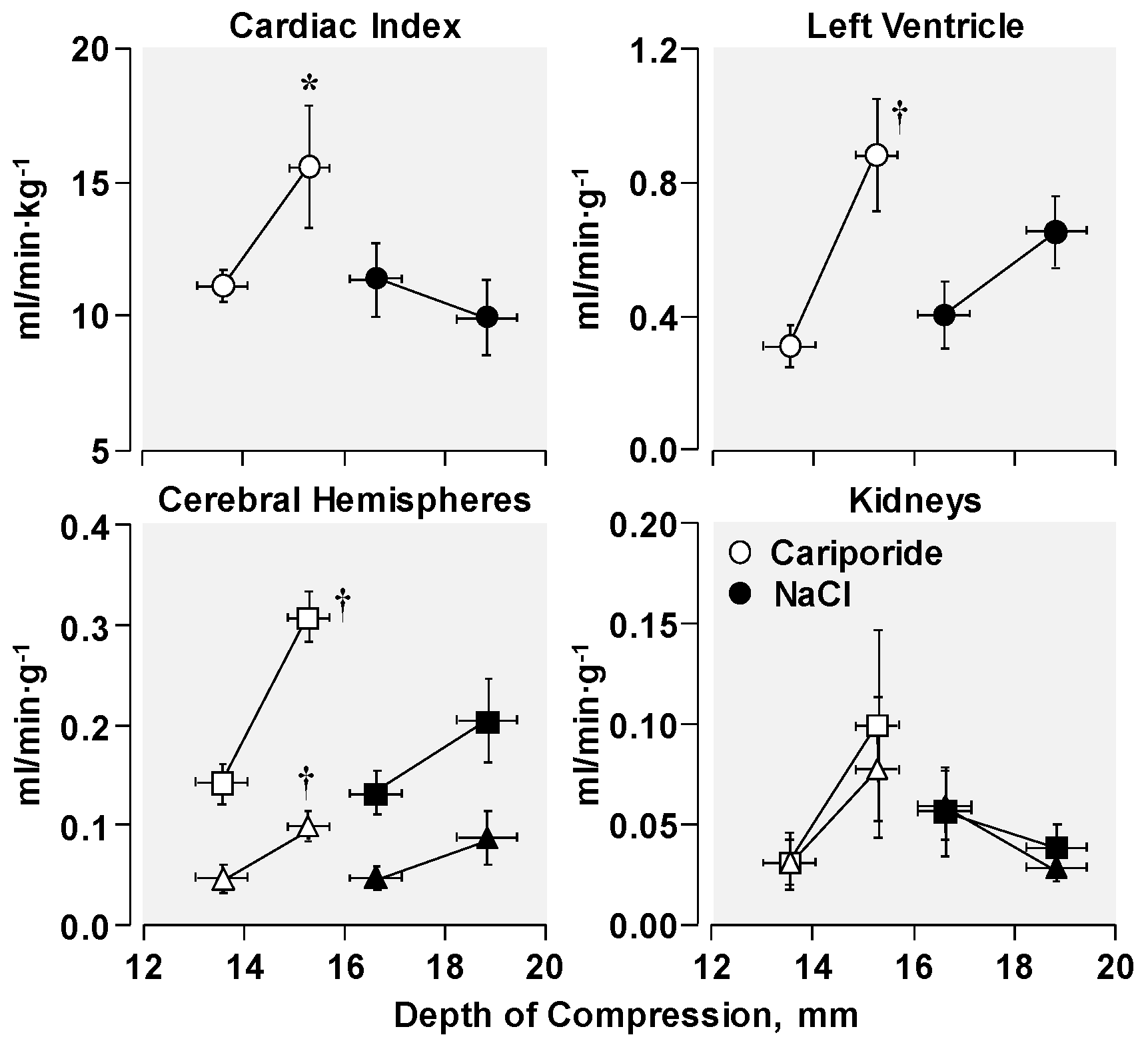
3. Functional Effects of NHE-1 Inhibition during Cardiac Resuscitation
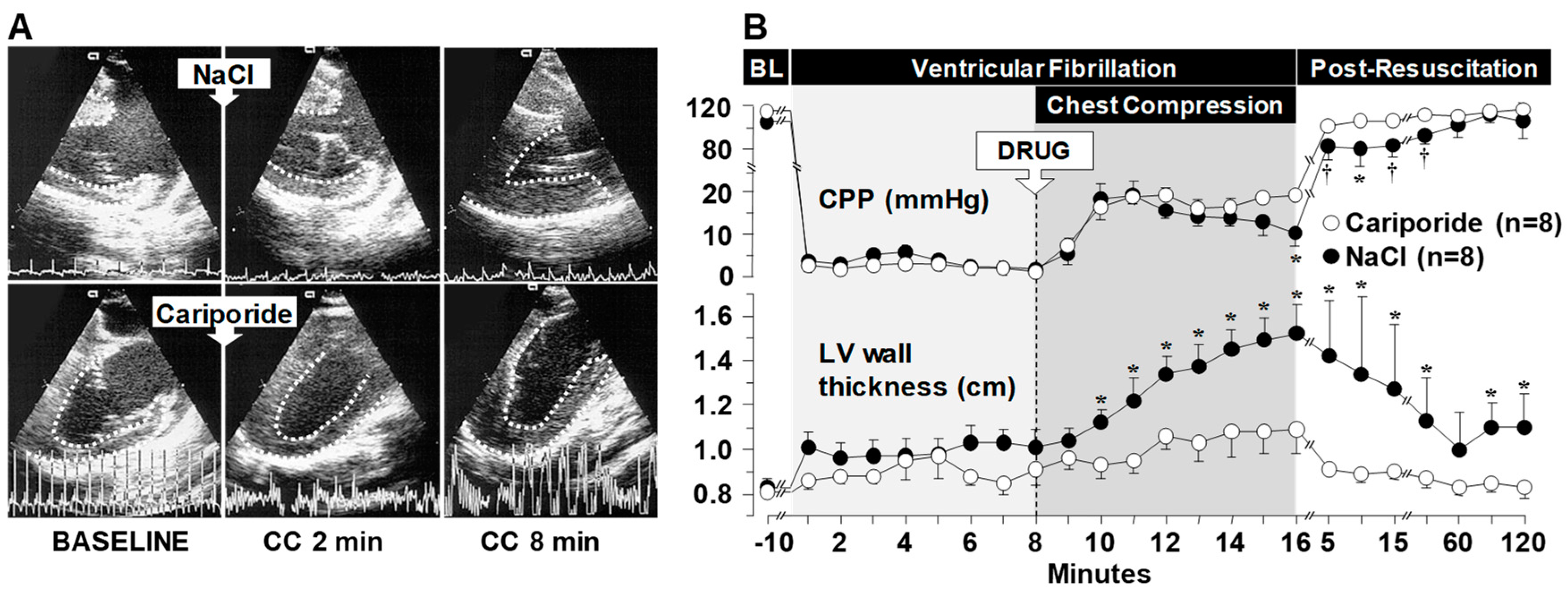
3.1. Effects on Left Ventricular Distensibility during VF-Induced Cardiac Arrest
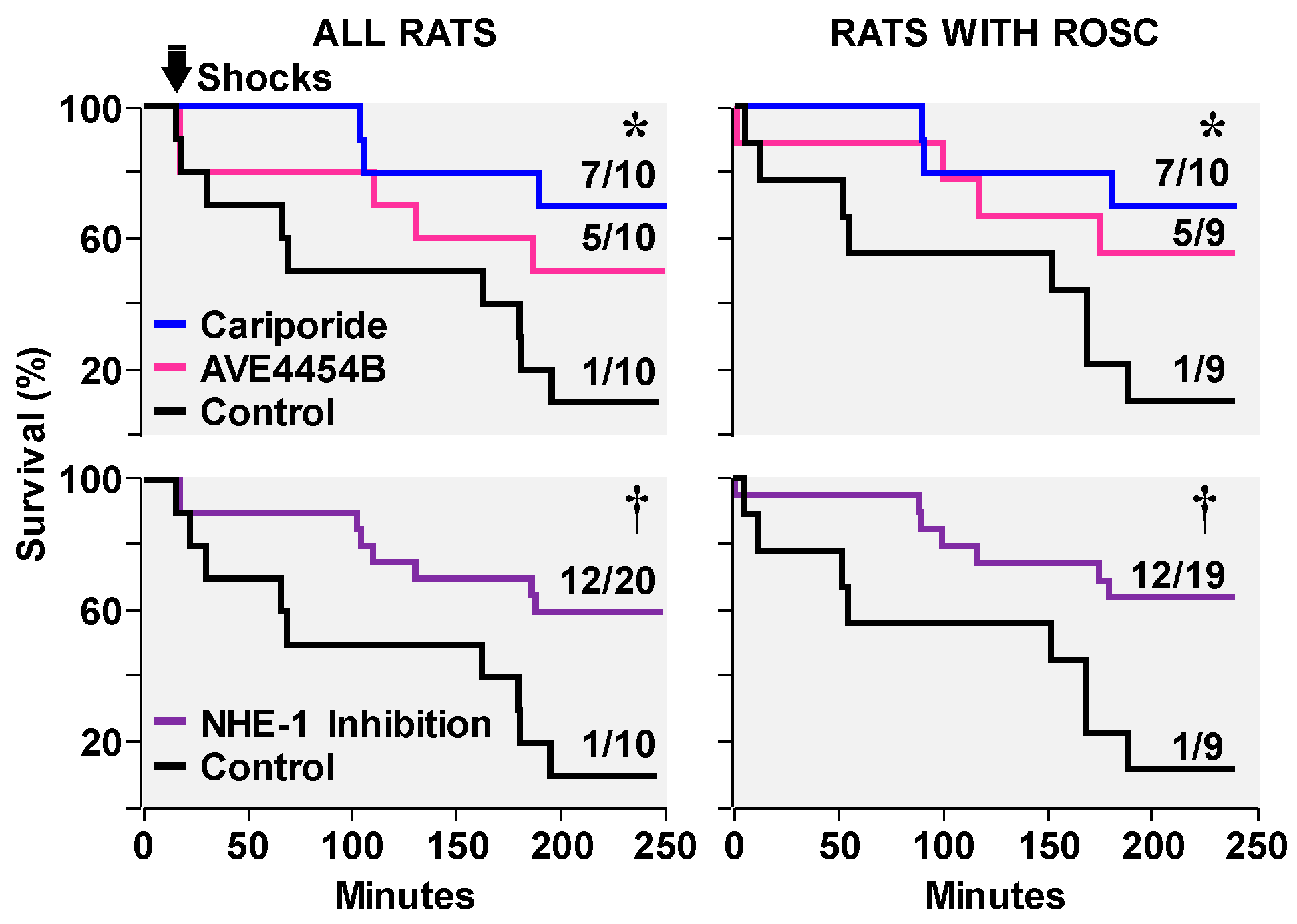
3.2. Effects on Ventricular Fibrillation, Defibrillation, and Post-Resuscitation Electrical Stability
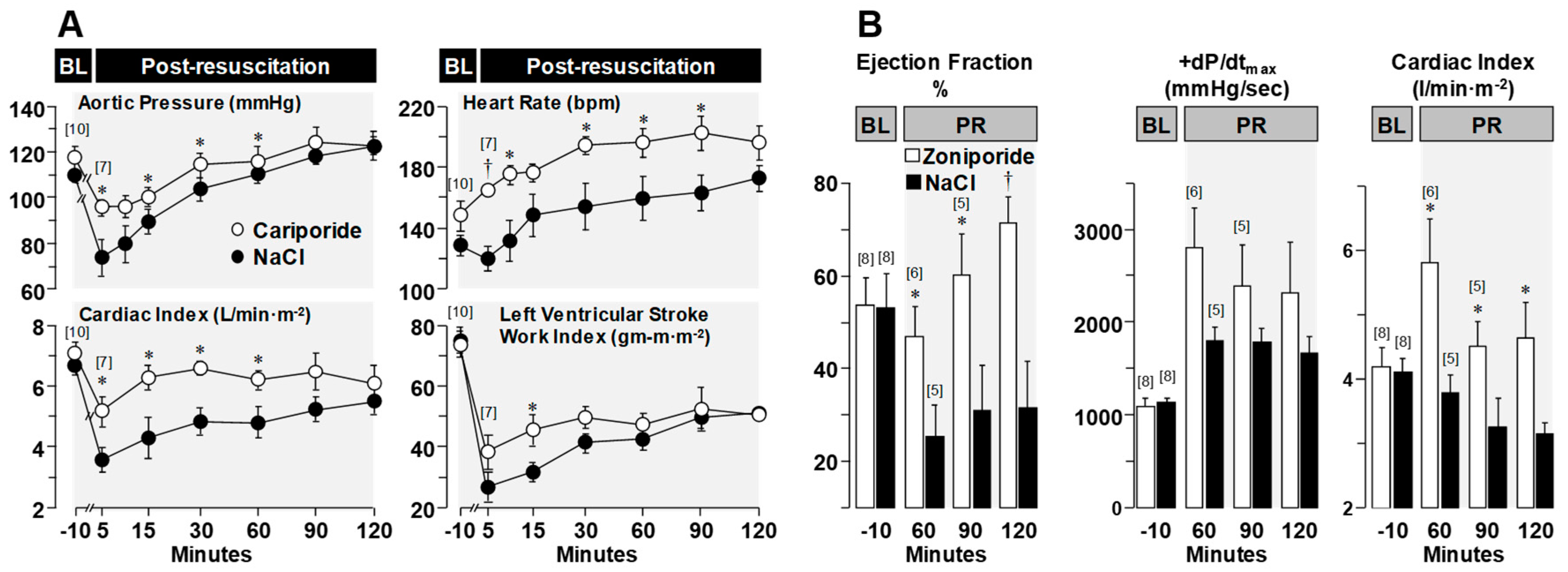
3.3. Effects on Post-Resuscitation Myocardial Function
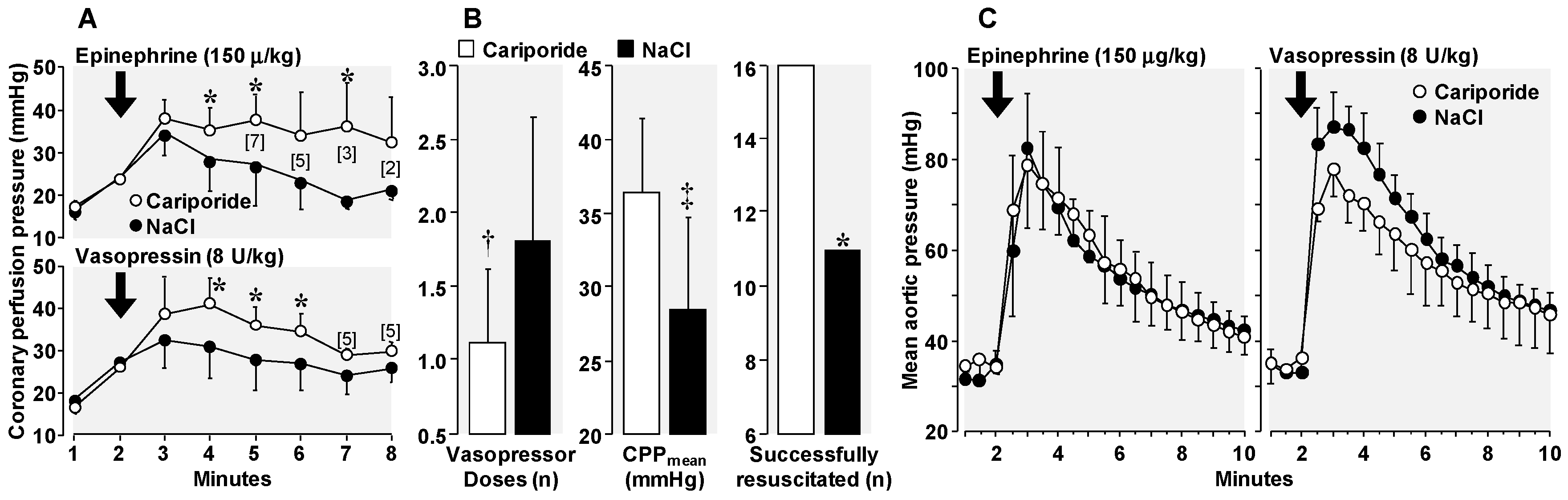
3.4. Amelioration of Adverse Epinephrine Effects
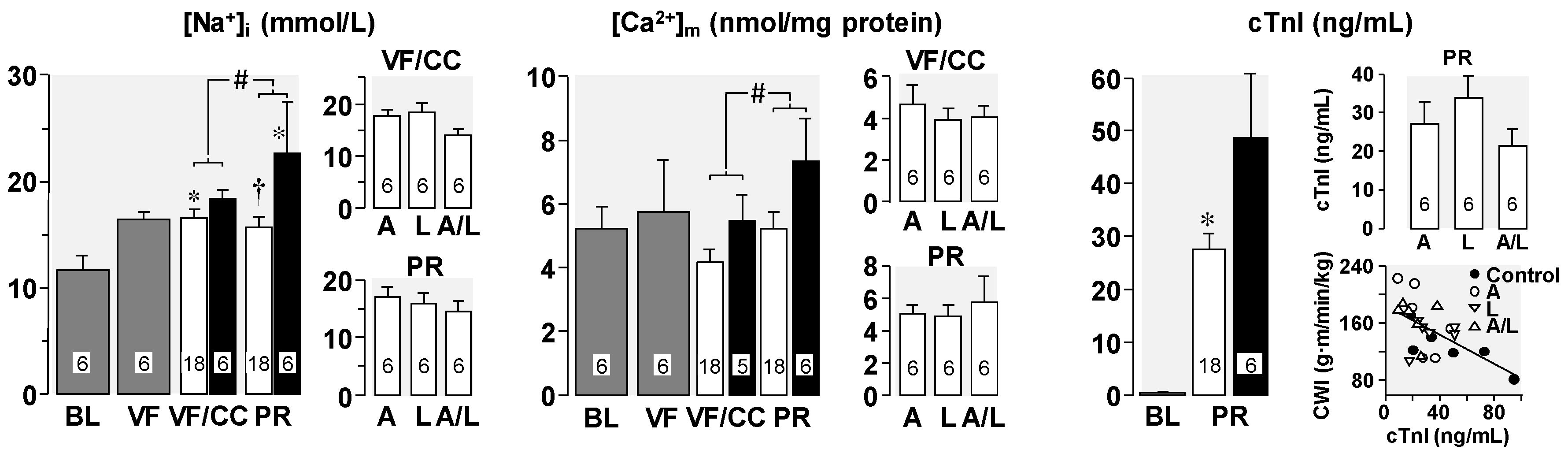
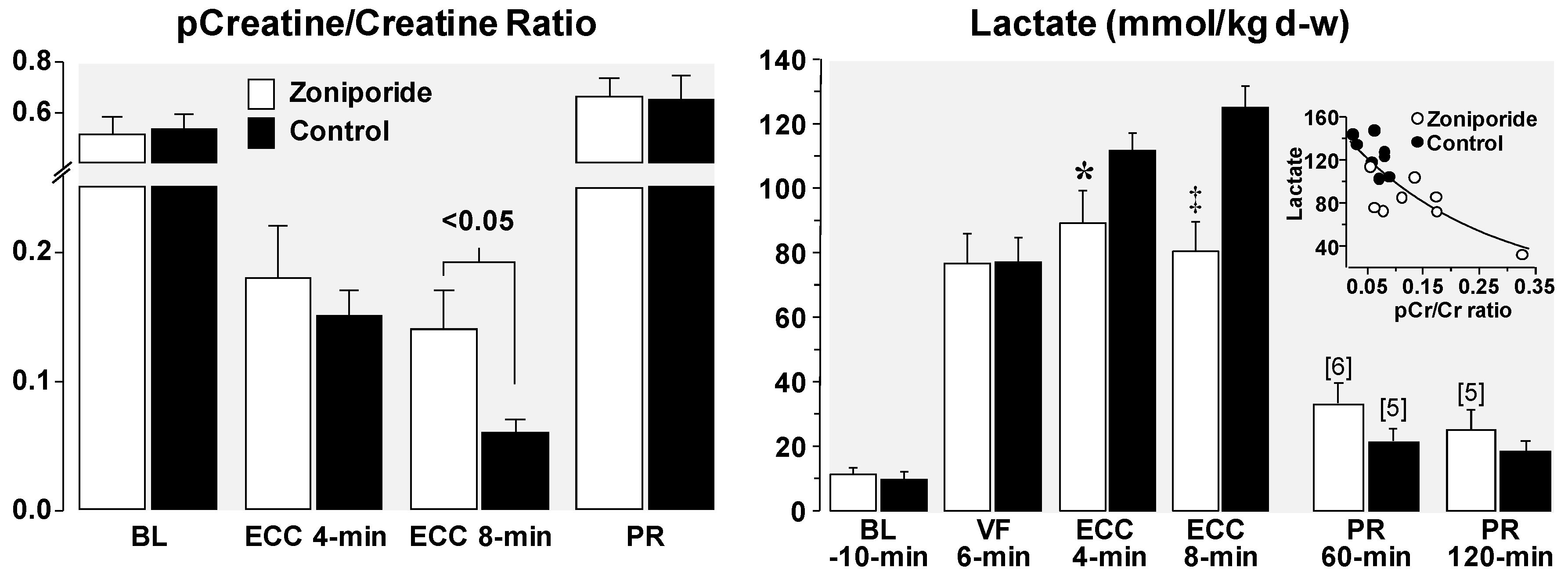
4. Cellular Mechanisms of the Observed Functional Benefits
5. Clinical Translation of NHE-1 Inhibitors
6. Conclusions
Funding
Conflicts of Interest
References
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef]
- Ditchey, R.V.; Horwitz, L.D. Metabolic evidence of inadequate coronary blood flow during closed-chest resuscitation in dogs. Cardiovasc. Res. 1985, 19, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Paradis, N.A.; Martin, G.B.; Rivers, E.P.; Goetting, M.G.; Appleton, T.J.; Feingold, M.; Nowak, R.M. Coronary perfusion pressure and the return of spontaneous circulation in human cardiopulmonary resuscitation. JAMA 1990, 263, 1106–1113. [Google Scholar] [CrossRef]
- Duggal, C.; Weil, M.H.; Gazmuri, R.J.; Tang, W.; Sun, S.; O’Connell, F.; Ali, M. Regional blood flow during closed-chest cardiac resuscitation in rats. J. Appl. Physiol. 1993, 74, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Kolarova, J.D.; Ayoub, I.M.; Gazmuri, R.J. Cariporide enables hemodynamically more effective chest compression by leftward shift of its flow-depth relationship. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2904–H2911. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, I.M.; Kolarova, J.; Kantola, R.; Radhakrishnan, J.; Gazmuri, R.J. Zoniporide preserves left ventricular compliance during ventricular fibrillation and minimizes post-resuscitation myocardial dysfunction through benefits on energy metabolism. Crit. Care Med. 2007, 35, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.M.; Korge, P.; Weiss, J.N. Mitochondria and ischemia/reperfusion injury. Ann. N. Y. Acad. Sci. 2005, 1047, 248–258. [Google Scholar] [CrossRef]
- Robin, E.; Guzy, R.D.; Loor, G.; Iwase, H.; Waypa, G.B.; Marks, J.D.; Vanden Hoek, T.L.; Schumacker, P.T. Oxidant stress during simulated ischemia primes cardiomyocytes for cell death during reperfusion. J. Biol. Chem. 2007, 282, 19133–19143. [Google Scholar] [CrossRef]
- Gazmuri, R.J.; Radhakrishnan, J. Protecting mitochondrial bioenergetic function during resuscitation from cardiac arrest. Crit. Care Clin. 2012, 28, 245–270. [Google Scholar] [CrossRef]
- Ayoub, I.M.; Radhakrishnan, J.; Gazmuri, R.J. In vivo opening of the mitochondrial permeability transition pore in a rat model of ventricular fibrillation and closed-chest resuscitation. Am. J. Transl. Res. 2017, 9, 3345–3359. [Google Scholar]
- Gazmuri, R.J.; Ayoub, I.M.; Hoffner, E.; Kolarova, J.D. Successful ventricular defibrillation by the selective sodium-hydrogen exchanger isoform-1 inhibitor cariporide. Circulation 2001, 104, 234–239. [Google Scholar] [CrossRef]
- Gazmuri, R.J.; Hoffner, E.; Kalcheim, J.; Ho, H.; Patel, M.; Ayoub, I.M.; Epstein, M.; Kingston, S.; Han, Y. Myocardial protection during ventricular fibrillation by reduction of proton-driven sarcolemmal sodium influx. J. Lab. Clin. Med. 2001, 137, 43–55. [Google Scholar] [CrossRef]
- Gazmuri, R.J.; Ayoub, I.M.; Kolarova, J.D.; Karmazyn, M. Myocardial protection during ventricular fibrillation by inhibition of the sodium-hydrogen exchanger isoform-1. Crit. Care Med. 2002, 30, S166–S171. [Google Scholar] [CrossRef]
- Ayoub, I.M.; Kolarova, J.D.; Yi, Z.; Trevedi, A.; Deshmukh, H.; Lubell, D.L.; Franz, M.R.; Maldonado, F.A.; Gazmuri, R.J. Sodium-hydrogen exchange inhibition during ventricular fibrillation: Beneficial effects on ischemic contracture, action potential duration, reperfusion arrhythmias, myocardial function, and resuscitability. Circulation 2003, 107, 1804–1809. [Google Scholar] [CrossRef]
- Kolarova, J.; Ayoub, I.M.; Yi, Z.; Gazmuri, R.J. Optimal timing for electrical defibrillation after prolonged untreated ventricular fibrillation. Crit. Care Med. 2003, 31, 2022–2028. [Google Scholar] [CrossRef]
- Ayoub, I.M.; Kolarova, J.; Kantola, R.L.; Sanders, R.; Gazmuri, R.J. Cariporide minimizes adverse myocardial effects of epinephrine during resuscitation from ventricular fibrillation. Crit. Care Med. 2005, 33, 2599–2605. [Google Scholar] [CrossRef]
- Kolarova, J.; Yi, Z.; Ayoub, I.M.; Gazmuri, R.J. Cariporide potentiates the effects of epinephrine and vasopressin by nonvascular mechanisms during closed-chest resuscitation. Chest 2005, 127, 1327–1334. [Google Scholar] [CrossRef]
- Singh, D.; Kolarova, J.D.; Wang, S.; Ayoub, I.M.; Gazmuri, R.J. Myocardial protection by erythropoietin during resuscitation from ventricular fibrillation. Am. J. Ther. 2007, 14, 361–368. [Google Scholar] [CrossRef]
- Wang, S.; Radhakrishnan, J.; Ayoub, I.M.; Kolarova, J.D.; Taglieri, D.M.; Gazmuri, R.J. Limiting sarcolemmal Na+ entry during resuscitation from VF prevents excess mitochondrial Ca2+ accumulation and attenuates myocardial injury. J. Appl. Physiol. 2007, 103, 55–65. [Google Scholar] [CrossRef]
- Radhakrishnan, J.; Ayoub, I.M.; Gazmuri, R.J. Activation of caspase-3 may not contribute to postresuscitation myocardial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1164–H1174. [Google Scholar] [CrossRef][Green Version]
- Ayoub, I.M.; Kolarova, J.; Gazmuri, R.J. Cariporide given during resuscitation promotes return of electrically stable and mechanically competent cardiac activity. Resuscitation 2010, 81, 106–110. [Google Scholar] [CrossRef][Green Version]
- Radhakrishnan, J.; Kolarova, J.D.; Ayoub, I.M.; Gazmuri, R.J. AVE4454B—A novel sodium-hydrogen exchanger isoform-1 inhibitor—compared less effective than cariporide for resuscitation from cardiac arrest. Transl. Res. 2011, 157, 71–80. [Google Scholar] [CrossRef][Green Version]
- Borovnik-Lesjak, V.; Whitehouse, K.; Baetiong, A.; Artin, B.; Radhakrishnan, J.; Gazmuri, R.J. High-dose erythropoietin during cardiac resuscitation lessens postresuscitation myocardial stunning in swine. Transl. Res. 2013, 162, 110–121. [Google Scholar] [CrossRef]
- Radhakrishnan, J.; Upadhyaya, M.P.; Ng, M.; Edelheit, A.; Moy, H.M.; Ayoub, I.M.; Gazmuri, R.J. Erythropoietin facilitates resuscitation from ventricular fibrillation by signaling protection of mitochondrial bioenergetic function in rats. Am. J. Transl. Res. 2013, 5, 316–326. [Google Scholar]
- Motl, J.; Radhakrishnan, J.; Ayoub, I.M.; Grmec, S.; Gazmuri, R.J. Vitamin C compromises cardiac resuscitability in a rat model of ventricular fibrillation. Am. J. Ther. 2014, 21, 352–357. [Google Scholar] [CrossRef]
- Miao, Y.; Edelheit, A.; Velmurugan, S.; Borovnik-Lesjak, V.; Radhakrishnan, J.; Gazmuri, R.J. Estrogen fails to facilitate resuscitation from ventricular fibrillation in male rats. Am. J. Transl. Res. 2015, 7, 522–534. [Google Scholar]
- Lamoureux, L.; Radhakrishnan, J.; Mason, T.G.; Kraut, J.A.; Gazmuri, R.J. Adverse postresuscitation myocardial effects elicited by buffer-induced alkalemia ameliorated by NHE-1 inhibition in a rat model of ventricular fibrillation. J. Appl. Physiol. (1985) 2016, 121, 1160–1168. [Google Scholar] [CrossRef]
- Avkiran, M. Rational basis for use of sodium-hydrogen exchange inhibitors in myocardial ischemia. Am. J. Cardiol. 1999, 83, 10G–17G. [Google Scholar] [CrossRef]
- Karmazyn, M.; Gan, X.T.; Humphreys, R.A.; Yoshida, H.; Kusumoto, K. The myocardial Na(+)-H(+) exchange: Structure, regulation, and its role in heart disease. Circ. Res. 1999, 85, 777–786. [Google Scholar] [CrossRef]
- Karmazyn, M.; Sostaric, J.V.; Gan, X.T. The myocardial Na+/H+ exchanger: A potential therapeutic target for the prevention of myocardial ischaemic and reperfusion injury and attenuation of postinfarction heart failure. Drugs 2001, 61, 375–389. [Google Scholar] [CrossRef]
- Karmazyn, M.; Sawyer, M.; Fliegel, L. The Na(+)/H(+) exchanger: A target for cardiac therapeutic intervention. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2005, 5, 323–335. [Google Scholar] [CrossRef]
- Fliegel, L.; Dyck, J.R. Molecular biology of the cardiac sodium/hydrogen exchanger. Cardiovasc. Res. 1995, 29, 155–159. [Google Scholar] [CrossRef]
- Karmazyn, M. NHE-1: Still a viable therapeutic target. J. Mol. Cell. Cardiol. 2013, 61, 77–82. [Google Scholar] [CrossRef]
- von Planta, M.; Weil, M.H.; Gazmuri, R.J.; Bisera, J.; Rackow, E.C. Myocardial acidosis associated with CO2 production during cardiac arrest and resuscitation. Circulation 1989, 80, 684–692. [Google Scholar] [CrossRef]
- Kette, F.; Weil, M.H.; Gazmuri, R.J.; Bisera, J.; Rackow, E.C. Intramyocardial hypercarbic acidosis during cardiac arrest and resuscitation. Crit. Care Med. 1993, 21, 901–906. [Google Scholar] [CrossRef]
- Noc, M.; Weil, M.H.; Gazmuri, R.J.; Sun, S.; Bisera, J.; Tang, W. Ventricular fibrillation voltage as a monitor of the effectiveness of cardiopulmonary resuscitation. J. Lab. Clin. Med. 1994, 124, 421–426. [Google Scholar]
- Imahashi, K.; Kusuoka, H.; Hashimoto, K.; Yoshioka, J.; Yamaguchi, H.; Nishimura, T. Intracellular sodium accumulation during ischemia as the substrate for reperfusion injury. Circ. Res. 1999, 84, 1401–1406. [Google Scholar] [CrossRef]
- Avkiran, M.; Ibuki, C.; Shimada, Y.; Haddock, P.S. Effects of acidic reperfusion on arrhythmias and Na(+)-K(+)-ATPase activity in regionally ischemic rat hearts. Am. J. Physiol. 1996, 270, H957–H964. [Google Scholar] [CrossRef]
- Allen, D.G.; Xiao, X.H. Role of the cardiac Na+/H+ exchanger during ischemia and reperfusion. Cardiovasc. Res. 2003, 57, 934–941. [Google Scholar] [CrossRef]
- Hunte, C.; Screpanti, E.; Venturi, M.; Rimon, A.; Padan, E.; Michel, H. Structure of a Na(+)/H(+) antiporter and insights into mechanism of action and regulation by pH. Nature 2005, 435, 1197–1202. [Google Scholar] [CrossRef]
- An, J.; Varadarajan, S.G.; Camara, A.; Chen, Q.; Novalija, E.; Gross, G.J.; Stowe, D.F. Blocking Na(+)/H(+) exchange reduces [Na(+)](i) and [Ca(2+)](i) load after ischemia and improves function in intact hearts. Am. J. Physiol. 2001, 281, H2398–H2409. [Google Scholar]
- Gunter, T.E.; Buntinas, L.; Sparagna, G.; Eliseev, R.; Gunter, K. Mitochondrial calcium transport: mechanisms and functions. Cell Calcium 2000, 28, 285–296. [Google Scholar] [CrossRef]
- Radhakrishnan, J.; Wang, S.; Ayoub, I.M.; Kolarova, J.D.; Levine, R.F.; Gazmuri, R.J. Circulating levels of cytochrome c after resuscitation from cardiac arrest: A marker of mitochondrial injury and predictor of survival. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H767–H775. [Google Scholar] [CrossRef] [PubMed]
- Wann, S.R.; Weil, M.H.; Sun, S.; Tang, W.; Yu, T. Cariporide for pharmacologic defibrillation after prolonged cardiac arrest. J. Cardiovasc. Pharmacol. Ther. 2002, 7, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Rabkin, D.G.; Cabreriza, S.E.; Cheema, F.H.; Hill, A.A.; Curtis, L.J.; Sciacca, R.R.; Mosca, R.S.; Spotnitz, H.M. Cariporide is cardioprotective after iatrogenic ventricular fibrillation in the intact swine heart. Ann. Thorac. Surg. 2003, 76, 1264–1269. [Google Scholar] [CrossRef]
- Fujii, M.; Avkiran, M.; Chambers, D.J. Experimental studies on myocardial protection with intermittent cross-clamp fibrillation: Additive effect of the sodium-hydrogen exchanger inhibitor, cariporide. Ann. Thorac. Surg. 2004, 77, 1398–1407. [Google Scholar] [CrossRef]
- Liakopoulos, O.J.; Hristov, N.; Buckberg, G.D.; Triana, J.; Trummer, G.; Allen, B.S. Resuscitation after prolonged cardiac arrest: Effects of cardiopulmonary bypass and sodium-hydrogen exchange inhibition on myocardial and neurological recovery. Eur. J. Cardiothorac. Surg. 2011, 40, 978–984. [Google Scholar] [CrossRef]
- Lin, X.; Lee, D.; Wu, D. Protective effects of NHE1 inhibition with sabiporide in an experimental model of asphyxia-induced cardiac arrest in piglets. Resuscitation 2013, 84, 520–525. [Google Scholar] [CrossRef]
- Takino, M.; Okada, Y. Firm myocardium in cardiopulmonary resuscitation. Resuscitation 1996, 33, 101–106. [Google Scholar] [CrossRef]
- Gazmuri, R.J.; Berkowitz, M.; Cajigas, H. Myocardial effects of ventricular fibrillation in the isolated rat heart. Crit. Care Med. 1999, 27, 1542–1550. [Google Scholar] [CrossRef]
- Wann, S.R., Sr.; Weil, M.H.; Sun, S.; Tang, W.; Pellis, T. Pharmacologic defibrillation. Crit. Care Med. 2002, 30, S154–S156. [Google Scholar] [CrossRef] [PubMed]
- von Planta, I.; Weil, M.H.; von Planta, M.; Bisera, J.; Bruno, S.; Gazmuri, R.J.; Rackow, E.C. Cardiopulmonary resuscitation in the rat. J. Appl. Physiol. 1988, 65, 2641–2647. [Google Scholar] [CrossRef] [PubMed]
- Gazmuri, R.J.; Weil, M.H.; Bisera, J.; Tang, W.; Fukui, M.; McKee, D. Myocardial dysfunction after successful resuscitation from cardiac arrest. Crit. Care Med. 1996, 24, 992–1000. [Google Scholar] [CrossRef]
- Kern, K.B.; Hilwig, R.W.; Rhee, K.H.; Berg, R.A. Myocardial dysfunction after resuscitation from cardiac arrest: An example of global myocardial stunning. J. Am. Coll. Cardiol. 1996, 28, 232–240. [Google Scholar] [CrossRef]
- Ruiz-Bailen, M.; Aguayo, D.H.; Ruiz-Navarro, S.; Diaz-Castellanos, M.A.; Rucabado-Aguilar, L.; Gomez-Jimenez, F.J.; Martinez-Escobar, S.; Moreno, R.M.; Fierro-Roson, J. Reversible myocardial dysfunction after cardiopulmonary resuscitation. Resuscitation 2005, 66, 175–181. [Google Scholar] [CrossRef]
- Ditchey, R.V.; Lindenfeld, J. Failure of epinephrine to improve the balance between myocardial oxygen supply and demand during closed-chest resuscitation in dogs. Circulation 1988, 78, 382–389. [Google Scholar] [CrossRef]
- Ditchey, R.V.; Rubio-Perez, A.; Slinker, B.K. Beta-adrenergic blockade reduces myocardial injury during experimental cardiopulmonary resuscitation. J. Am. Coll. Cardiol. 1994, 24, 804–812. [Google Scholar] [CrossRef]
- Perkins, G.D.; Ji, C.; Deakin, C.D.; Quinn, T.; Nolan, J.P.; Scomparin, C.; Regan, S.; Long, J.; Slowther, A.; Pocock, H.; et al. A Randomized Trial of Epinephrine in Out-of-Hospital Cardiac Arrest. N. Engl. J. Med. 2018, 379, 711–721. [Google Scholar] [CrossRef]
- Radhakrishnan, J.; Origenes, R.; Littlejohn, G.; Nikolich, S.; Choi, E.; Smite, S.; Lamoureux, L.; Baetiong, A.; Shah, M.; Gazmuri, R.J. Plasma Cytochrome c Detection Using a Highly Sensitive Electrochemiluminescence Enzyme-Linked Immunosorbent Assay. Biomark Insights 2017, 12, 1177271917746972. [Google Scholar] [CrossRef]
- Trivedi, B.; Danforth, W.H. Effect of pH on the kinetics of frog muscle phosphofructokinase. J. Biol. Chem. 1966, 241, 4110–4112. [Google Scholar]
- Scheuer, J.; Berry, M.N. Effect of alkalosis on glycolysis in the isolated rat heart. Am. J. Physiol. 1967, 213, 1143–1148. [Google Scholar] [CrossRef]
- Bristow, J.; Bier, D.M.; Lange, L.G. Regulation of adult and fetal myocardial phosphofructokinase. Relief of cooperativity and competition between fructose 2,6-bisphosphate, ATP, and citrate. J. Biol. Chem. 1987, 262, 2171–2175. [Google Scholar]
- Meert, K.L.; Donaldson, A.; Nadkarni, V.; Tieves, K.S.; Schleien, C.L.; Brilli, R.J.; Clark, R.S.; Shaffner, D.H.; Levy, F.; Statler, K.; et al. Multicenter cohort study of in-hospital pediatric cardiac arrest. Pediatr. Crit. Care Med. 2009, 10, 544–553. [Google Scholar] [CrossRef]
- Raymond, T.T.; Stromberg, D.; Stigall, W.; Burton, G.; Zaritsky, A. Sodium bicarbonate use during in-hospital pediatric pulseless cardiac arrest—A report from the American Heart Association Get With The Guidelines((R))-Resuscitation. Resuscitation 2015, 89, 106–113. [Google Scholar] [CrossRef]
- Rupprecht, H.J.; vom Dahl, J.; Terres, W.; Seyfarth, K.M.; Richardt, G.; Schultheibeta, H.P.; Buerke, M.; Sheehan, F.H.; Drexler, H. Cardioprotective effects of the Na(+)/H(+) exchange inhibitor cariporide in patients with acute anterior myocardial infarction undergoing direct PTCA. Circulation 2000, 101, 2902–2908. [Google Scholar] [CrossRef]
- Boyce, S.W.; Bartels, C.; Bolli, R.; Chaitman, B.; Chen, J.C.; Chi, E.; Jessel, A.; Kereiakes, D.; Knight, J.; Thulin, L.; et al. Impact of sodium-hydrogen exchange inhibition by cariporide on death or myocardial infarction in high-risk CABG surgery patients: results of the CABG surgery cohort of the GUARDIAN study. J. Thorac. Cardiovasc. Surg. 2003, 126, 420–427. [Google Scholar] [CrossRef]
- Zeymer, U.; Suryapranata, H.; Monassier, J.P.; Opolski, G.; Davies, J.; Rasmanis, G.; Linssen, G.; Tebbe, U.; Schroder, R.; Tiemann, R.; et al. The Na(+)/H(+) exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (ESCAMI) trial. J. Am. Coll. Cardiol. 2001, 38, 1644–1650. [Google Scholar] [CrossRef]
- Mentzer, R.M., Jr.; Bartels, C.; Bolli, R.; Boyce, S.; Buckberg, G.D.; Chaitman, B.; Haverich, A.; Knight, J.; Menasche, P.; Myers, M.L.; et al. Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: results of the EXPEDITION study. Ann. Thorac. Surg. 2008, 85, 1261–1270. [Google Scholar] [CrossRef]
- Gazmuri, R.J.; Karmazyn, M. Letter by Gazmuri and Karmazyn Regarding Article, “Activation and Inhibition of Sodium-Hydrogen Exchanger Is a Mechanism That Links the Pathophysiology and Treatment of Diabetes Mellitus With That of Heart Failure”. Circulation 2018, 137, 1979–1980. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gazmuri, R.J.; Radhakrishnan, J.; Ayoub, I.M. Sodium-Hydrogen Exchanger Isoform-1 Inhibition: A Promising Pharmacological Intervention for Resuscitation from Cardiac Arrest. Molecules 2019, 24, 1765. https://doi.org/10.3390/molecules24091765
Gazmuri RJ, Radhakrishnan J, Ayoub IM. Sodium-Hydrogen Exchanger Isoform-1 Inhibition: A Promising Pharmacological Intervention for Resuscitation from Cardiac Arrest. Molecules. 2019; 24(9):1765. https://doi.org/10.3390/molecules24091765
Chicago/Turabian StyleGazmuri, Raúl J., Jeejabai Radhakrishnan, and Iyad M. Ayoub. 2019. "Sodium-Hydrogen Exchanger Isoform-1 Inhibition: A Promising Pharmacological Intervention for Resuscitation from Cardiac Arrest" Molecules 24, no. 9: 1765. https://doi.org/10.3390/molecules24091765
APA StyleGazmuri, R. J., Radhakrishnan, J., & Ayoub, I. M. (2019). Sodium-Hydrogen Exchanger Isoform-1 Inhibition: A Promising Pharmacological Intervention for Resuscitation from Cardiac Arrest. Molecules, 24(9), 1765. https://doi.org/10.3390/molecules24091765