
Silymarin’s Inhibition and Treatment Effects for Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
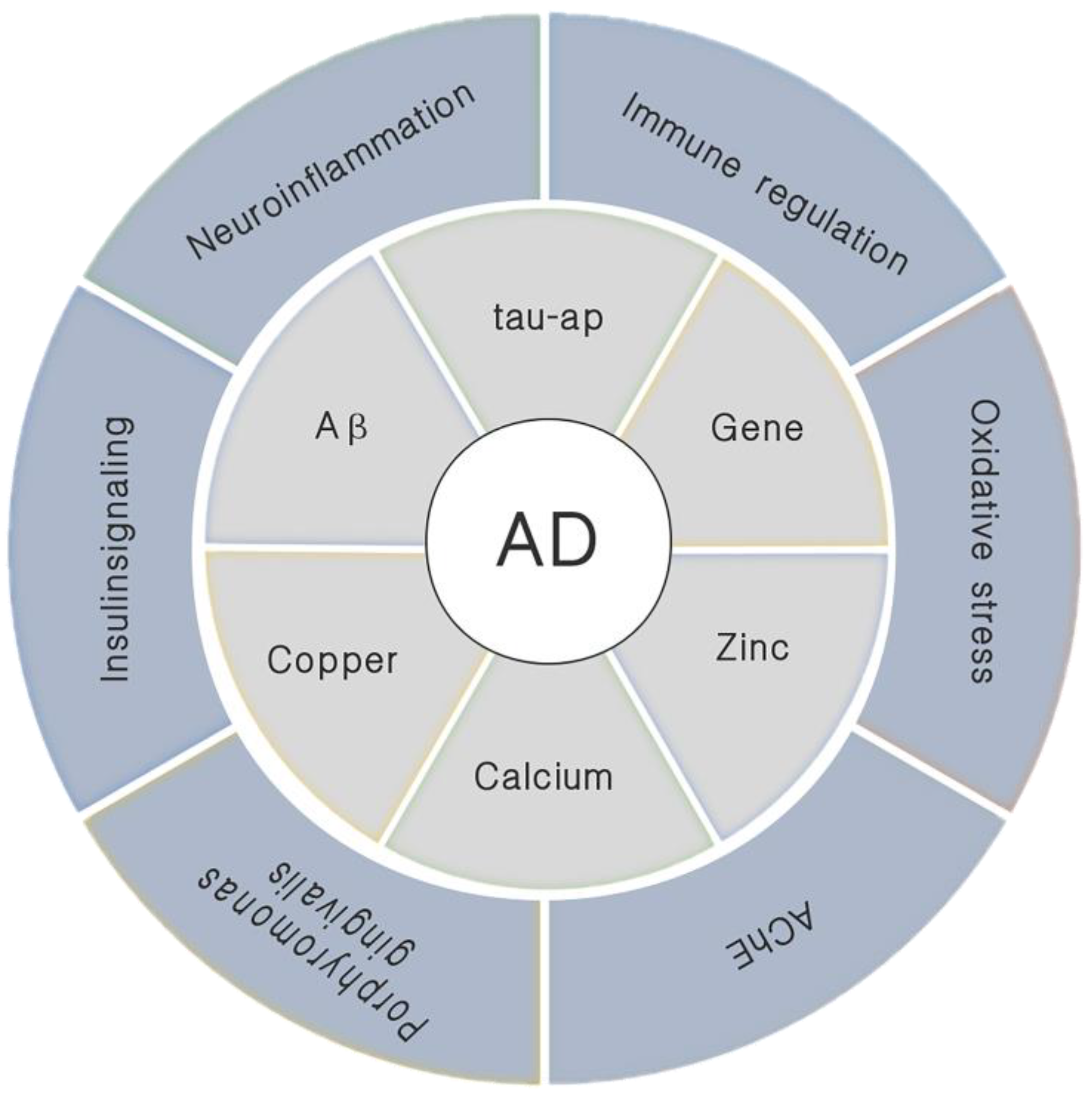
1. Introduction
2. Aβ Inhibitor
2.1. Aβ and the Onset of AD
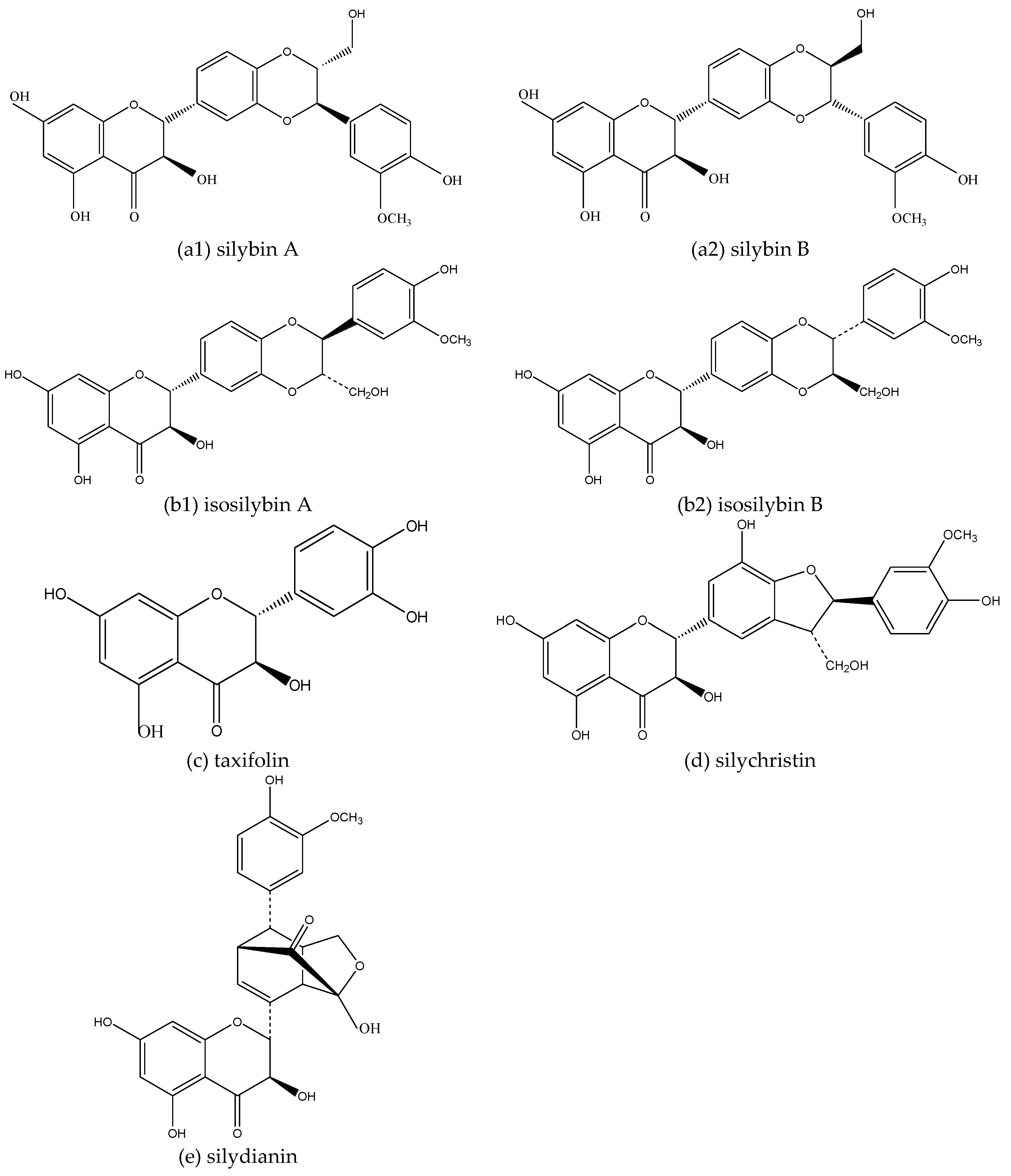
2.2. Inhibition of Silymarin on Aβ and Aβ Aggregate Products
3. Enhancing Cholinergic Energy
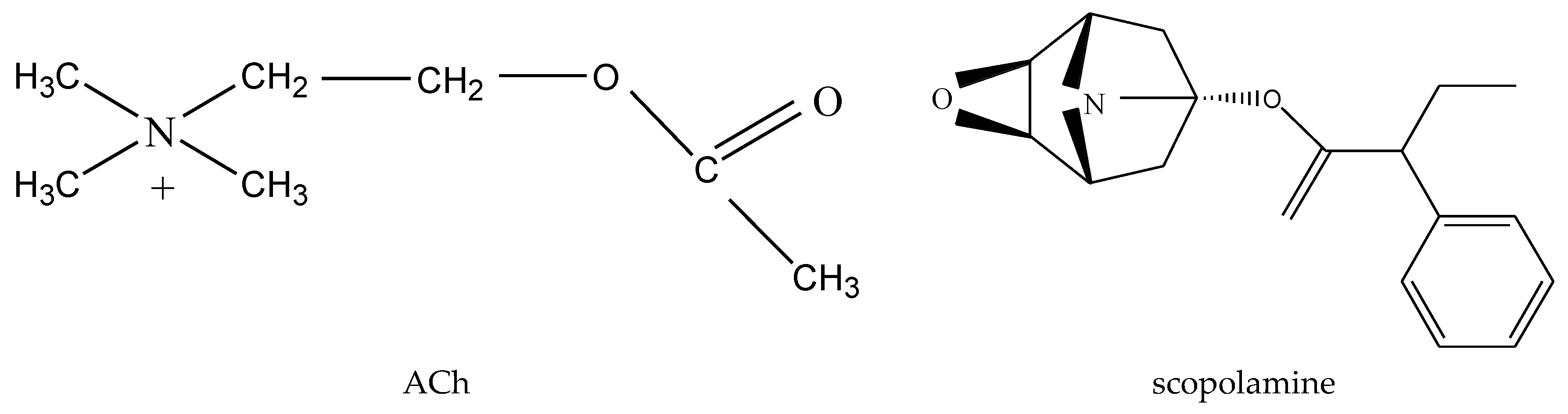
3.1. ACh Deficiency and the Onset of AD
3.2. Silymarin Inhibits AChE and BChE
4. Oxidative Stress Protectant
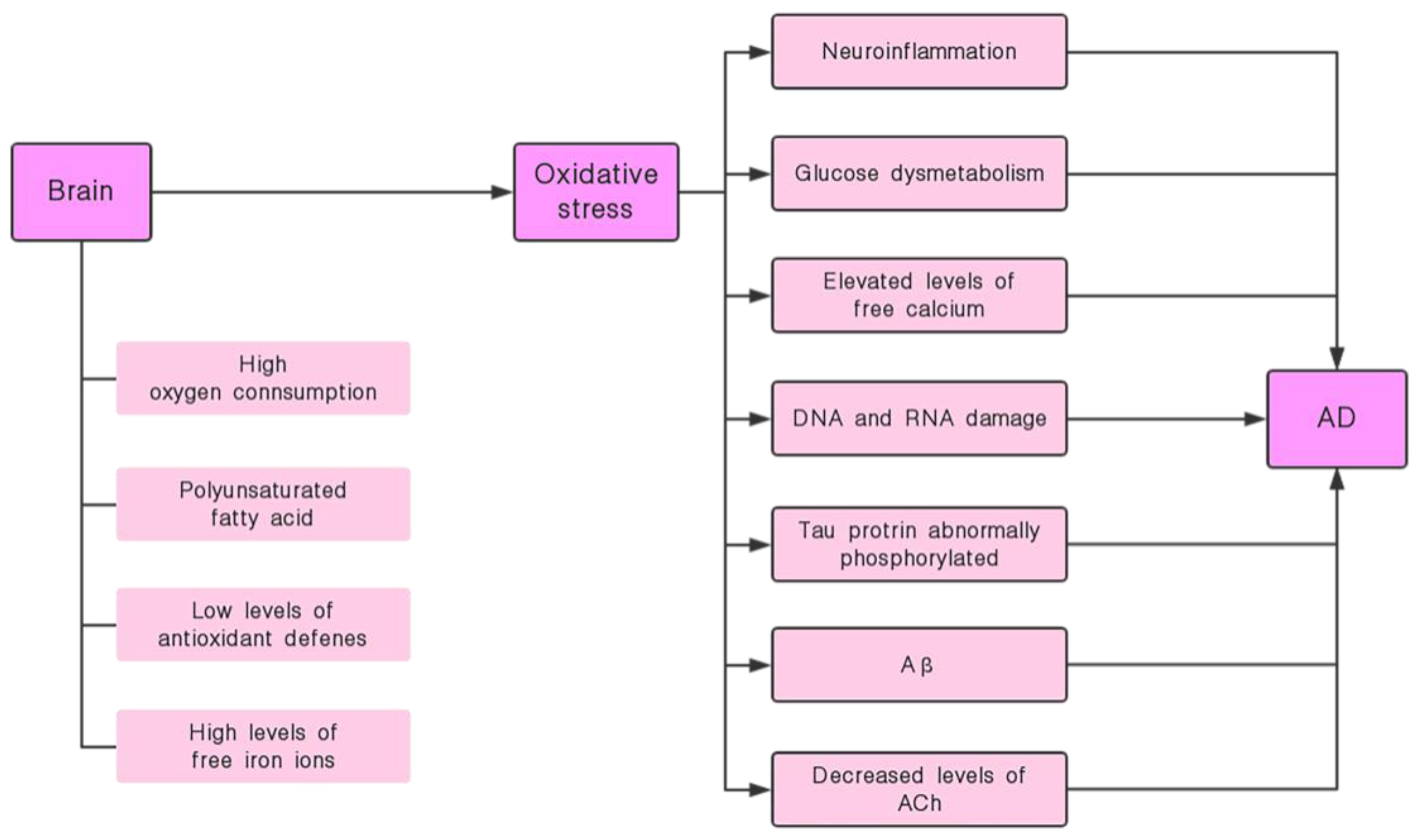
4.1. Oxidative Stress and the Onset of AD
4.2. Silymarin has the Effect of Inhibiting Oxidative Stress and its Induced Nerve Damage
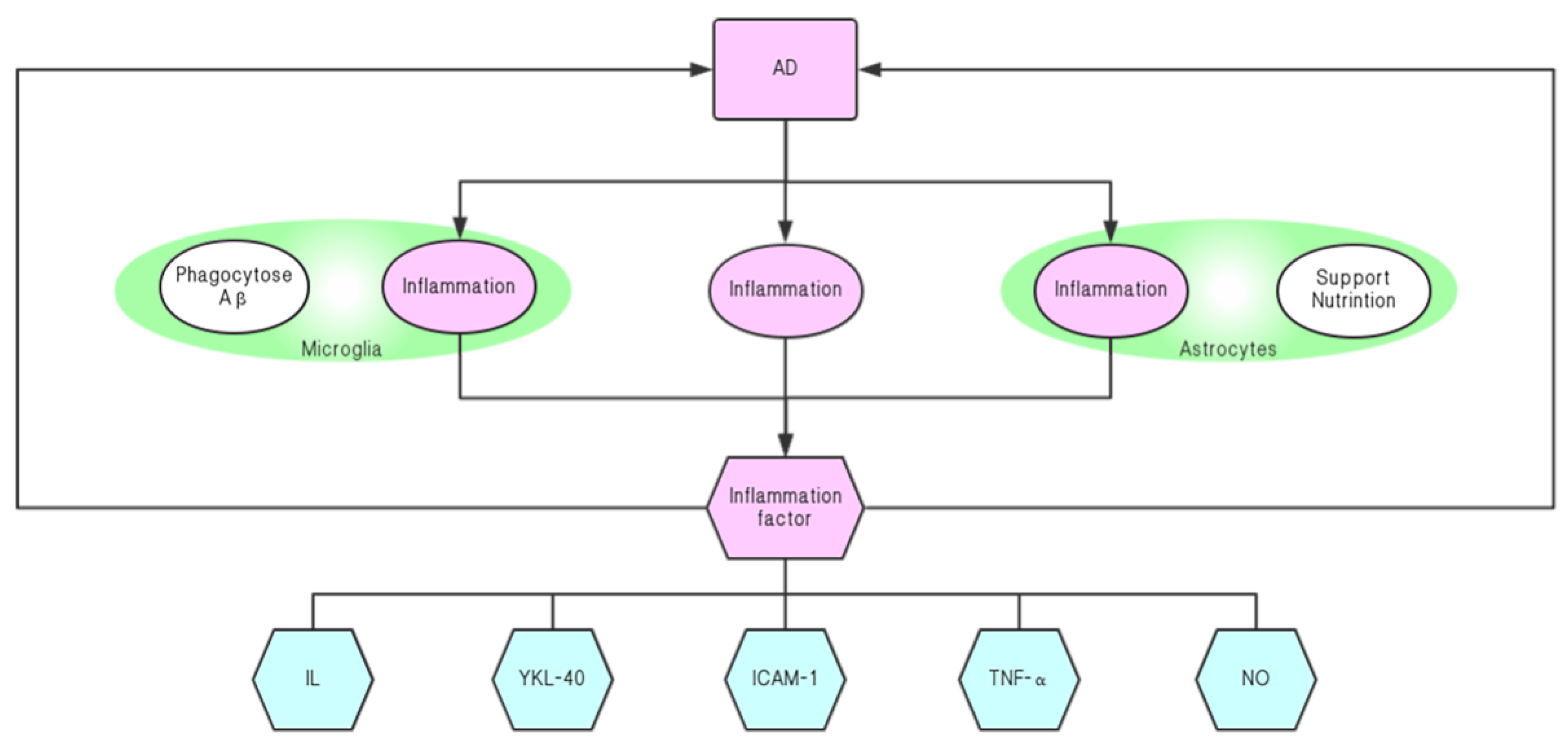
5. Neuroinflammatory Resistance Agent
5.1. Neuroinflammatory Response and the Onset of AD
5.2. Silymarin Relieves the Progression of AD by Inhibiting Neuroinflammation
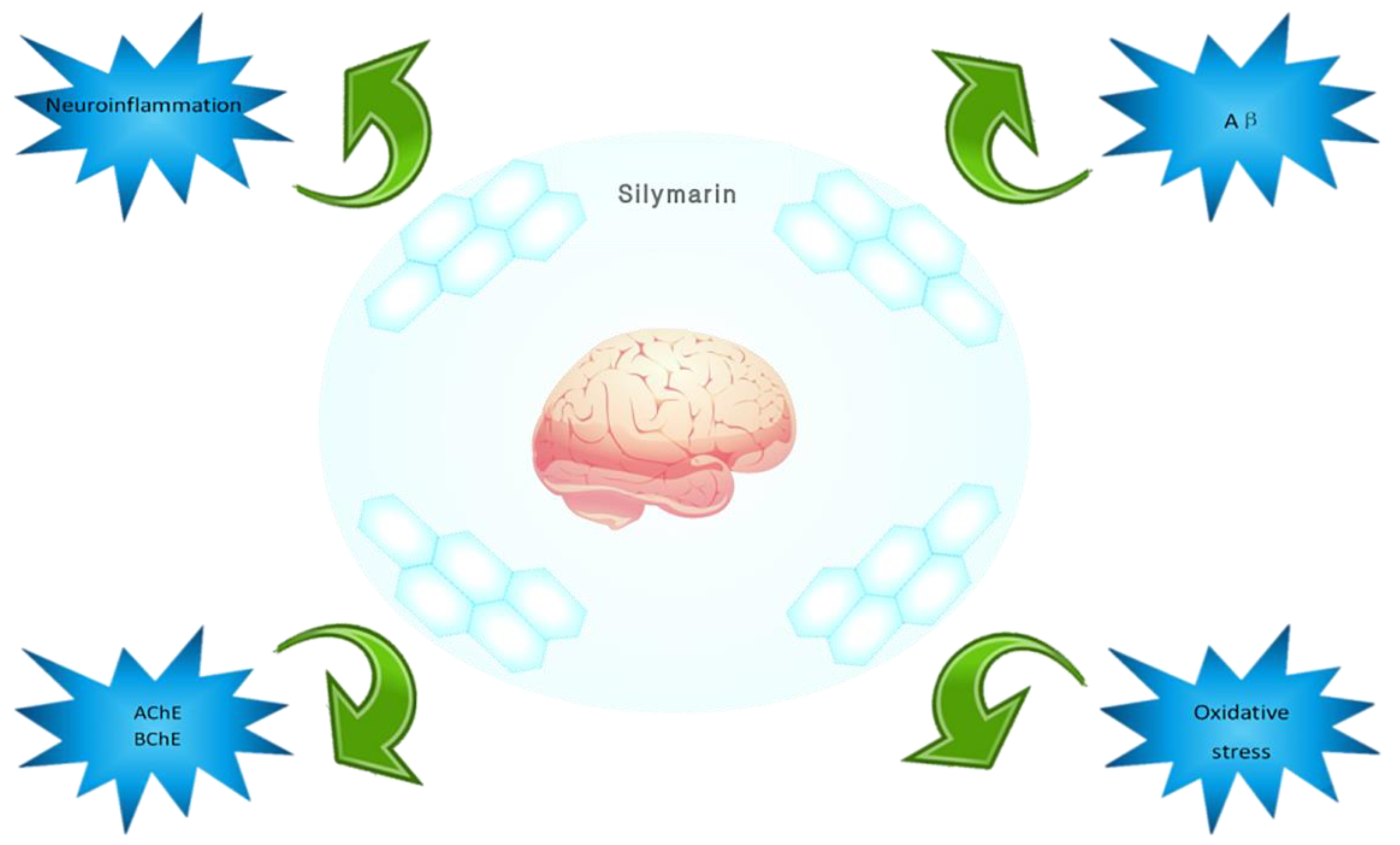
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Rauk, A. The chemistry of Alzheimer’s disease. Chem. Soc. Rev. 2009, 38, 2698–2715. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Sciacca, M.F.; Kotler, S.A.; Brender, J.R.; Chen, J.; Lee, D.K.; Ramamoorthy, A. Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys. J. 2012, 103, 702–710. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Tempra, C.; Scollo, F.; Milardi, D.; La Rosa, C. Amyloid growth and membrane damage: Current themes and emerging perspectives from theory and experiments on Aβ and hIAPP. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1625–1638. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Singhrao, S.K.; Olsen, I. Assessing the role of Porphyromonas gingivalis in periodontitis to determine a causative relationship with Alzheimer’s disease. J. Oral Microbiol. 2019, 11, 1563405. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; Bush, A.I. Biometals and their therapeutic implications in Alzheimer’s disease. Neurotherapeutics 2015, 12, 109–120. [Google Scholar] [CrossRef]
- Atrián-Blasco, E.; Gonzalez, P.; Santoro, A.; Alies, B.; Faller, P.; Hureau, C. Cu and Zn coordination to amyloid peptides: From fascinating chemistry to debated pathological relevance. Coord. Chem. Rev. 2018, 375, 38–55. [Google Scholar] [CrossRef]
- Tamano, H.; Takeda, A. Is interaction of amyloid β-peptides with metals involved in cognitive activity? Metallomics 2015, 7, 1205–1212. [Google Scholar] [CrossRef]
- Lanza, V.; Milardi, D.; Di Natale, G.; Pappalardo, G. Repurposing of Copper(II)-chelating Drugs for the Treatment of Neurodegenerative Diseases. Curr. Med. Chem. 2018, 25, 525–539. [Google Scholar] [CrossRef]
- Gu, M.; Bode, D.C.; Viles, J.H. Copper Redox Cycling Inhibits Aβ Fibre Formation and Promotes Fibre Fragmentation, while Generating a Dityrosine Aβ Dimer. Sci. Rep. 2018, 8, 16190. [Google Scholar] [CrossRef]
- Křen, V.; Walterová, D. Silybin and silymarin-new effects and applications. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc. Czech Repub. 2005, 149, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Laferla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Castellani, R.J.; Plascencia-Villa, G.; Perry, G. The amyloid cascade and Alzheimer’s disease therapeutics: Theory versus observation. Lab. Investig. 2019. [Google Scholar] [CrossRef]
- Shen, Y.; Chen, Y.; Fu, J.; Ye, Y. Molecular mechanisms underlying the pathophysiology of Alzheimer’s disease. Chin. Bull. Life Sci. 2014, 26, 550–559. [Google Scholar]
- Viola, K.L.; Velasco, P.T.; Klein, W.L. Why Alzheimer’s is a disease of memory: The attack on synapses by A beta oligomers (ADDLs). J. Nutr. Health Aging 2008, 12, S51–S57. [Google Scholar] [CrossRef]
- Masliah, E.; Mallory, M.; Alford, M.; DeTeresa, R.; Hansen, L.A.; McKeel, D.W., Jr.; Morris, J.C. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 2001, 56, 127–129. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; DeKosky, S.T.; Mufson, E.J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007, 68, 1501–1508. [Google Scholar] [CrossRef]
- Klyubin, I.; Walsh, D.M.; Lemere, C.A.; Cullen, W.K.; Shankar, G.M.; Betts, V.; Spooner, E.T.; Jiang, L.; Anwyl, R.; Selkoe, D.J.; et al. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat. Med. 2005, 11, 556–561. [Google Scholar] [CrossRef]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef]
- Knobloch, M.; Mansuy, I.M. Dendritic Spine Loss and Synaptic Alterations in Alzheimer’s Disease. Mol. Neurobiol. 2008, 37, 73–82. [Google Scholar] [CrossRef]
- Li, S.; Li, W.; Sun, A. Tau propagation and its modulation in Alzheimer’s disease. Chin. J. Pathophysiol. 2019, 35, 571–576. [Google Scholar]
- Scollo, F.; Tempra, C.; Lolicato, F.; Sciacca, M.F.M.; Raudino, A.; Milardi, D.; La Rosa, C. Phospholipids Critical Micellar Concentrations Trigger Different Mechanisms of Intrinsically Disordered Proteins Interaction with Model Membranes. J. Phys. Chem. Lett. 2018, 9, 5125–5129. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Monaco, I.; La Rosa, C.; Milardi, D. The active role of Ca2+ ions in Aβ-mediated membrane damage. Chem. Commun. (Camb) 2018, 54, 3629–3631. [Google Scholar] [CrossRef]
- Cai, Z.; Zhao, B.; Ratka, A. Oxidative Stress and β-Amyloid Protein in Alzheimer’s Disease. Neuromol. Med. 2011, 13, 223–250. [Google Scholar] [CrossRef]
- Combs, C.K.; Johnson, D.E.; Karlo, J.C.; Cannady, S.B.; Landreth, G.E. Inflammatory mechanisms in Alzheimer’s disease: Inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J. Neurosci. 2000, 20, 558–567. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Zhang, L.; Fiala, M.; Cashman, J.; Sayre, J.; Espinosa, A.; Mahanian, M.; Zaghi, J.; Badmaev, V.; Graves, M.C.; Bernard, G.; et al. Curcuminoids enhance amyloid-beta uptake by macrophages of Alzheimer’s disease patients. J. Alzheimers Dis. 2006, 10, 1–7. [Google Scholar] [CrossRef]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Murata, N.; Murakami, K.; Ozawa, Y.; Kinoshita, N.; Irie, K.; Shirasawa, T.; Shimizu, T. Silymarin attenuated the amyloid β plaque burden and improved behavioral abnormalities in an Alzheimer’s disease mouse model. Biosci. Biotechnol. Biochem. 2010, 74, 2299–2306. [Google Scholar] [CrossRef]
- Yin, F.; Liu, J.; Ji, X.; Wang, Y.; Zidichouski, J.; Zhang, J. Silibinin: A novel inhibitor of Ab aggregation. Neurochem. Int. 2011, 58, 399–403. [Google Scholar] [CrossRef]
- Theuns, J.; Brouwers, N.; Engelborghs, S.; Sleegers, K.; Bogaerts, V.; Corsmit, E.; De Pooter, T.; van Duijn, C.M.; De Deyn, P.P.; Van Broeckhoven, C. Promoter mutations that increase amyloid precursor-protein expression are associated with Alzheimer disease. Am. J. Hum. Genet. 2006, 78, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Howlett, D.R.; Richardson, J.C. The pathology of APP transgenic mice: A model of Alzheimer’s disease or simply overexpression of APP? Histol. Histopathol. 2009, 24, 83–100. [Google Scholar]
- Lukiw, W.J. Gene expression profiling in fetal, aged, and Alzheimer hippocampus: A continuum of stress-related signaling. Neurochem. Res. 2004, 29, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Yaghmaei, P.; Azarfar, K.; Dezfulian, M.; Ebrahim-Habibi, A. Silymarin effect on amyloid-β plaque accumulation and gene expression of APP in an Alzheimer’s disease rat model. Daru 2014, 22, 24. [Google Scholar] [CrossRef]
- Sato, M.; Murakami, K.; Uno, M.; Ikubo, H.; Nakagawa, Y.; Katayama, S.; Akagi, K.; Irie, K. Structure-activity relationship for (+)-taxifolin isolated from silymarin as an inhibitor of amyloid β aggregation. Biosci. Biotechnol. Biochem. 2013, 77, 1100–1103. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Romanucci, V.; Zarrelli, A.; Monaco, I.; Lolicato, F.; Spinella, N.; Galati, C.; Grasso, G.; D’Urso, L.; Romeo, M.; et al. Inhibition of Aβ Amyloid Growth and Toxicity by Silybins: The Crucial Role of Stereochemistry. ACS Chem. Neurosci. 2017, 8, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Liu, F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog. Neurobiol. 2008, 85, 148–175. [Google Scholar] [CrossRef] [PubMed]
- Koleske, A.J. Molecular mechanisms of dendrite stability. Nat. Rev. Neurosci. 2013, 14, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.L.; Wang, Y.H. Injury and main mechanism of central cholinergic system in Alzheimer’s disease. Chin. J. Gerontol. 2010, 30, 3840–3842. [Google Scholar]
- Guix, F.X.; Uribesalgo, I.; Coma, M.; Muñoz, F.J. The Physiology and pathophysiology of nitric oxide in the brain. Prog. Neurobiol. 2005, 76, 126–152. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Wang, Z. The changes of praxiology, choline acetyltransferase and synaptophsin in model rats with Alzheimer’s disease. Chin. J. Gerontol. 2004, 24, 658–659. [Google Scholar]
- Loizzo, M.R.; Tundis, R.; Menichini, F.; Menichini, F. Natural products and their derivatives as cholinesterase inhibitors in the treatment of neurodegenerative disorders: An update. Curr. Med. Chem. 2008, 15, 1209–1228. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, M.; Bertucci, C.; Cavrini, V.; Andrisano, V. β-Amyloid aggregation induced by human acetylcholinesterase: Inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. [Google Scholar] [CrossRef]
- Chacon, M.A.; Reyes, A.E.; Inestrosa, N.C. Acetylcholinesterase induces neuronal cell loss, astrocyte hypertrophy and behavioral deficits in mammalian hippocampus. J. Neurochem. 2003, 87, 195–204. [Google Scholar] [CrossRef]
- Rees, T.M.; Berson, A.; Sklan, E.H.; Younkin, L.; Younkin, S.; Brimijoin, S.; Soreq, H. Memory deficits correlating with acetylcholinesterase splice shift and amyloid burden in doubly transgenic mice. Curr. Alzheimer Res. 2005, 2, 291–300. [Google Scholar] [CrossRef]
- Shi, Q.; Xue, J.; Liu, Y.; Li, J. Research advances in multi-target directed ligands based on dual binding site acetylcholinesterase inhibition for the treatment of Alzheimer’s disease. Chin. J. Clin. Pharmacol. Ther. 2016, 21, 943–949. [Google Scholar]
- Inestrosa, N.C.; Dinamarca, M.C.; Alvarez, A. Amyloid-cholinesterase interactions: Implications for Alzheimer’s disease. FEBS J. 2008, 275, 625–632. [Google Scholar] [CrossRef]
- Perry, E.K. The cholinergic system in old age and Alzheimer’s disease. Age Ageing 1980, 9, 1–8. [Google Scholar] [CrossRef]
- Orhan, I.; Kartal, M.; Tosun, F.; Sener, B. Screening of various phenolic acids and flavonoid derivatives for their anticholinesterase potential. Z. Naturforsch. C 2007, 62, 829–832. [Google Scholar] [CrossRef]
- Nazir, N.; Karim, N.; Abdel-Halim, H.; Khan, I.; Wadood, S.F.; Nisar, M. Phytochemical analysis, molecular docking and antiamnesic effects of methanolic extract of Silybum marianum (L.) Gaertn seeds in scopolamine induced memory impairment in mice. J. Ethnopharmacol. 2018, 210, 198–208. [Google Scholar] [CrossRef]
- Kiruthiga, P.V.; Karutha Pandian, S.; Pandima Devi, K. Silymarin prevents the toxicity induced by benzo(a)pyrene in human erythrocytes by preserving its membrane integrity: An in vitro study. Environ. Toxicol. 2014, 29, 165–175. [Google Scholar] [CrossRef]
- El-Marasy, S.A.; Abd-Elsalam, R.M.; Ahmed-Farid, O.A. Ameliorative Effect of Silymarin on Scopolamine-induced Dementia in Rats. Open Access Maced. J. Med. Sci. 2018, 6, 1215–1224. [Google Scholar] [CrossRef]
- Qiao, D.; Seidler, F.J.; Slotkin, A.T. Oxidative mechanisms contributing to the developmental neurotoxicity of nicotine and chlorpyrifos. Toxicol. Appl. Pharmacol. 2005, 206, 17–26. [Google Scholar] [CrossRef]
- Balu, M.; Sangeetha, P.; Haripriya, D.; Panneerselvam, C. Rejuvenation of the antioxidant system in the central nervous system of aged rats by grape seed extract. Neurosci. Lett. 2005, 383, 295–300. [Google Scholar] [CrossRef]
- Praticò, D. Evidence of oxidative stress in Alzheimer’s disease brain and antioxidant therapy: Lights and shadows. Ann. N. Y. Acad. Sci. 2008, 1147, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Pupo, G.; Tramutola, A.; Giorgi, A.; Schininà, M.E.; Coccia, R.; Head, E.; Butterfield, D.A.; Perluigi, M. Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: Clues for understanding the development of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Good, P.F.; Werner, P.; Hsu, A.; Olanow, C.W.; Perl, D.P. Evidence of neuronal oxidative damage in Alzheimer’s disease. Am. J. Pathol. 1996, 149, 21–28. [Google Scholar]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef]
- Da Cruz e Silva, E.F.; Henriques, A.G.; Vintém, A.P.; da Cruz e Silva, O.A.B. PP1 Inhibition By Aβ Peptide And Aluminum As A Potential Pathological Mechanism In Alzheimer’s Disease. Alzheimer’s Dement. 2009, 5, e16. [Google Scholar]
- Smith, M.A.; Perry, G.; Richey, P.L.; Sayre, L.M.; Anderson, V.E.; Beal, M.F.; Kowall, N. Oxidative damage in Alzheimer’s. Nature 1996, 382, 120–121. [Google Scholar] [CrossRef]
- Austen, B.M.; Allsop, D.; Gibson, G.L. Induction of Cellular Oxidative Stress by the β-amyloid Peptide Involved in Alzheimer’s disease. Protein Pept. Lett. 2004, 11, 257–270. [Google Scholar]
- Fang, W.; Hu, G.Z.; Liu, N.J. Advances in research on Alzheimer’s disease and oxidative stress. Chin. J. Gerontol. 2017, 37, 5205–5207. [Google Scholar]
- Polidori, M.C. Oxidative stress and risk factors for Alzheimer’s disease: Clues to prevention and therapy. J. Alzheimers Dis. 2004, 6, 185–191. [Google Scholar] [CrossRef]
- Korshavn, K.J.; Satriano, C.; Lin, Y.; Zhang, R.; Dulchavsky, M.; Bhunia, A.; Ivanova, M.I.; Lee, Y.H.; La Rosa, C.; Lim, M.H.; et al. Reduced Lipid Bilayer Thickness Regulates the Aggregation and Cytotoxicity of Amyloid-β. J. Biol. Chem. 2017, 292, 4638–4650. [Google Scholar] [CrossRef]
- La Rosa, C.; Scalisi, S.; Lolicato, F.; Pannuzzo, M.; Raudino, A. Lipid-assisted protein transport: A diffusion-reaction model supported by kinetic experiments and molecular dynamics simulations. J. Chem. Phys. 2016, 144, 184901. [Google Scholar] [CrossRef]
- Chtourou, Y.; Fetoui, H.; Garoui, E.M.; Boudawara, T.; Zeghal, N. Improvement of cerebellum redox states and cholinergic functions contribute to the beneficial effects of silymarin against manganese-induced neurotoxicity. Neurochem. Res. 2012, 37, 469–479. [Google Scholar] [CrossRef]
- Nencini, C.; Giorgi, G.; Micheli, L. Protective effect of silymarin on oxidative stress in rat brain. Phytomedicine 2007, 14, 129–135. [Google Scholar] [CrossRef]
- Sun, T.M.; Li, X. Advances in Pharmacological Studies of Silymarin. Chin. Tradit. Herb. Drugs 2000, 229–231. [Google Scholar] [CrossRef]
- Kiruthiga, P.V.; Karutha Pandian, S. Pandima Devi, K. Silymarin protects PBMC against B(a)P induced toxicity by replenishing redox status and modulating glutathione metabolizing enzymes—An in vitro study. Toxicol. Appl. Pharmacol. 2010, 247, 116–128. [Google Scholar] [CrossRef]
- Alidoost, F.; Gharagozloo, M.; Bagherpour, B.; Jafarian, A.; Sajjadi, S.E.; Hourfar, H.; Moayedi, B. Effects of silymarin on the proliferation and glutathione levels of peripheral blood mononuclear cells from β-thalassemia major patients. Int. Immunopharmacol. 2006, 6, 1305–1310. [Google Scholar] [CrossRef]
- Kim, Y.C.; Na, J.D.; Kwon, D.Y.; Park, J.H. Silymarin prevents acetaminophen-induced hepatotoxicity via up-regulation of the glutathione conjugation capacity in mice. J. Funct. Foods 2018, 49, 235–240. [Google Scholar] [CrossRef]
- Chauhan, V.; Chauhan, A. Oxidative stress in Alzheimer’s disease. Pathophysiology 2006, 13, 195–208. [Google Scholar] [CrossRef]
- Galhardi, F.; Mesquita, K.; Monserrat, J.M.; Barros, D.M. Effect of silymarin on biochemical parameters of oxidative stress in aged and young rat brain. Food Chem. Toxicol. 2009, 47, 2655–2660. [Google Scholar] [CrossRef]
- Thakare, V.N.; Dhakane, V.D.; Patel, B.M. Potential antidepressant-like activity of silymarin in the acute restraint stress in mice: Modulation of corticosterone and oxidative stress response in cerebral cortex and hippocampus. Pharmacol. Rep. 2016, 68, 1020–1027. [Google Scholar] [CrossRef]
- Onaolapo, A.Y.; Abdusalam, S.Z.; Onaolapo, O.J. Silymarin attenuates aspartame-induced variation in mouse behaviour, cerebrocortical morphology and oxidative stress markers. Pathophysiology 2017, 24, 51–62. [Google Scholar] [CrossRef]
- Trovato, A.; Siracusa, R.; Di Paola, R.; Scuto, M.; Ontario, M.L.; Bua, O.; Di Mauro, P.; Toscano, M.A.; Petralia, C.C.T.; Maiolino, L.; et al. Redox modulation of cellular stress response and lipoxin A4 expression by Hericium Erinaceus in rat brain: Relevance to Alzheimer’s disease pathogenesis. Immun. Ageing 2016, 13, 23. [Google Scholar] [CrossRef]
- Trovato Salinaro, A.; Pennisi, M.; Di Paola, R.; Scuto, M.; Crupi, R.; Cambria, M.T.; Ontario, M.L.; Tomasello, M.; Uva, M.; Maiolino, L.; et al. Neuroinflammation and neurohormesis in the pathogenesis of Alzheimer’s disease and Alzheimer-linked pathologies: Modulation by nutritional mushrooms. Immun. Ageing 2018, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Meraz-Ríos, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernández, J.; Campos-Peña, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef]
- Prati, F.; Bartolini, M.; Simoni, E.; De Simone, A.; Pinto, A.; Andrisano, V.; Bolognesi, M.L. Quinones bearing non-steroidal anti-inflammatory fragments as multitarget ligands for Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2013, 23, 6254–6258. [Google Scholar] [CrossRef]
- Zhang, H.B.; Lin, A.H.; Gong, P.; Zhang, Y.; Ye, D.Q.; Yu, Y. The effect of neuroinflammation on tau pathology in Alzheimer’s disease. Chin. J. New Drugs 2018, 27, 2245–2252. [Google Scholar]
- Sawikr, Y.; Yarla, N.S.; Peluso, I.; Kamal, M.A.; Aliev, G.; Bishayee, A. Neuroinflammation in Alzheimer’s Disease: The Preventive and Therapeutic Potential of Polyphenolic Nutraceuticals. Adv. Protein Chem. Struct. Biol. 2017, 108, 33–57. [Google Scholar]
- Miklossy, J.; McGeer, P.L. Common mechanisms involved in Alzheimer’s disease and type 2 diabetes: A key role of chronic bacterial infection and inflammation. Aging (Albany NY) 2016, 8, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Lijun, Z.; Wei, Z. Neuroimmune inflammation and Alzheimer’s disease. Chin. J. Clin. (Electron. Ed.) 2013, 7, 6547–6550. [Google Scholar]
- Renard, D.; Wacongne, A.; Ayrignac, X.; Charif, M.; Fourcade, G.; Azakri, S.; Le Floch, A.; Bouly, S.; Marelli, C.; Arquizan, C.; et al. Cerebrospinal Fluid Alzheimer’s Disease Biomarkers in Cerebral Amyloid Angiopathy-Related Inflammation. J. Alzheimers Dis. 2016, 50, 759–764. [Google Scholar] [CrossRef]
- Gabbita, S.P.; Srivastava, M.K.; Eslami, P.; Johnson, M.F.; Kobritz, N.K.; Tweedie, D.; Greig, N.H.; Zemlan, F.P.; Sharma, S.P.; Harris-White, M.E. Early intervention with a small molecule inhibitor for tumor necrosis factor-α prevents cognitive deficits in a triple transgenic mouse model of Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 99. [Google Scholar] [CrossRef]
- Szekely, C.A.; Town, T.; Zandi, P.P. NSAIDs for the chemoprevention of Alzheimer’s disease. In Inflammation in the Pathogenesis of Chronic Diseases; Harris, R.E., Bittman, R., Dasgupta, D., Engelhardt, H., Flohe, L., Herrmann, H., Holzenburg, A., Nasheuer, H.-P., Rottem, S., Wyss, M., et al., Eds.; Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2007; Volume 42, pp. 229–248. [Google Scholar]
- Shu, Y.W.; Qu, Y.; Zhang, J.Y. Advances in research on the relationship between inflammation and Alzheimer’s disease. J. Xinxiang Med. Univ. 2018, 35, 1130–1133. [Google Scholar]
- Shadfar, S.; Hwang, C.J.; Lim, M.S.; Choi, D.Y.; Hong, J.T. Involvement of inflammation in Alzheimer’s disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch. Pharm. Res. 2015, 38, 2106–2119. [Google Scholar] [CrossRef]
- Browne, T.C.; McQuillan, K.; McManus, R.M.; O’Reilly, J.A.; Mills, K.H.; Lynch, M.A. IFN-γ Production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J. Immunol. 2013, 190, 2241–2251. [Google Scholar] [CrossRef]
- Ma, W.P.; Zhang, X.; Wu, Q. Research Advances in the Neuroinflammation in Alzheimer’s Disease. Acta Academiae Medicinae Sinicae 2017, 39, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Aβ production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Hatami, A.; Albay, R., III; Monjazeb, S.; Milton, S.; Glabe, C. Monoclonal antibodies against Aβ42 fibrils distinguish multiple aggregation state polymorphisms in vitro and in Alzheimer disease brain. J. Biol. Chem. 2014, 289, 32131–32143. [Google Scholar] [CrossRef]
- Janelidze, S.; Mattsson, N.; Stomrud, E.; Lindberg, O.; Palmqvist, S.; Zetterberg, H.; Blennow, K.; Hansson, O. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology 2018, 91, e867–e877. [Google Scholar] [CrossRef]
- Mudò, G.; Frinchi, M.; Nuzzo, D.; Scaduto, P.; Plescia, F.; Massenti, M.F.; Di Carlo, M.; Cannizzaro, C.; Cassata, G.; Cicero, L.; et al. Anti-inflammatory and cognitive effects of interferon-β1a (IFNβ1a) in a rat model of Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 44. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, S.; Wang, Y.; Zhu, T. Silymarin improved diet-induced liver damage and insulin resistance by decreasing inflammation in mice. Pharm. Biol. 2016, 54, 2995–3000. [Google Scholar] [CrossRef]
- Haddadi, R.; Mohajjel Nayebi, A.; Brooshghalan, S.E. Pre-treatment with silymarin reduces brain myeloperoxidase activity and inflammatory cytokines in 6-OHDA hemi-parkinsonian rats. Neurosci. Lett. 2013, 555, 106–111. [Google Scholar] [CrossRef]
- Thakare, V.N.; Aswar, M.K.; Kulkarni, Y.P.; Patil, R.R.; Patel, B.M. Silymarin ameliorates experimentally induced depressive like behavior in rats: Involvement of hippocampal BDNF signaling, inflammatory cytokines and oxidative stress response. Physiol. Behav. 2017, 179, 401–410. [Google Scholar] [CrossRef]
- Kim, S.H.; Oh, D.S.; Oh, J.Y.; Son, T.G.; Yuk, D.Y.; Jung, Y.S. Silymarin Prevents Restraint Stress-Induced Acute Liver Injury by Ameliorating Oxidative Stress and Reducing Inflammatory Response. Molecules 2016, 21, 443. [Google Scholar] [CrossRef]
- Lovelace, E.S.; Wagoner, J.; MacDonald, J.; Bammler, T.; Bruckner, J.; Brownell, J.; Beyer, R.P.; Zink, E.M.; Kim, Y.M.; Kyle, J.E.; et al. Silymarin Suppresses Cellular Inflammation By Inducing Reparative Stress Signaling. J. Nat. Prod. 2015, 78, 1990–2000. [Google Scholar] [CrossRef]
- Avila-Muñoz, E.; Arias, C. When astrocytes become harmful: Functional and inflammatory responses that contribute to Alzheimer’s disease. Ageing Res. Rev. 2014, 18, 29–40. [Google Scholar] [CrossRef]
- Levit, A.; Regis, A.M.; Gibson, A.; Hough, O.H.; Maheshwari, S.; Agca, Y.; Agca, C.; Hachinski, V.; Allman, B.L.; Whitehead, S.N. Impaired behavioural flexibility related to white matter microgliosis in the TgAPP21 rat model of Alzheimer disease. Brain Behav. Immun. 2019, in press. [Google Scholar] [CrossRef]
- Tsai, M.J.; Liao, J.F.; Lin, D.Y.; Huang, M.C.; Liou, D.Y.; Yang, H.C.; Lee, H.J.; Chen, Y.T.; Chi, C.W.; Huang, W.C.; et al. Silymarin protects spinal cord and cortical cells against oxidative stress and lipopolysaccharide stimulation. Neurochem. Int. 2010, 57, 867–875. [Google Scholar] [CrossRef]
- Lee, Y.; Chun, H.J.; Lee, K.M.; Jung, Y.S.; Lee, J. Silibinin suppresses astroglial activation in a mouse model of acute Parkinson’s disease by modulating the ERK and JNK signaling pathways. Brain Res. 2015, 1627, 233–242. [Google Scholar] [CrossRef]
- Wang, M.J.; Lin, W.W.; Chen, H.L.; Chang, Y.H.; Ou, H.C.; Kuo, J.S.; Hong, J.S.; Jeng, K.C. Silymarin protects dopaminergic neurons against lipopolysaccharide-induced neurotoxicity by inhibiting microglia activation. Eur. J. Neurosci. 2002, 16, 2103–2112. [Google Scholar] [CrossRef]
- Deleidi, M.; Gasser, T. The role of inflammation in sporadic and familial Parkinson’s disease. Cell Mol. Life Sci. 2013, 70, 4259–4273. [Google Scholar] [CrossRef]
- Han, W.; Li, C. Linking type 2 diabetes and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 6557–6558. [Google Scholar] [CrossRef] [PubMed]
- Andreetto, E.; Yan, L.M.; Tatarek-Nossol, M.; Velkova, A.; Frank, R.; Kapurniotu, A. Identification of hot regions of the Abeta-IAPP interaction interface as high-affinity binding sites in both cross- and self-association. Angew. Chem. Int. Ed. Engl. 2010, 49, 3081–3085. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, H.; Cao, H.; Cui, X.; Zheng, W.; Wang, S.; Yu, J.; Chen, Z. Silymarin’s Inhibition and Treatment Effects for Alzheimer’s Disease. Molecules 2019, 24, 1748. https://doi.org/10.3390/molecules24091748
Guo H, Cao H, Cui X, Zheng W, Wang S, Yu J, Chen Z. Silymarin’s Inhibition and Treatment Effects for Alzheimer’s Disease. Molecules. 2019; 24(9):1748. https://doi.org/10.3390/molecules24091748
Chicago/Turabian StyleGuo, Hong, Hui Cao, Xiaowei Cui, Wenxiu Zheng, Shanshan Wang, Jiyang Yu, and Zhi Chen. 2019. "Silymarin’s Inhibition and Treatment Effects for Alzheimer’s Disease" Molecules 24, no. 9: 1748. https://doi.org/10.3390/molecules24091748
APA StyleGuo, H., Cao, H., Cui, X., Zheng, W., Wang, S., Yu, J., & Chen, Z. (2019). Silymarin’s Inhibition and Treatment Effects for Alzheimer’s Disease. Molecules, 24(9), 1748. https://doi.org/10.3390/molecules24091748