
Analyzing Secondary Structure Patterns in DNA Aptamers Identified via CompELS


Abstract
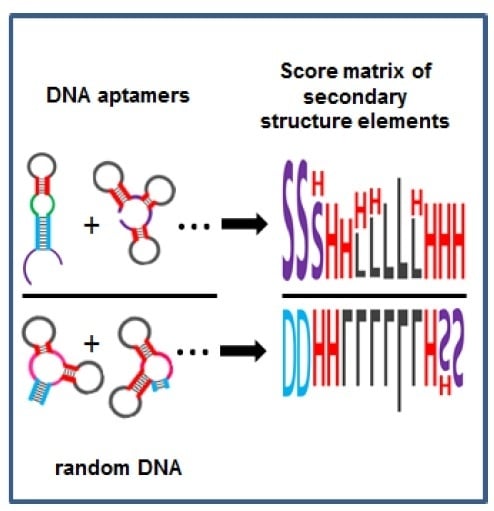
1. Introduction
2. Results and Discussion
2.1. DNA Aptamers for AuNR Identified Using CompELS Screening Platform
2.2. Assigning Subgroups of Aptamer sequeNces to a Secondary Structure Family (SSF)
2.3. Mapping Secondary Structure Elements (SSE) at Each Base Position
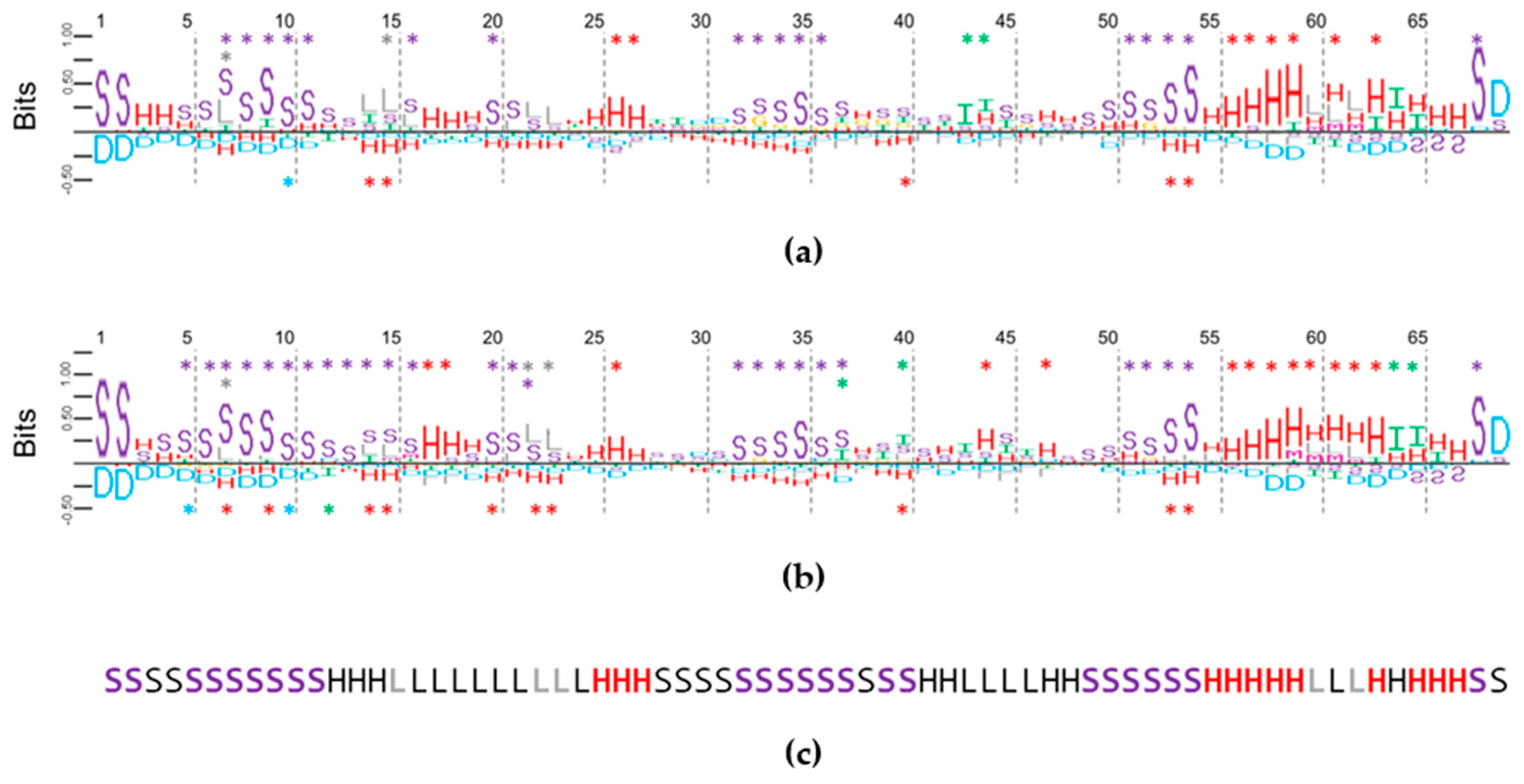
2.4. Using Random Sequence Populations to Generate Position-Specific Score Matrices (PSSM) of SSE in Aptamers
2.5. Multiple Secondary Structure String Alignment (MSS$A) in Aptamers and Random Sequences
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Synthesis of Gold Nanorods (AuNR)
4.3. Preparation of ssDNA Library for CompELS Screening
4.4. Competition-Enhanced Selection of Ligands (CompELS) Screening for DNA Aptamers against AuNR Targets
4.5. Primary Structure Analysis of DNA Aptamers
4.6. Predictions of Dominant and Suboptimal Secondary Structures for DNA Aptamer Sequences and Random DNA Sequences
4.7. Defining Secondary Structure Elements (SSE), Secondary Structure Families (SSF), and Secondary Structure Strings (SS$) for Aptamers and Random DNA Sequences
4.8. Using SS$ to Generate Position Specific Score Matrices (PSSM)
4.9. Using SS$ to Perform Multiple Secondary Structure String Alignment (MSS$A)
4.10. Defining Secondary Structure Consensus Domains in MSS$A with Gaps Included
4.11. Removing Gaps to Determine Distribution of Consensus Domains in MSS$A
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABB | aptamer binding buffer |
| AuNR | gold nanorod(s) |
| CompELS | competition-enhanced ligands selection |
| dsDNA | double-stranded DNA |
| MEME | Multiple EM (Expectation Maximization) for Motif Elicitation |
| MSS$A | multiple secondary structure string alignment |
| P/C/I | phenol:chloroform:isoamyl alcohol |
| PSSM | position specific score matrix(ces) |
| SELEX | systematic evolution of ligands by exponential enrichment |
| SOC | super optimal bath |
| ssDNA | single-stranded DNA |
| SSE | secondary structure element(s) |
| SSF | secondary structure family(ies) |
| SS$ | secondary structure string(s) |
References
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Van Noorden, R.; Maher, B.; Nuzzo, R. The top 100 papers. Nature 2014, 514, 550–553. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Moretti, S.; Xenarios, I.; Orobitg, M.; Montanyola, A.; Chang, J.-M.; Taly, J.-F.; Notredame, C. T-Coffee: A web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011, 39, W13–W17. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.J.; Kececioglu, J.D. Multiple alignment by aligning alignments. Bioinformatics 2007, 23, i559–i568. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, Z.; Lu, Y. Functional nucleic acid sensors. Chem. Rev. 2009, 109, 1948–1998. [Google Scholar] [CrossRef]
- Soukup, G.A.; Breaker, R.R. Engineering precision RNA molecular switches. Proc. Natl. Acad. Sci. USA 1999, 96, 3584–3589. [Google Scholar] [CrossRef] [PubMed]
- Chávez, J.L.; Lyon, W.; Kelley-Loughnane, N.; Stone, M.O. Theophylline detection using an aptamer and DNA-gold nanoparticle conjugates. Biosens. Bioelectron. 2010, 26, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Lubin, A.A.; Heeger, A.J.; Plaxco, K.W. Label-free electronic detection of thrombin in blood serum using an aptamer-based sensor. Angew. Chem. Int. Ed. 2005, 44, 5456–5459. [Google Scholar] [CrossRef]
- Green, L.S.; Jellinek, D.; Jenison, R.D.; Östman, A.; Heldin, C.-H.; Janjic, N. Inhibitory DNA ligands to platelet-derived growth factor B-chain. Biochemistry 1996, 35, 14413–14424. [Google Scholar] [CrossRef]
- Ng, E.W.M.; Shima, D.T.; Calias, P.; Cunningham, E.T.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef]
- Rusconi, C.P.; Scardino, E.; Layzer, J.; Pitoc, G.A.; Ortei, T.L.; Monroe, D.; Sullenger, B.A. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature 2002, 419, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.L.; Joyce, G.F. Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 1990, 344, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Hoinka, J.; Zotenko, E.; Friedman, A.; Sauna, Z.E.; Przytycka, T.M. Identification of sequence-structure RNA binding motifs for SELEX-derived aptamers. Bioinformatics 2012, 28, 215–223. [Google Scholar] [CrossRef]
- Pei, S.; Slinger, B.L.; Meyer, M.M. Recognizing RNA structural motifs in HT-SELEX data for ribosomal protein S15. BMC Bioinform. 2017, 18, 298. [Google Scholar] [CrossRef]
- Caroli, J.; Taccioli, C.; De La Fuente, A.; Serafini, P.; Bicciato, S. APTANI: A computational tool to select aptamers through sequence-structure motif analysis of HT-SELEX data. Bioinformatics 2016, 32, 161–164. [Google Scholar] [CrossRef]
- Dao, P.; Hoinka, J.; Takahashi, M.; Zhou, J.; Ho, M.; Wang, Y.; Costa, F.; Rossi, J.J.; Backofen, R.; Burnett, J.; et al. AptaTRACE elucidates RNA sequence-structure motifs from selection trends in HT-SELEX experiments. Cell Syst. 2016, 3, 62–70. [Google Scholar] [CrossRef]
- Berezhnoy, A.; Stewart, C.A.; Mcnamara, J.O.; Thiel, W.; Giangrande, P.; Trinchieri, G.; Gilboa, E. Isolation and optimization of murine IL-10 receptor blocking oligonucleotide aptamers using high-throughput sequencing. Mol. Ther. 2012, 20, 1242–1250. [Google Scholar] [CrossRef]
- Dunaway, A.B.; Sullivan, R.S.; Siegel, K.J.; Milam, V.T. Evaluating the dual target binding capabilities of immobilized aptamers using flow cytometry. Biointerphases 2015, 10, 019015. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Bing, T.; Yang, X.; Mei, H.; Cao, Z.; Shangguan, D. Conservative secondary structure motif of streptavidin-binding aptamers generated by different laboratories. Bioorg. Med. Chem. 2010, 18, 1798–1805. [Google Scholar] [CrossRef]
- Shangguan, D.; Tang, Z.; Mallikaratchy, P.; Xiao, Z.; Tan, W. Optimization and modifications of aptamers selected from live cancer cell lines. ChemBioChem 2007, 8, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.A.; Feignon, J.; Yeates, T.O. Reconciliation of the x-ray and NMR structures of the thrombin-binding aptamer d(GGTTGGTGTGGTTGG). J. Mol. Biol. 1996, 256, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Macaya, R.F.; Schultze, P.; Smith, F.W.; Roe, J.A.; Feignon, J. Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc. Natl. Acad. Sci. USA 1993, 90, 3745–3749. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, K.; Padmanabhan, K.P.; Ferrara, J.D.; Sadler, J.E.; Tulinsky, A. The structure of alpha-thrombin inhibited by a 15-mer single-stranded DNA aptamer. J. Biol. Chem. 1993, 268, 17651–17654. [Google Scholar] [PubMed]
- Le, T.T.; Chumphukan, O.; Cass, A.E.G. Determination of minimal sequence for binding of an aptamer. A comparison of truncation and hybridization inhibition methods. RSC Adv. 2014, 4, 47227–47233. [Google Scholar] [CrossRef]
- Nonaka, Y.; Sode, K.; Ikebukuro, K. Screening and improvement of an anti-VEGF DNA aptamer. Molecules 2010, 15, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Yung, L.Y. Probing high affinity sequences of DNA aptamer against VEGF165. PLoS ONE 2012, 7, e31196. [Google Scholar] [CrossRef] [PubMed]
- Klosterman, P.S.; Hendrix, D.K.; Tamura, M.; Holbrook, S.R.; Brenner, S.E. Three-dimensional motifs from the SCOR, structural classification of RNA database: Extruded strands, base triples, tetraloops and U-turns. Nucleic Acids Res. 2004, 32, 2342–2352. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bindewald, E.; Hayes, R.; Yingling, Y.G.; Kasprzak, W.; Shapiro, B.A. RNAJunction: A database of RNA junctions and kissing loops for three-dimensional structural analysis and nanodesign. Nucleic Acids Res. 2007, 36, D392–D397. [Google Scholar] [CrossRef]
- Shulman-Peleg, A.; Nussinov, R.; Wolfson, H.J. RsiteDB: A database of protein binding pockets that interact with RNA nucleotide bases. Nucleic Acids Res. 2009, 37, D369–D373. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Zhao, Y.; Li, C.; Zhang, L.; Huang, H.; Li, Y.; Liu, L.; Hou, P.; Cui, T.; Tan, P.; et al. RAID v2.0: An updated resource of RNA-associated interactions across organisms. Nucleic Acids Res. 2017, 45, D115–D118. [Google Scholar] [CrossRef]
- Rockey, W.M.; Hernandez, F.J.; Huang, S.-Y.; Cao, S.; Howell, C.A.; Thomas, G.S.; Liu, X.Y.; Lapteva, N.; Spencer, D.M.; McNamara II, J.O.; et al. Rational truncation of an RNA aptamer to prostate-specific membrane antigen using computational structural modeling. Nucleic Acid Ther. 2011, 21, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Kumar, R.A.; Jones, R.A.; Patel, D.J. Structural basis of RNA folding and recognition in an AMP-RNA aptamer complex. Nature 1996, 382, 183–186. [Google Scholar] [CrossRef]
- Yang, Y.; Kochayan, M.; Burgstaller, P.; Westhof, E.; Famulok, M. Structural basis of ligand discrimination by two related RNA aptamers resolved by NMR spectroscopy. Science 1996, 272, 1343–1347. [Google Scholar] [CrossRef]
- Tapp, M.J.N.; Slocik, J.M.; Dennis, P.B.; Naik, R.R.; Milam, V.T. Competition-enhanced ligand selection to identify DNA aptamers. ACS Comb. Sci. 2018, 20, 585–593. [Google Scholar] [CrossRef]
- Mathews, D.H.; Turner, D.H. Dyalign: An algorithm for finding the secondary structure common to two RNA sequences. J. Mol. Biol. 2002, 317, 191–203. [Google Scholar] [CrossRef]
- Mathews, D.H. Predicting a set of minimal free energy RNA secondary structures common to two sequences. Bioinformatics 2005, 21, 2246–2253. [Google Scholar] [CrossRef]
- Carothers, J.M.; Oestreich, S.C.; Davis, J.H.; Szostak, J.W. Informational complexity and functional activity of RNA structures. J. Am. Chem. Soc. 2004, 126, 5130–5137. [Google Scholar] [CrossRef]
- Tabei, Y.; Kiryu, H.; Kin, T.; Asai, K. A fast structural multiple alignment method for long RNA sequences. BMC Bioinform. 2008, 9, 33. [Google Scholar] [CrossRef]
- Will, S.; Otto, C.; Miladi, M.; Möhl, M.; Backofen, R. SPARSE: Quadratic time simultaneous alignment and folding of RNAs without sequence-based heuristics. Bioinformatics 2015, 31, 2489–2496. [Google Scholar] [CrossRef]
- Rogers, E.; Heitsch, C. New insights from cluster analysis methods for RNA secondary structure prediction. WIREs RNA 2016, 7, 278–294. [Google Scholar] [CrossRef]
- Rogers, E.; Heitsch, C.E. Profiling small RNA reveals multimodal substructural signals in a Boltzmann ensemble. Nucleic Acids Res. 2014, 42, e171. [Google Scholar] [CrossRef]
- Kemena, C.; Bussotti, G.; Capriotti, E.; Marti-Renom, M.A.; Notredame, C. Using tertiary structure for the computation of highly accurate multiple RNA alignments with the SARA-Coffee package. Bioinformatics 2013, 29, 1112–1119. [Google Scholar] [CrossRef]
- Sorescu, D.A.; Mohl, M.; Mann, M.; Backofen, R.; Will, S. CARNA-alignment of RNA structure ensembles. Nucleic Acids Res. 2012, 40, W49–W53. [Google Scholar] [CrossRef]
- Schneider, T.D.; Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef] [PubMed]
- Stormo, G.D.; Zhao, Y. Determining the specificity of protein-DNA interactions. Nat. Rev. Genet. 2010, 11, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Nikoobakht, B.; El-Sayed, M.A. Preparation and growth mechanism of gold nanorods (NRs) using seed-mediated growth method. Chem. Mater. 2003, 15, 1957–1962. [Google Scholar] [CrossRef]
- Huang, X.; El-Sayed, I.H.; Qian, W.; El-Sayed, M.A. Cancer cell imaging and photothermal therapy in the near-infrared region by using gold nanorods. J. Am. Chem. Soc. 2006, 128, 2115–2120. [Google Scholar] [CrossRef]
- Nusz, G.J.; Marinakos, S.M.; Curry, A.C.; Dahlin, A.; Hook, F.; Wax, A.; Chilkoti, A. Label-free plasmonic detection of biomolecular binding by a single gold nanorod. Anal. Chem. 2008, 80, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Funston, A.M.; Novo, C.; Davis, T.J.; Mulvaney, P. Plasmon coupling of gold nanorods at short distances and in different geometries. Nano Lett. 2009, 9, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Neretina, S.; El-Sayed, M.A. Gold nanorods: From synthesis and properties to biological and biomedical applications. Adv. Mater. 2009, 21, 4880–4910. [Google Scholar] [CrossRef] [PubMed]
- Lohse, S.E.; Murphy, C.J. The quest for shape control: A history of gold nanorod synthesis. Chem. Mater. 2013, 25, 1250–1261. [Google Scholar] [CrossRef]
- Huang, J.; Wang, W.; Murphy, C.J.; Cahill, D.G. Resonant secondary light emission from plasmonic Au nanostructures at high electron temperatures created by pulsed-laser excitation. Proc. Natl. Acad. Sci. USA 2014, 111, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Tapp, M.; Sullivan, R.; Dennis, P.; Naik, R.R.; Milam, V.T. Spectroscopic studies of nucleic acid additions during seed-mediated growth of gold nanoparticles. J. Mater. Res. 2015, 30, 666–676. [Google Scholar] [CrossRef][Green Version]
- Mirau, P.A.; Smith, J.E.; Chavez, J.L.; Hagen, J.A.; Kelley-Loughnane, N.; Naik, R. Structured DNA aptamer interactions with gold nanoparticles. Langmuir 2018, 34, 2139–2146. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, Y.; Hayakawa, T.; Kawamura, G.; Nogami, M. Tuned longitudinal surface plasmon resonance and third-order nonlinear optical properties of gold nanorods. Nanotechnology 2011, 22, 275203. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in bipolymers. In Proceedings of the 4th International Conference on Intelligent Systems for Molecular Biology, Palo Alto, CA, USA, 14–17 August 1994; Volume 2, pp. 28–36. [Google Scholar]
- Bailey, T.L.; Gribskov, M. Combining evidence using p-values: Application to sequence homology searches. Bioinformatics 1998, 14, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Markham, N.R.; Zuker, M. UNAFold: Software for nucleic acid folding and hybridization. Methods Mol. Biol. 2008, 453, 3–31. [Google Scholar] [CrossRef] [PubMed]
- Mélard, G. On the accuracy of statistical procedures in Microsoft Excel 2010. Comput. Stat. 2014, 29, 1095–1128. [Google Scholar] [CrossRef]
- Farrance, I.; Frenkel, R. Uncertainty in measurement: A review of Monte Carlo simulation using Microsoft Excel for the calculation of uncertainties through functional relationships, including uncertainties in empirically derived constants. Clin. Biochem. Rev. 2014, 35, 37–61. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
Sample Availability: Sample information for the compounds is available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SSF No. | Structural Element Composition | Aptamer Nomenclature 1,2 of SSF Members | No. of SSF Members | No. Dominant (Suboptimal) Self-Hybridized Structures |
|---|---|---|---|---|
| SSF1 | 2S 2H/L 1I 0G 1D 0M | 111.S1, 116.S1, 116.S2 | 3 | 2 (1) |
| SSF2 | 2S 2H/L 1I 1G 2D 0M | 101.S1, 117.S1, 418.S1 | 3 | 3 (0) |
| SSF3 | 3S 2H/L 0I 0G 0D 0M | 111.S2, 111.S3, 120.S1, 214.S1, 403.S1, 412.S2 | 6 | 3 (3) |
| SSF4 | 3S 2H/L 0I 1G 1D 0M | 401.S1, 420.S1 | 2 | 2 (0) |
| SSF5 | 3S 2H/L 1I 0G 1D 0M | 108.S2, 112.S1, 406.S1 | 3 | 2 (1) |
| SSF6 | 3S 2H/L 2I 0G 2D 0M | 110.S1, 115.S1, 416.S1 | 3 | 3 (0) |
| SSF7 | 4S 3H/L 0I 0G 0D 0M | 102.S1, 212.S1, 217.S1, 219.S1, 404.S1, 412.S1 | 6 | 6 (0) |
| SSF8 | 4S 3H/L 0I 1G 1D 0M | 101.S2, 118.S1, 201.S2, 210.S1 | 4 | 2 (2) |
| SSF9 | 4S 3H/L 1I 0G 1D 0M | 105.S1, 108.S1, 114.S1, 119.S1, 204.S1, 204.S2, 402.S1, 413.S1, 415.S2, 421.S1 | 10 | 8 (2) |
| SSF10 | 5S 4H/L 0I 0G 0D 0M | 217.S2, 410.S1, 417.S1, 419.S1 | 4 | 3 (1) |
| SSF11 | 5S 4H/L 1I 0G 1D 0M | 409.S1, 414.S1 | 2 | 2 (0) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sullivan, R.; Adams, M.C.; Naik, R.R.; Milam, V.T. Analyzing Secondary Structure Patterns in DNA Aptamers Identified via CompELS. Molecules 2019, 24, 1572. https://doi.org/10.3390/molecules24081572
Sullivan R, Adams MC, Naik RR, Milam VT. Analyzing Secondary Structure Patterns in DNA Aptamers Identified via CompELS. Molecules. 2019; 24(8):1572. https://doi.org/10.3390/molecules24081572
Chicago/Turabian StyleSullivan, Richard, Mary Catherine Adams, Rajesh R. Naik, and Valeria T. Milam. 2019. "Analyzing Secondary Structure Patterns in DNA Aptamers Identified via CompELS" Molecules 24, no. 8: 1572. https://doi.org/10.3390/molecules24081572
APA StyleSullivan, R., Adams, M. C., Naik, R. R., & Milam, V. T. (2019). Analyzing Secondary Structure Patterns in DNA Aptamers Identified via CompELS. Molecules, 24(8), 1572. https://doi.org/10.3390/molecules24081572