Effect of Protein Conformation and AMP Protonation State on Fireflies’ Bioluminescent Emission


Abstract
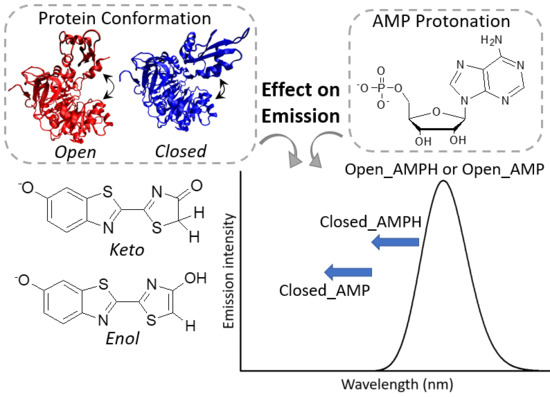
{kind=link}
{kind=link}
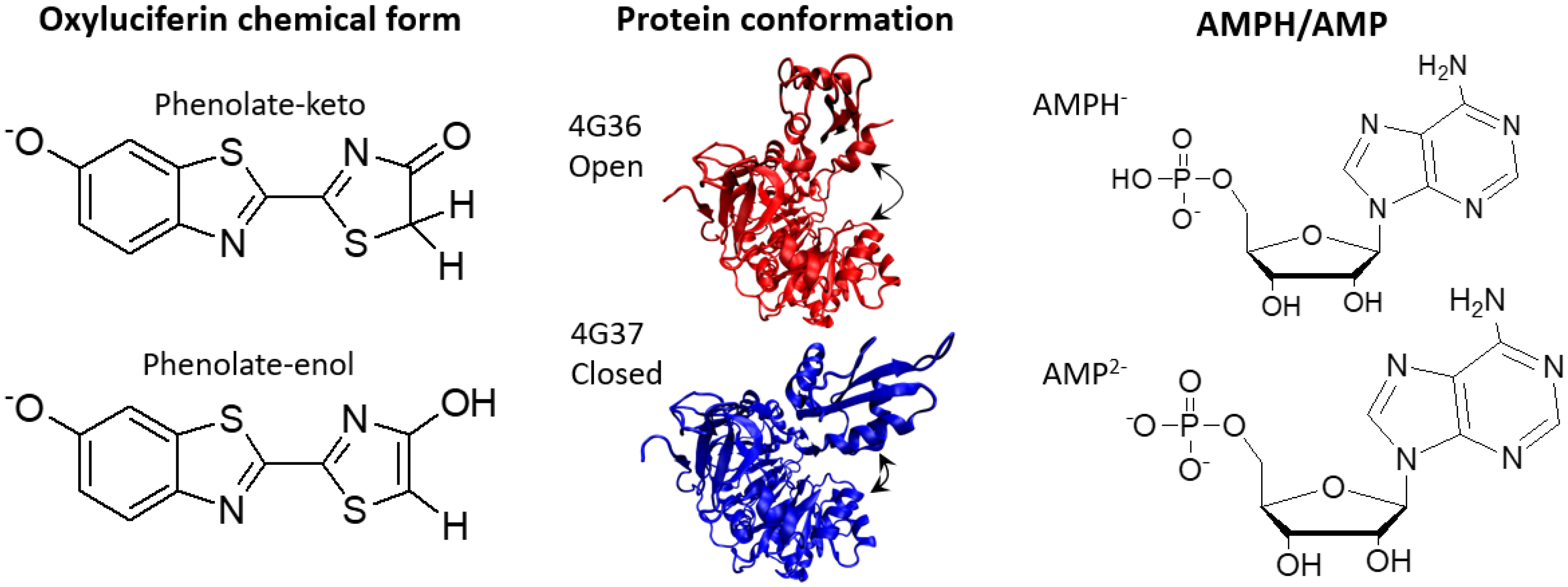
{kind=link}
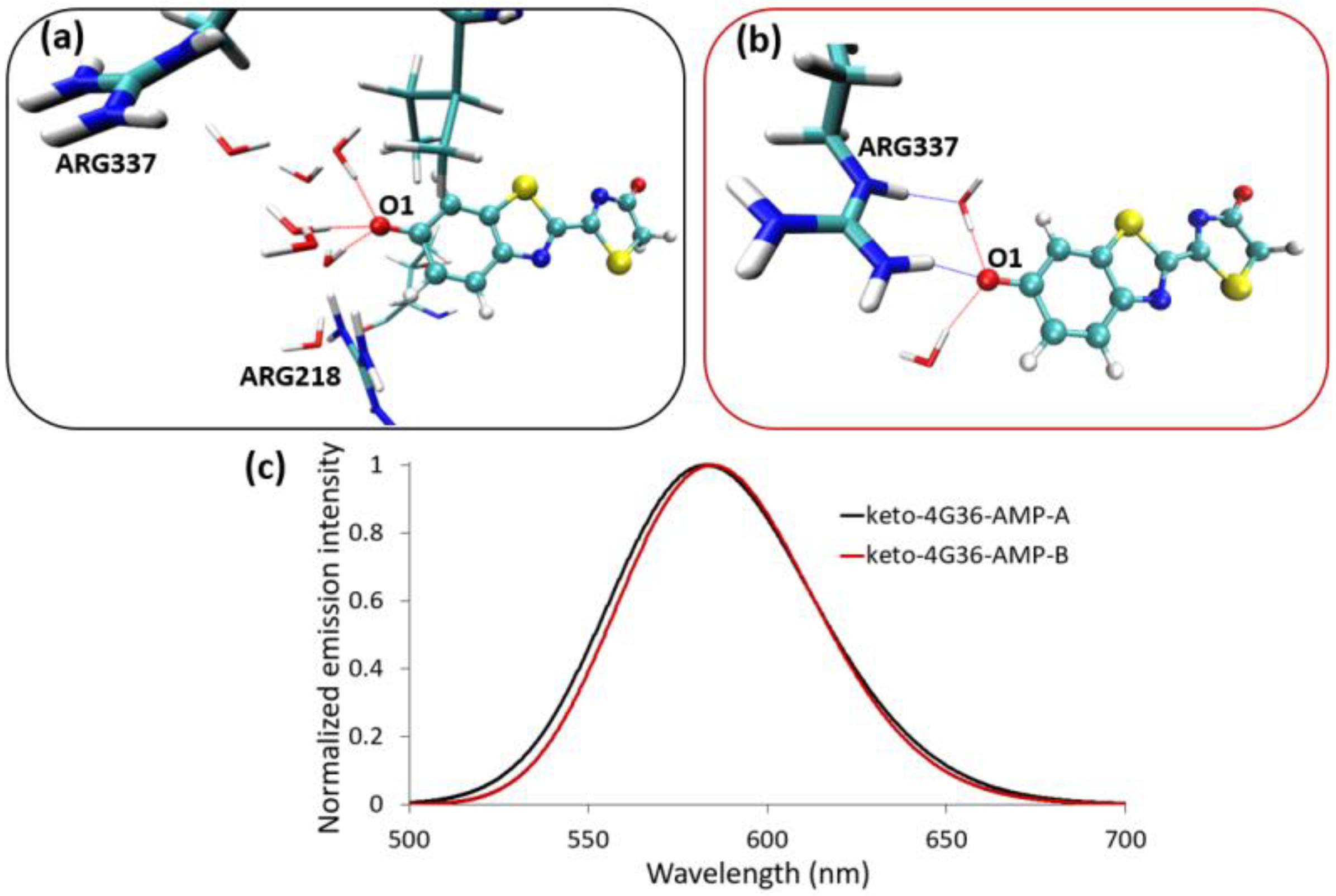
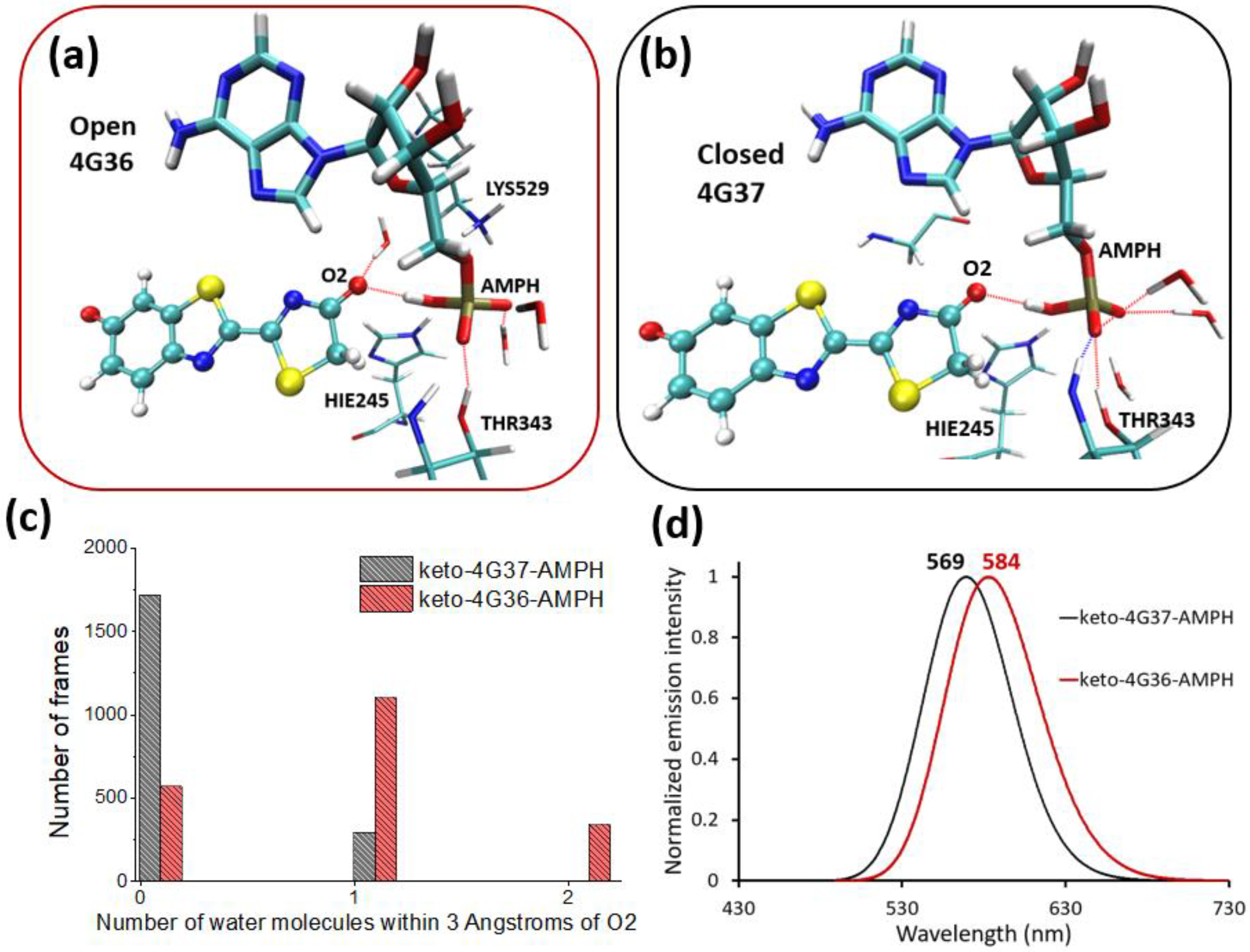{kind=link}
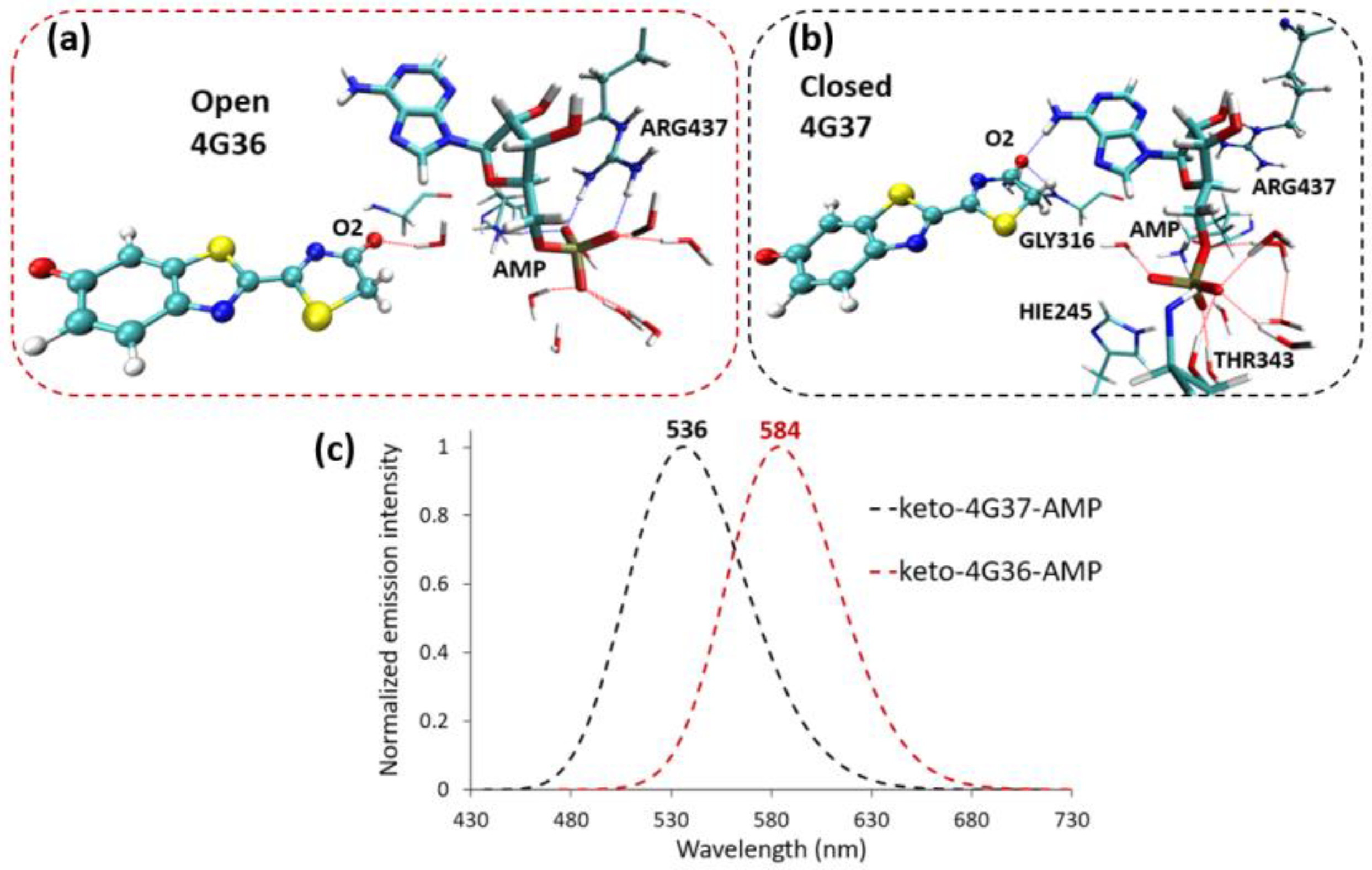{kind=link}
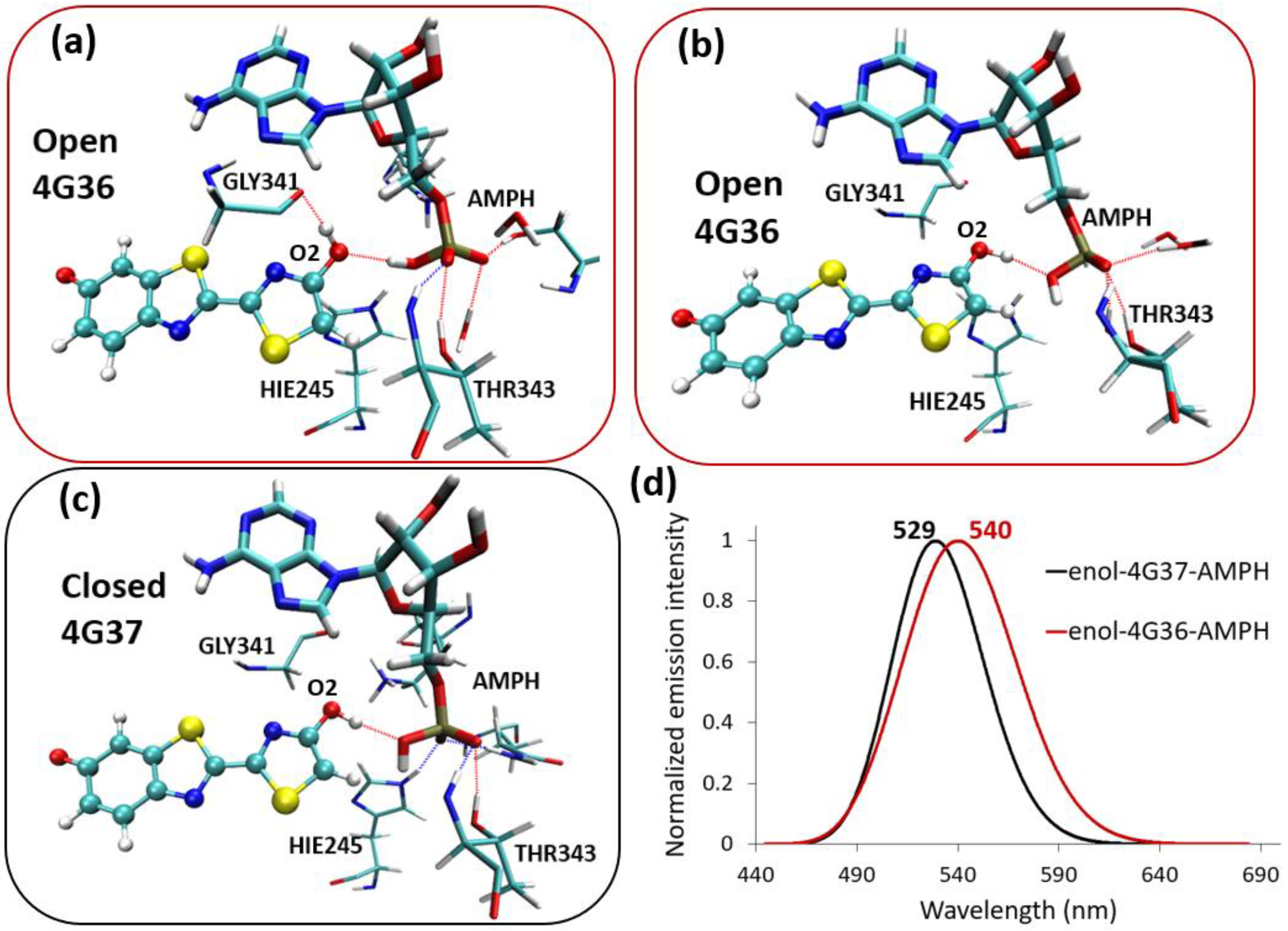{kind=link}
{kind=link}
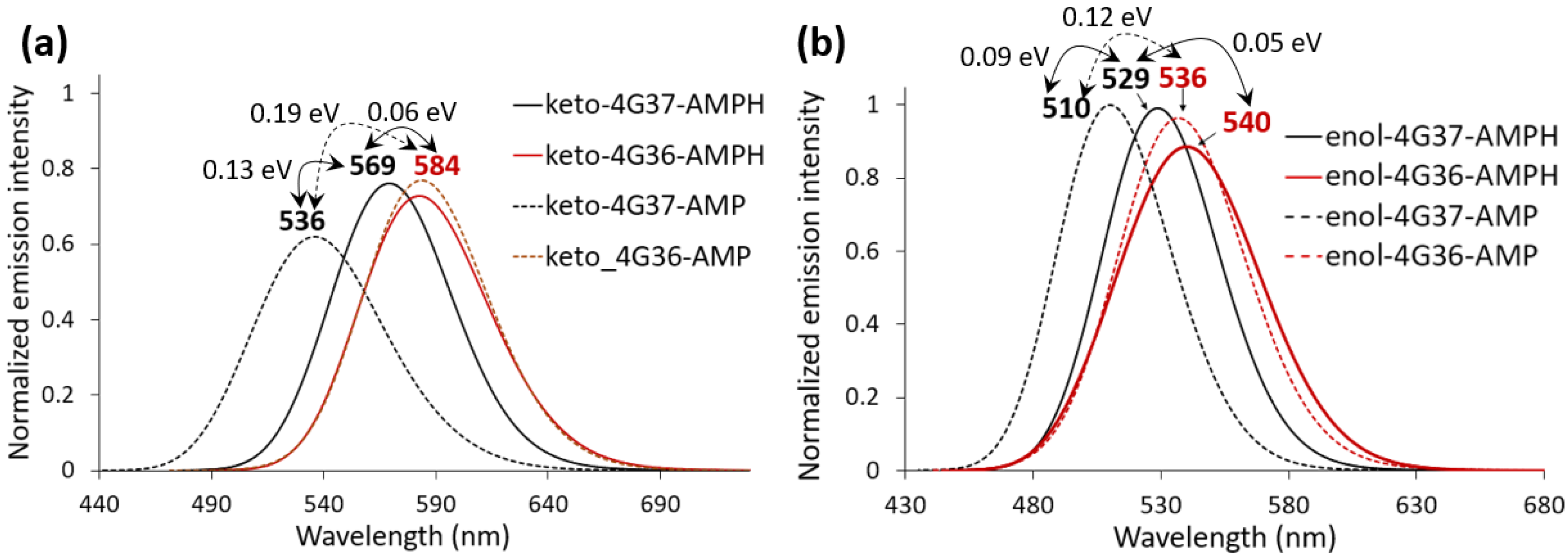{kind=link}
{kind=link}
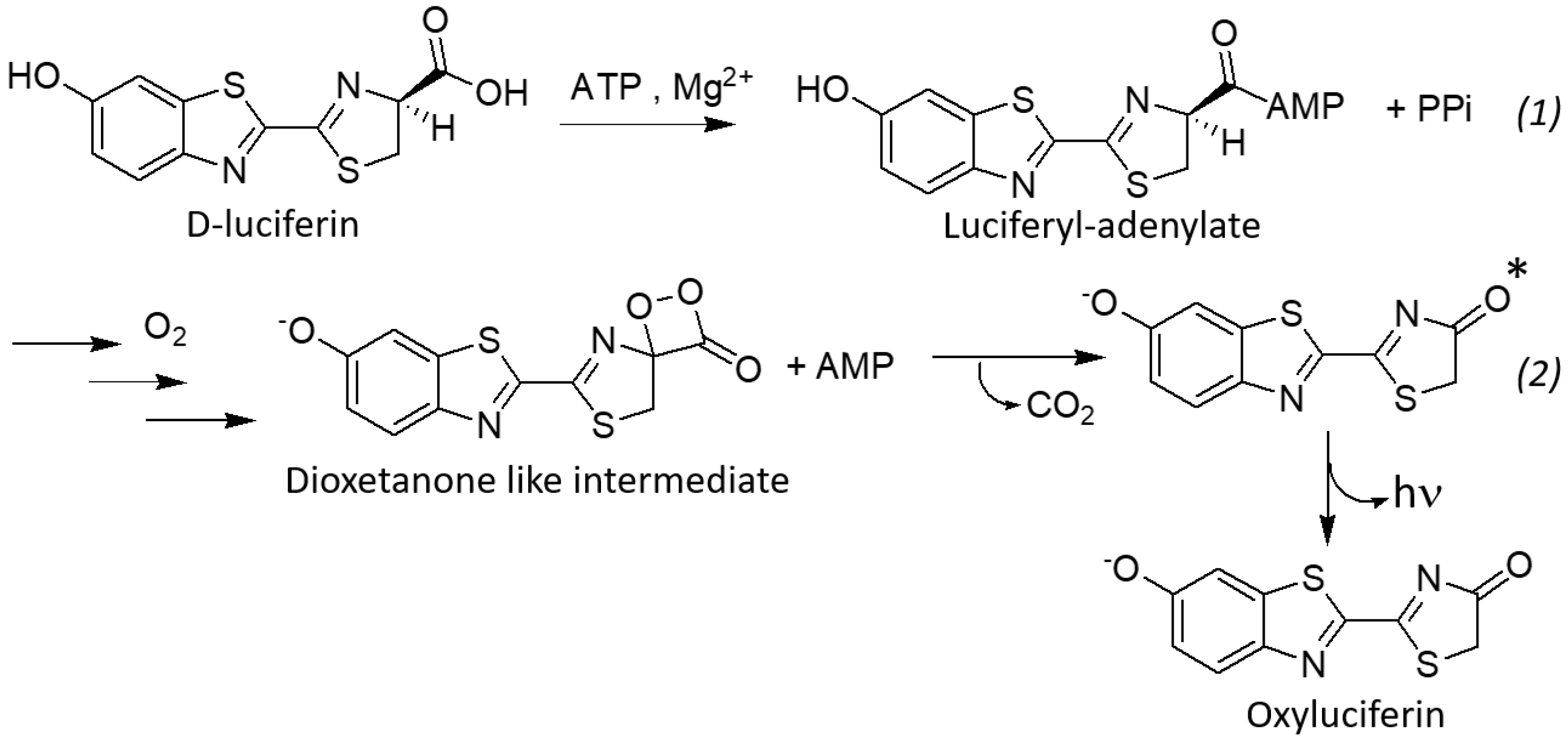
1. Introduction
2. Results
2.1. Phenolate-Environment Interactions
2.2. Keto-(Protein and AMP) Interactions
2.3. Enol-(Protein and AMP) Interactions
3. Discussion
4. Materials and Methods
4.1. Model Setup
4.2. MD Simulations
4.3. QM/MM Calculations
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Griffiths, M. The role of ATP bioluminescence in the food industry: new light on old problems. Food Technol. 1996, 50, 62–73. [Google Scholar]
- Shinde, R.; Perkins, J.; Contag, C.H. Luciferin Derivatives for Enhanced in Vitro and in Vivo Bioluminescence Assays. Biochemistry 2006, 45, 11103–11112. [Google Scholar] [CrossRef] [PubMed]
- Greer, L.F.; Szalay, A.A. Imaging of light emission from the expression of luciferases in living cells and organisms: a review. Luminescence 2002, 17, 43–74. [Google Scholar] [CrossRef]
- Kricka, L.J. Application of bioluminescence and chemiluminescence in biomedical sciences. Methods Enzymol. 2000, 305, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Ohkuma, H.; Abe, K.; Kosaka, Y.; Maeda, M. Detection of luciferase having two kinds of luminescent colour based on optical filter procedure: Application to an enzyme immunoassay. Luminescence 2000, 15, 21–27. [Google Scholar] [CrossRef]
- Shao, Q.; Jiang, T.; Ren, G.; Cheng, Z.; Xing, B. Photoactivable bioluminescent probes for imaging luciferase activity. Chem. Commun. 2009, 27, 4028. [Google Scholar] [CrossRef] [PubMed]
- Doyle, T.C.; Burns, S.M.; Contag, C.H. In vivo bioluminescence imaging for integrated studies of infection. Cell. Microbiol. 2004, 6, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Wood, K.V. Bioluminescent Assays for High-Throughput Screening. Assay Drug Dev. Technol. 2007, 5, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Niwa, K.; Yamada, N.; Enomoto, T.; Irie, T.; Kubota, H.; Ohmiya, Y.; Akiyama, H. Firefly bioluminescence quantum yield and colour change by pH-sensitive green emission. Nat. Photonics 2008, 2, 44–47. [Google Scholar] [CrossRef]
- Wang, Y.; Kubota, H.; Yamada, N.; Irie, T.; Akiyama, H. Quantum Yields and Quantitative Spectra of Firefly Bioluminescence with Various Bivalent Metal Ions. Photochem. Photobiol. 2011, 87, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, T.; Wang, Y.; Hiyama, M.; Akiyama, H. Robust red-emission spectra and yields in firefly bioluminescence against temperature changes. Appl. Phys. Lett. 2014, 104, 213704. [Google Scholar] [CrossRef]
- Biggley, W.H.; Lloyd, J.E.; Seliger, H.H. The spectral distribution of firefly light. J. Gen. Physiol. 1967, 50, 1681–1692. [Google Scholar] [CrossRef] [PubMed]
- Viviani, V.R. Fireflies (Coleoptera: Lampyridae) from Southeastern Brazil: Habitats, Life History, and Bioluminescence. Ann. Entomol. Soc. Am. 2001, 94, 129–145. [Google Scholar] [CrossRef]
- Branchini, B.R.; Magyar, R.A.; Murtiashaw, M.H.; Anderson, S.M.; Helgerson, L.C.; Zimmer, M. Site-directed mutagenesis of firefly luciferase active site amino acids: A proposed model for bioluminescence color. Biochemistry 1999, 38, 13223–13230. [Google Scholar] [CrossRef] [PubMed]
- Tafreshi, N.K.; Sadeghizadeh, M.; Emamzadeh, R.; Ranjbar, B.; Naderi-Manesh, H.; Hosseinkhani, S. Site-directed mutagenesis of firefly luciferase: implication of conserved residue(s) in bioluminescence emission spectra among firefly luciferases. Biochem. J. 2008, 412, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Akiyama, H.; Terakado, K.; Nakatsu, T. Impact of site-directed mutant luciferase on quantitative green and orange/red emission intensities in firefly bioluminescence. Sci. Rep. 2013, 3, 2490. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, A.; Yoshizaki, H.; Ohmiya, Y.; Ueda, H.; Nagamune, T. Creation of a Thermostable Firefly Luciferase with pH-insensitive Luminescent Color¶. Photochem. Photobiol. 2003, 77, 333. [Google Scholar] [CrossRef]
- Wu, R.; Cao, J.; Lu, X.; Reger, A.S.; Gulick, A.M.; Dunaway-Mariano, D. Mechanism of 4-chlorobenzoate: Coenzyme A ligase catalysis. Biochemistry 2008, 47, 8026–8039. [Google Scholar] [CrossRef] [PubMed]
- Kochan, G.; Pilka, E.S.; von Delft, F.; Oppermann, U.; Yue, W.W. Structural Snapshots for the Conformation-dependent Catalysis by Human Medium-chain Acyl-coenzyme A Synthetase ACSM2A. J. Mol. Biol. 2009, 388, 997–1008. [Google Scholar] [CrossRef]
- Sundlov, J.A.; Shi, C.; Wilson, D.J.; Aldrich, C.C.; Gulick, A.M. Structural and functional investigation of the intermolecular interaction between NRPS adenylation and carrier protein domains. Chem. Biol. 2012, 19, 188–198. [Google Scholar] [CrossRef]
- Branchini, B.R.; Murtiashaw, M.H.; Magyar, R.A.; Anderson, S.M. The role of lysine 529, a conserved residue of the acyl-adenylate- forming enzyme superfamily, in firefly luciferase. Biochemistry 2000, 39, 5433–5440. [Google Scholar] [CrossRef] [PubMed]
- Branchini, B.R.; Magyar, R.A.; Murtiashaw, M.H.; Anderson, S.M.; Zimmer, M. Site-directed mutagenesis of histidine 245 in firefly luciferase: A proposed model of the active site. Biochemistry 1998, 37, 15311–15319. [Google Scholar] [CrossRef] [PubMed]
- Branchini, B.R.; Rosenberg, J.C.; Fontaine, D.M.; Southworth, T.L.; Behney, C.E.; Uzasci, L. Bioluminescence is produced from a trapped firefly luciferase conformation predicted by the domain alternation mechanism. J. Am. Chem. Soc. 2011, 133, 11088–11091. [Google Scholar] [CrossRef] [PubMed]
- Sundlov, J.A.; Fontaine, D.M.; Southworth, T.L.; Branchini, B.R.; Gulick, A.M. Crystal structure of firefly luciferase in a second catalytic conformation supports a domain alternation mechanism. Biochemistry 2012, 51, 6493–6495. [Google Scholar] [CrossRef]
- Berraud-Pache, R.; Garcia-Iriepa, C.; Navizet, I. Modelling chemical reactions by QM/MM calculations: The case of the tautomerization in fireflies bioluminescent systems. Front. Chem. 2018, 6, 116. [Google Scholar] [CrossRef]
- García-Iriepa, C.; Gosset, P.; Berraud-Pache, R.; Zemmouche, M.; Taupier, G.; Dorkenoo, K.D.; Didier, P.; Léonard, J.; Ferré, N.; Navizet, I. Simulation and Analysis of the Spectroscopic Properties of Oxyluciferin and Its Analogues in Water. J. Chem. Theory Comput. 2018, 14, 2117–2126. [Google Scholar] [CrossRef]
- Pinto Da Silva, L.; Esteves Da Silva, J.C.G. Computational investigation of the effect of pH on the color of firefly bioluminescence by DFT. ChemPhysChem 2011, 12, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Branchini, B.R.; Martha, H.; Magyar, R.A.; Portier, N.C.; Ruggiero, M.C.; Stroh, J.G. Yellow-Green and Red Firefly Bioluminescence from 5,5-Dimethyloxyluciferin. J. Am. Chem. Soc. 2002, 124, 2112–2113. [Google Scholar] [CrossRef]
- Chen, S.F.; Liu, Y.J.; Navizet, I.; Ferré, N.; Fang, W.H.; Lindh, R. Systematic theoretical investigation on the light emitter of firefly. J. Chem. Theory Comput. 2011, 7, 798–803. [Google Scholar] [CrossRef]
- Solntsev, K.M.; Laptenok, S.P.; Naumov, P. Photoinduced dynamics of oxyluciferin analogues: Unusual enol “super”photoacidity and evidence for keto-enol isomerization. J. Am. Chem. Soc. 2012, 134, 16452–16455. [Google Scholar] [CrossRef]
- Naumov, P.; Kochunnoonny, M. Spectral−Structural Effects of the Keto−Enol−Enolate and Phenol−Phenolate Equilibria of Oxyluciferin. J. Am. Chem. Soc. 2010, 132, 11566–11579. [Google Scholar] [CrossRef] [PubMed]
- Rebarz, M.; Kukovec, B.M.; Maltsev, O.V.; Ruckebusch, C.; Hintermann, L.; Naumov, P.; Sliwa, M. Deciphering the protonation and tautomeric equilibria of firefly oxyluciferin by molecular engineering and multivariate curve resolution. Chem. Sci. 2013, 4, 3803–3809. [Google Scholar] [CrossRef]
- Snellenburg, J.J.; Laptenok, S.P.; DeSa, R.J.; Naumov, P.; Solntsev, K.M. Excited-State Dynamics of Oxyluciferin in Firefly Luciferase. J. Am. Chem. Soc. 2016, 138, 16252–16258. [Google Scholar] [CrossRef]
- Ghose, A.; Rebarz, M.; Maltsev, O.V.; Hintermann, L.; Ruckebusch, C.; Fron, E.; Hofkens, J.; Mély, Y.; Naumov, P.; Sliwa, M.; Didier, P. Emission Properties of Oxyluciferin and Its Derivatives in Water: Revealing the Nature of the Emissive Species in Firefly Bioluminescence. J. Phys. Chem. B 2015, 119, 2638–2649. [Google Scholar] [CrossRef]
- Milne, B.F.; Marques, M.A.L.; Nogueira, F. Fragment molecular orbital investigation of the role of AMP protonation in firefly luciferase pH-sensitivity. Phys. Chem. Chem. Phys. 2010, 12, 14285–14293. [Google Scholar] [CrossRef]
- Garcia Iriepa, C.; Zemmouche, M.; Ponce-Vargas, M.; Navizet, I. The role of solvation models on the computed absorption and emission spectra: The case of fireflies oxyluciferin. Phys. Chem. Chem. Phys. 2019. [Google Scholar] [CrossRef]
- Viviani, V.R.; Simões, A.; Bevilaqua, V.R.; Gabriel, G.V.M.; Arnoldi, F.G.C.; Hirano, T. Glu311 and Arg337 Stabilize a Closed Active-site Conformation and Provide a Critical Catalytic Base and Countercation for Green Bioluminescence in Beetle Luciferases. Biochemistry 2016, 55, 4764–4776. [Google Scholar] [CrossRef] [PubMed]
- Navizet, I.; Liu, Y.-J.; Ferré, N.; Xiao, H.-Y.; Fang, W.-H.; Lindh, R. Color-Tuning Mechanism of Firefly Investigated by Multi-Configurational Perturbation Method. J. Am. Chem. Soc. 2010, 132, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Zhang, J.; Liang, J. Fast Protein Loop Sampling and Structure Prediction Using Distance-Guided Sequential Chain-Growth Monte Carlo Method. PLoS Comput. Biol. 2014, 10. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. Amber 14. Available online: https://orbilu.uni.lu/handle/10993/16614 (accessed on 19 April 2019).
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.V.; van Gunsteren, W.F.; DiNola, A.R.H.J.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684. [Google Scholar] [CrossRef]
- Navizet, I.; Liu, Y.-J.; Ferré, N.; Roca-Sanjuán, D.; Lindh, R. The Chemistry of Bioluminescence: An Analysis of Chemical Functionalities. ChemPhysChem 2011, 12, 3064–3076. [Google Scholar] [CrossRef]
- Navizet, I.; Roca-Sanjuán, D.; Yue, L.; Liu, Y.J.; Ferré, N.; Lindh, R. Are the bio- and chemiluminescence states of the firefly oxyluciferin the same as the fluorescence state? Photochem. Photobiol. 2013, 89, 319–325. [Google Scholar] [CrossRef]
- Ferré, N.; Ángyán, J.G. Approximate electrostatic interaction operator for QM/MM calculations. Chem. Phys. Lett. 2002, 356, 331–339. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09 Revision D.01. Available online: http://www.rsc.org/suppdata/c5/sc/c5sc02423d/c5sc02423d1.pdf (accessed on 19 April 2019).
- Ponder, J.W. TINKER, Software Tools for Molecular Design, Version 6.3; Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine: St. Louis, MO, USA, 2004. [Google Scholar]
- Berraud-Pache, R.; Navizet, I. QM/MM calculations on a newly synthesised oxyluciferin substrate: New insights into the conformational effect. Phys. Chem. Chem. Phys. 2016, 18, 27460–27467. [Google Scholar] [CrossRef]
- Min, C.G.; Leng, Y.; Zhu, Y.Q.; Yang, X.K.; Huang, S.J.; Ren, A.M. Modification of firefly cyclic amino oxyluciferin analogues emitting multicolor light for OLED and near-Infrared biological window light for bioluminescence imaging: A theoretical study. J. Photochem. Photobiol. A Chem. 2017, 336, 115–122. [Google Scholar] [CrossRef]
- Melaccio, F.; Olivucci, M.; Lindh, R.; Ferré, N. Unique QM/MM potential energy surface exploration using microiterations. Int. J. Quantum Chem. 2011, 111, 3339–3346. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Iriepa, C.; Navizet, I. Effect of Protein Conformation and AMP Protonation State on Fireflies’ Bioluminescent Emission. Molecules 2019, 24, 1565. https://doi.org/10.3390/molecules24081565
Garcia-Iriepa C, Navizet I. Effect of Protein Conformation and AMP Protonation State on Fireflies’ Bioluminescent Emission. Molecules. 2019; 24(8):1565. https://doi.org/10.3390/molecules24081565
Chicago/Turabian StyleGarcia-Iriepa, Cristina, and Isabelle Navizet. 2019. "Effect of Protein Conformation and AMP Protonation State on Fireflies’ Bioluminescent Emission" Molecules 24, no. 8: 1565. https://doi.org/10.3390/molecules24081565
APA StyleGarcia-Iriepa, C., & Navizet, I. (2019). Effect of Protein Conformation and AMP Protonation State on Fireflies’ Bioluminescent Emission. Molecules, 24(8), 1565. https://doi.org/10.3390/molecules24081565