The Distribution of Tryptophan-Dependent Indole-3-Acetic Acid Synthesis Pathways in Bacteria Unraveled by Large-Scale Genomic Analysis


 , , and
, , and 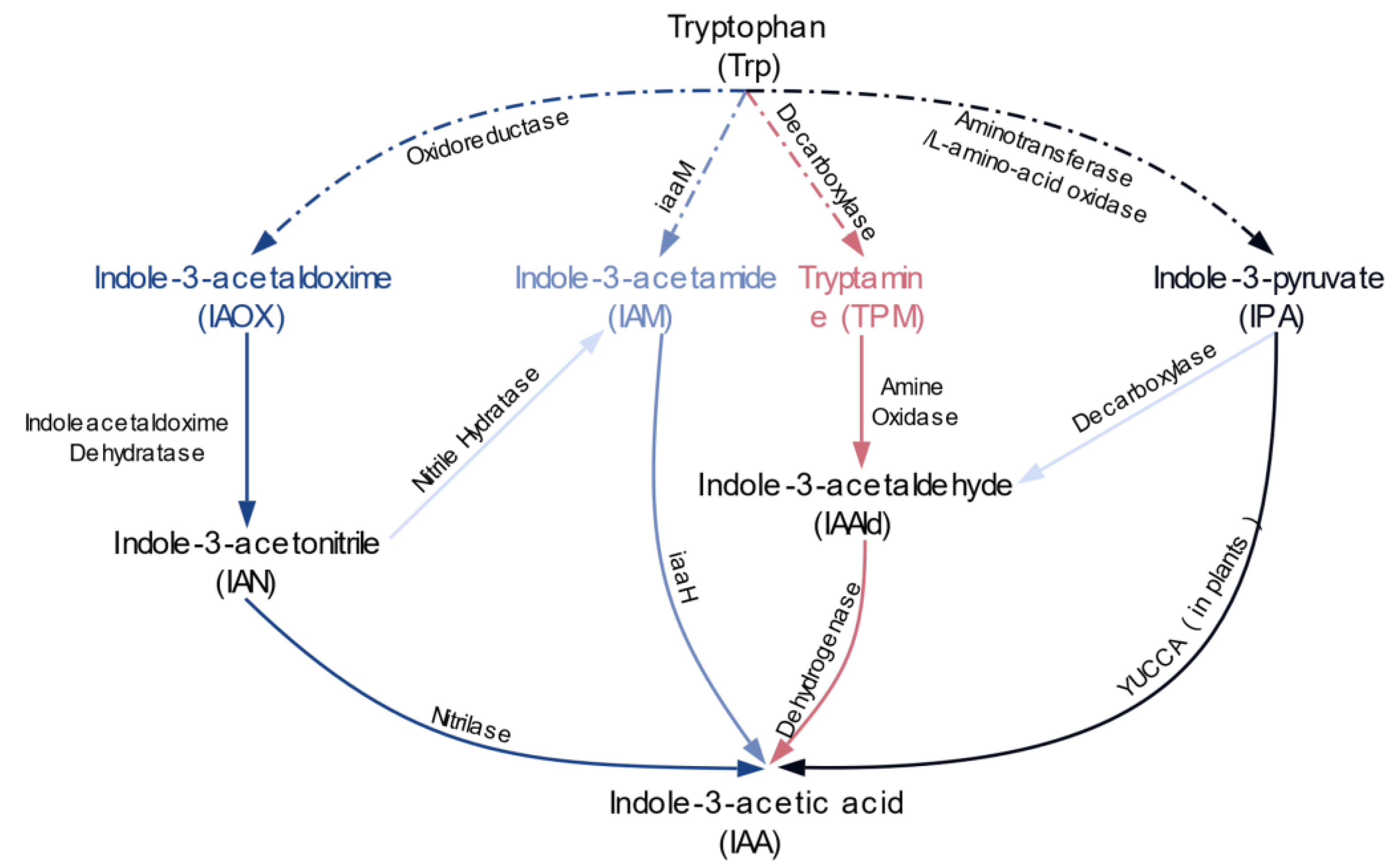{kind=link}
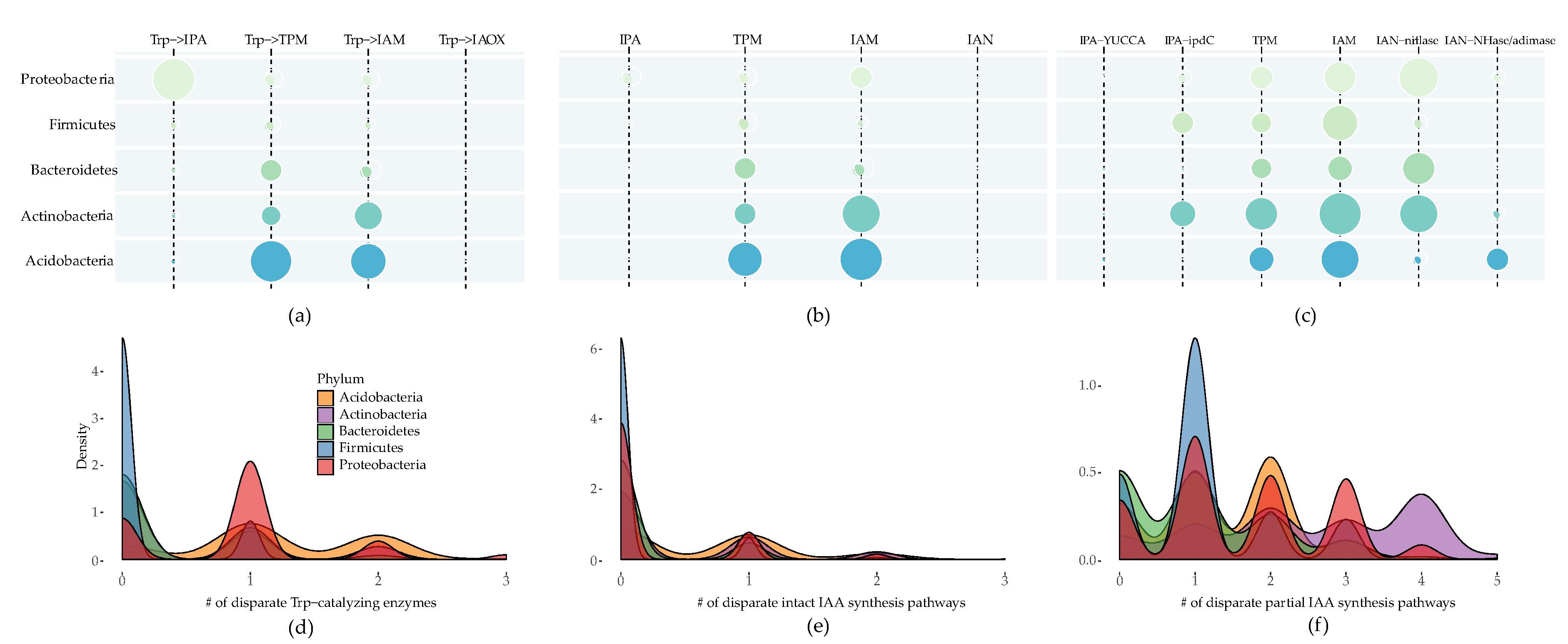{kind=link}
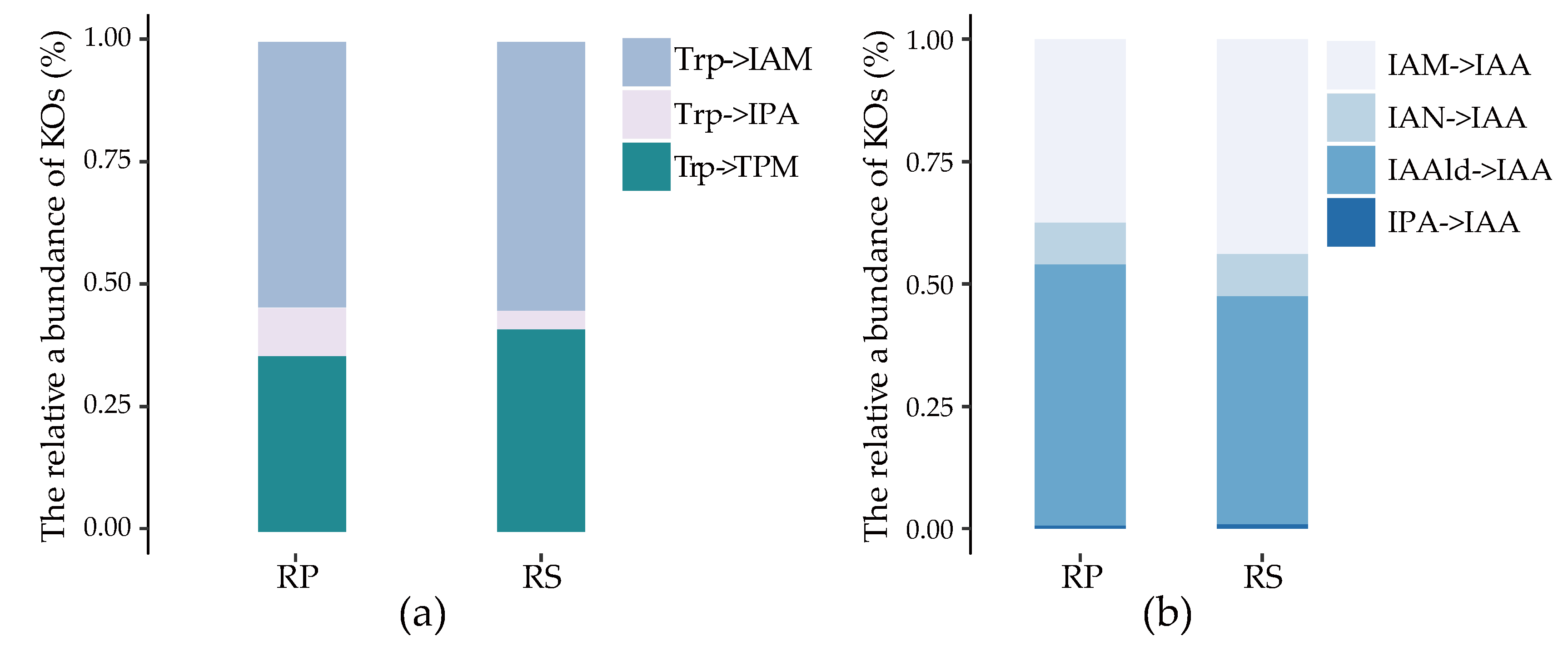{kind=link}
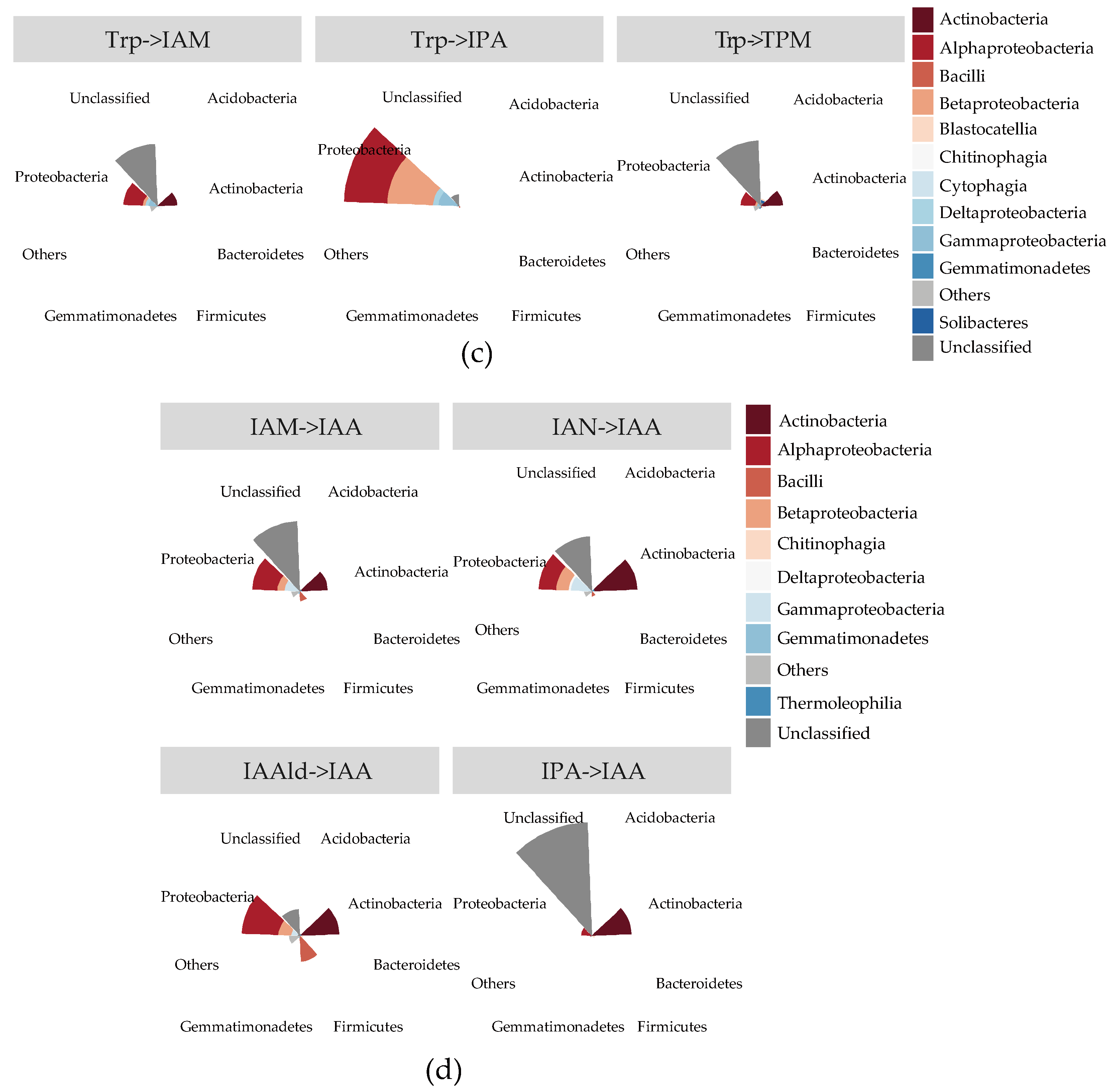{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
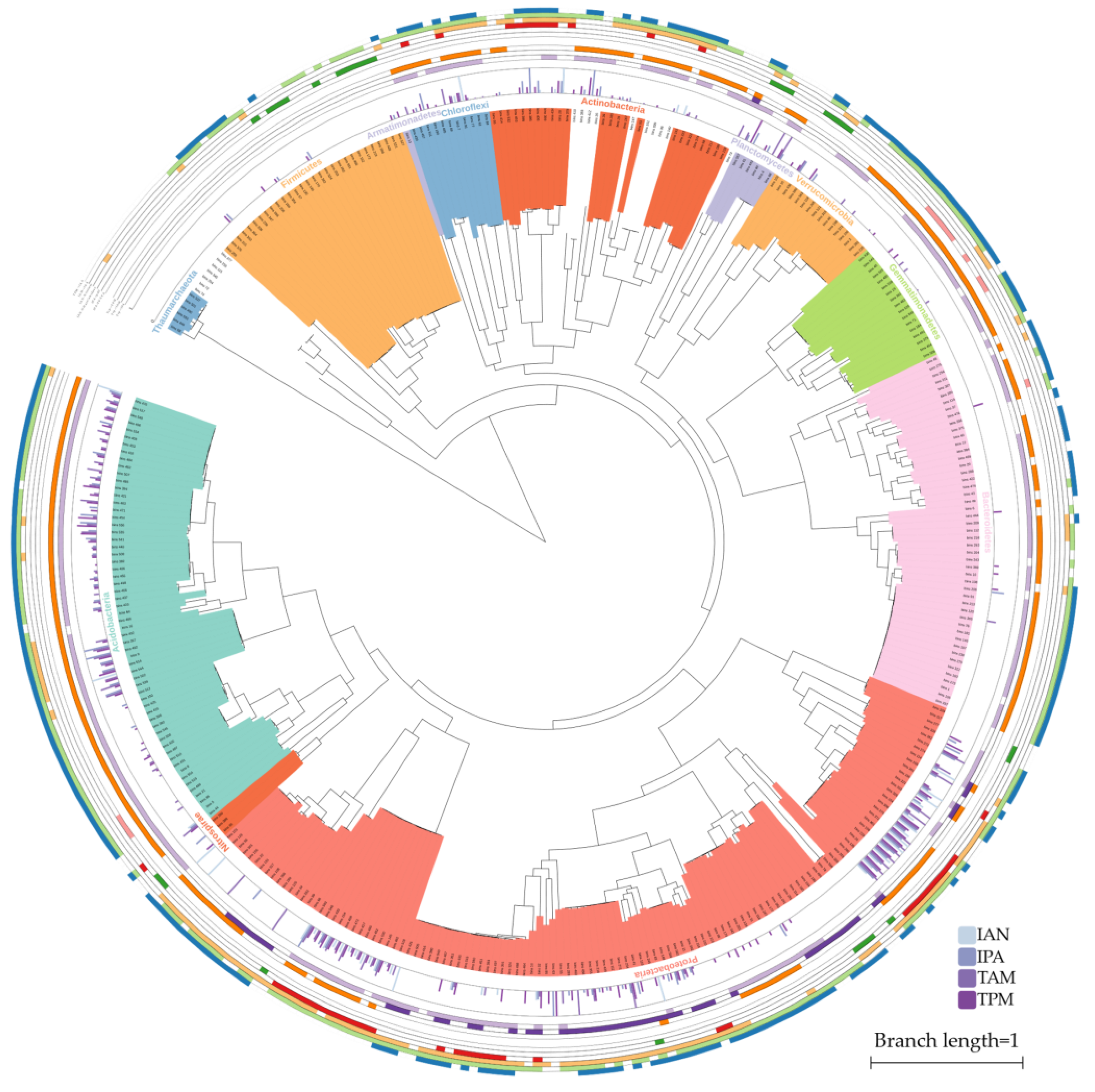
2.1. IAA Biosynthesis is Widely Distributed among Different Bacterial Phyla
2.2. Multiple Distinct Biosynthetic Pathways Coexist in Bacteria
2.3. Quantification of IAA Synthetic Genes in Root Environments
2.4. The Stronger Capacity of IAA Synthesis for Rhizobacteria
3. Discussion
4. Materials and Methods
4.1. Genome Retrieval and Analysis
4.2. Sample Collection for Metagenomic Sequencing
4.3. Metagenomic Sequencing and Processing
4.4. Metagenomic Binning
4.5. Phylogenetic Annotations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhao, Y. Auxin biosynthesis and its role in plant development. Annu. Rev. Plant Biol. 2010, 61, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Duca, D.; Lorv, J.; Patten, C.L.; Rose, D.; Glick, B.R. Indole-3-acetic acid in plant–microbe interactions. Antonie Van Leeuwenhoek 2014, 106, 85–125. [Google Scholar] [CrossRef] [PubMed]
- Fierro-Coronado, R.A.; Quiroz-Figueroa, F.R.; García-Pérez, L.M.; Ramírez-Chávez, E.; Molina-Torres, J.; Maldonado-Mendoza, I.E. IAA-producing rhizobacteria from chickpea (Cicer arietinum L.) induce changes in root architecture and increase root biomass. Can. J. Microbiol. 2014, 60, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Gamalero, E.; Glick, B.R. Mechanisms used by plant growth-promoting bacteria. In Bacteria in Agrobiology: Plant Nutrient Management; Springer: Berlin, Germany, 2011; pp. 17–46. [Google Scholar]
- Patten, C.L.; Glick, B.R. Role of Pseudomonas putida indoleacetic acid in development of the host plant root system. Appl. Environ. Microbiol. 2002, 68, 3795–3801. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Defez, R. Improvement of phosphate solubilization and Medicago plant yield by an indole-3-acetic acid-overproducing strain of Sinorhizobium meliloti. Appl. Environ. Microbiol. 2010, 76, 4626–4632. [Google Scholar] [CrossRef] [PubMed]
- Hooykaas, P.; Beijersbergen, A.G. The virulence system of Agrobacterium tumefaciens. Annu. Rev. Phytopathol. 1994, 32, 157–181. [Google Scholar] [CrossRef]
- Chalupowicz, L.; Barash, I.; Schwartz, M.; Aloni, R.; Manulis, S. Comparative anatomy of gall development on Gypsophila paniculata induced by bacteria with different mechanisms of pathogenicity. Planta 2006, 224, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-C.; Liu, P.; Saenkham, P.; Kerr, K.; Nester, E.W. Transcriptome profiling and functional analysis of Agrobacterium tumefaciens reveals a general conserved response to acidic conditions (pH 5.5) and a complex acid-mediated signaling involved in Agrobacterium-plant interactions. J. Bacteriol. 2008, 190, 494–507. [Google Scholar] [CrossRef]
- Imperlini, E.; Bianco, C.; Lonardo, E.; Camerini, S.; Cermola, M.; Moschetti, G.; Defez, R. Effects of indole-3-acetic acid on Sinorhizobium meliloti survival and on symbiotic nitrogen fixation and stem dry weight production. Appl. Microbiol. Biotechnol. 2009, 83, 727. [Google Scholar] [CrossRef]
- Spaepen, S.; Vanderleyden, J.; Remans, R. Indole-3-acetic acid in microbial and microorganism-plant signaling. FEMS Microbiol. Rev. 2007, 31, 425–448. [Google Scholar] [CrossRef]
- Mano, Y.; Nemoto, K. The pathway of auxin biosynthesis in plants. J. Exp. Botany 2012, 63, 2853–2872. [Google Scholar] [CrossRef]
- Hartmann, A.; Singh, M.; Klingmüller, W. Isolation and characterization of Azospirillum mutants excreting high amounts of indoleacetic acid. Can. J. Microbiol. 1983, 29, 916–923. [Google Scholar] [CrossRef]
- Suzuki, S.; He, Y.; Oyaizu, H. Indole-3-acetic acid production in Pseudomonas fluorescens HP72 and its association with suppression of creeping bentgrass brown patch. Curr. Microbiol. 2003, 47, 0138–0143. [Google Scholar] [CrossRef]
- Oberhänsli, T.; Défago, G.; Haas, D. Indole-3-acetic acid (IAA) synthesis in the biocontrol strain CHA0 of Pseudomonas fluorescens: Role of tryptophan side chain oxidase. Microbiology 1991, 137, 2273–2279. [Google Scholar] [CrossRef]
- Whistler, C.A.; Corbell, N.A.; Sarniguet, A.; Ream, W.; Loper, J.E. The Two-Component Regulators GacS and GacA Influence Accumulation of the Stationary-Phase Sigma Factor ςS and the Stress Response in Pseudomonas fluorescensPf-5. J. Bacteriol. 1998, 180, 6635–6641. [Google Scholar]
- Spaepen, S.; Vanderleyden, J. Auxin and plant-microbe interactions. Cold Spring Harbor Perspect. Biol. 2010, 3, a001438. [Google Scholar] [CrossRef]
- Patten, C.L.; Blakney, A.J.; Coulson, T.J. Activity, distribution and function of indole-3-acetic acid biosynthetic pathways in bacteria. Crit. Rev. Microbiol. 2013, 39, 395–415. [Google Scholar] [CrossRef]
- Sun, S.-L.; Yang, W.-L.; Fang, W.-W.; Zhao, Y.-X.; Guo, L.; Dai, Y.-J. The plant growth-promoting rhizobacterium Variovorax boronicumulans CGMCC 4969 regulates the level of indole-3-acetic acid synthesized from indole-3-acetonitrile. Appl. Environ. Microbiol. 2018, 84, e00298-18. [Google Scholar] [CrossRef]
- Manulis, S.; Haviv-Chesner, A.; Brandl, M.T.; Lindow, S.E.; Barash, I. Differential involvement of indole-3-acetic acid biosynthetic pathways in pathogenicity and epiphytic fitness of Erwinia herbicola pv. gypsophilae. Mol. Plant Microbe Int. 1998, 11, 634–642. [Google Scholar] [CrossRef]
- Jin, T.; Wang, Y.; Huang, Y.; Xu, J.; Zhang, P.; Wang, N.; Liu, X.; Chu, H.; Liu, G.; Jiang, H. Taxonomic structure and functional association of foxtail millet root microbiome. GigaScience 2017, 6. [Google Scholar] [CrossRef]
- Kittell, B.L.; Helinski, D.R.; Ditta, G.S. Aromatic aminotransferase activity and indoleacetic acid production in Rhizobium meliloti. J. Bacteriol. 1989, 171, 5458–5466. [Google Scholar] [CrossRef]
- Law, D.M. Gibberellin-enhanced indole-3-acetic acid biosynthesis: D-Tryptophan as the precursor of indole-3-acetic acid. Physiol. Plantarum 1987, 70, 626–632. [Google Scholar] [CrossRef]
- Podar, M.; Eads, J.R.; Richardson, T.H. Evolution of a microbial nitrilase gene family: A comparative and environmental genomics study. BMC Evolutionary Biol. 2005, 5, 42. [Google Scholar] [CrossRef]
- Follin, A.; Inzé, D.; Budar, F.; Genetello, C.; Van Montagu, M.; Schell, J. Genetic evidence that the tryptophan 2-mono-oxygenase gene of Pseudomonas savastonoi is functionally equivalent to one of the T-DNA genes involved in plant tumour formation by Agrobacterium tumefaciens. Mol. Gene. Gene. MGG 1985, 201, 178–185. [Google Scholar] [CrossRef]
- Krawczyk, P.S.; Lipinski, L.; Dziembowski, A. PlasFlow: Predicting plasmid sequences in metagenomic data using genome signatures. Nucl. Acids Res. 2018, 46, e35. [Google Scholar] [CrossRef]
- Bianco, C.; Imperlini, E.; Calogero, R.; Senatore, B.; Amoresano, A.; Carpentieri, A.; Pucci, P.; Defez, R. Indole-3-acetic acid improves Escherichia coli’s defences to stress. Arch. Microbiol. 2006, 185, 373–382. [Google Scholar] [CrossRef]
- Van Puyvelde, S.; Cloots, L.; Engelen, K.; Das, F.; Marchal, K.; Vanderleyden, J.; Spaepen, S. Transcriptome analysis of the rhizosphere bacterium Azospirillum brasilense reveals an extensive auxin response. Microbial Ecol. 2011, 61, 723–728. [Google Scholar] [CrossRef]
- Tromas, A.; Perrot-Rechenmann, C. Recent progress in auxin biology. Comptes Rendus Biol. 2010, 333, 297–306. [Google Scholar] [CrossRef]
- Shelton, A.N.; Seth, E.C.; Mok, K.C.; Han, A.W.; Jackson, S.N.; Haft, D.R.; Taga, M.E. Uneven distribution of cobamide biosynthesis and dependence in bacteria predicted by comparative genomics. BioRxiv 2019, 13, 789. [Google Scholar] [CrossRef]
- Mathesius, U. Goldacre paper: Auxin: At the root of nodule development? Funct. Plant Biol. 2008, 35, 651–668. [Google Scholar] [CrossRef]
- Xie, H.; Pasternak, J.; Glick, B.R. Isolation and characterization of mutants of the plant growth-promoting rhizobacterium Pseudomonas putida GR12-2 that overproduce indoleacetic acid. Curr. Microbiol. 1996, 32, 67–71. [Google Scholar] [CrossRef]
- Finkel, O.M.; Castrillo, G.; Paredes, S.H.; González, I.S.; Dangl, J.L. Understanding and exploiting plant beneficial microbes. Curr. Opin. Plant Biol. 2017, 38, 155–163. [Google Scholar] [CrossRef]
- Fu, J.; Liu, H.; Li, Y.; Yu, H.; Li, X.; Xiao, J.; Wang, S. Manipulating broad-spectrum disease resistance by suppressing pathogen-induced auxin accumulation in rice. Plant Physiol. 2011, 155, 589–602. [Google Scholar] [CrossRef]
- Patten, C.L.; Glick, B.R. Bacterial biosynthesis of indole-3-acetic acid. Can. J. Microbiol. 1996, 42, 207–220. [Google Scholar] [CrossRef]
- Ryu, R.J.; Patten, C.L. Aromatic amino acid-dependent expression of indole-3-pyruvate decarboxylase is regulated by TyrR in Enterobacter cloacae UW5. J. Bacteriol. 2008, 190, 7200–7208. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, Q.; Guo, J.; Charkowski, A.O.; Glick, B.R.; Ibekwe, A.M.; Cooksey, D.A.; Yang, C.-H. Global effect of indole-3-acetic acid biosynthesis on multiple virulence factors of Erwinia chrysanthemi 3937. Appl. Environ. Microbiol. 2007, 73, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Matsukawa, E.; Nakagawa, Y.; Iimura, Y.; Hayakawa, M. Stimulatory effect of indole-3-acetic acid on aerial mycelium formation and antibiotic production in Streptomyces spp. Actinomycetologica 2007, 21, 32–39. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucl. Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z. SOAPnuke: A MapReduce Acceleration supported Software for integrated Quality Control and Preprocessing of High-Throughput Sequencing Data. Gigascience 2017, 7. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, X.; Quan, Z.; Cheng, S.; Xu, X.; Pan, S.; Xie, M.; Zeng, P.; Yue, Z.; Wang, W. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 2012, 30, 549. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal prokaryotic dynamic programming genefinding algorithm. Bmc Bioinform. 2010, 11, 119. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucl. Acids Res. 2000, 27, 29–34. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1. 0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Varghese, N.J.; Mukherjee, S.; Ivanova, N.; Konstantinidis, K.T.; Mavrommatis, K.; Kyrpides, N.C.; Pati, A. Microbial species delineation using whole genome sequences. Nucl. Acids Res. 2015, 43, 6761–6771. [Google Scholar] [CrossRef]
- Anantharaman, K.; Brown, C.T.; Hug, L.A.; Sharon, I.; Castelle, C.J.; Probst, A.J.; Thomas, B.C.; Singh, A.; Wilkins, M.J.; Karaoz, U. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 2016, 7, 13219. [Google Scholar] [CrossRef]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucl. Acids Res. 2006, 35, D61–D65. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucl. Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2006, 23, 127–128. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610. [Google Scholar] [CrossRef]
Sample Availability: Not available. |





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, P.; Jin, T.; Kumar Sahu, S.; Xu, J.; Shi, Q.; Liu, H.; Wang, Y. The Distribution of Tryptophan-Dependent Indole-3-Acetic Acid Synthesis Pathways in Bacteria Unraveled by Large-Scale Genomic Analysis. Molecules 2019, 24, 1411. https://doi.org/10.3390/molecules24071411
Zhang P, Jin T, Kumar Sahu S, Xu J, Shi Q, Liu H, Wang Y. The Distribution of Tryptophan-Dependent Indole-3-Acetic Acid Synthesis Pathways in Bacteria Unraveled by Large-Scale Genomic Analysis. Molecules. 2019; 24(7):1411. https://doi.org/10.3390/molecules24071411
Chicago/Turabian StyleZhang, Pengfan, Tao Jin, Sunil Kumar Sahu, Jin Xu, Qiong Shi, Huan Liu, and Yayu Wang. 2019. "The Distribution of Tryptophan-Dependent Indole-3-Acetic Acid Synthesis Pathways in Bacteria Unraveled by Large-Scale Genomic Analysis" Molecules 24, no. 7: 1411. https://doi.org/10.3390/molecules24071411
APA StyleZhang, P., Jin, T., Kumar Sahu, S., Xu, J., Shi, Q., Liu, H., & Wang, Y. (2019). The Distribution of Tryptophan-Dependent Indole-3-Acetic Acid Synthesis Pathways in Bacteria Unraveled by Large-Scale Genomic Analysis. Molecules, 24(7), 1411. https://doi.org/10.3390/molecules24071411