
In Silico Peptide Ligation: Iterative Residue Docking and Linking as a New Approach to Predict Protein-Peptide Interactions

 ,
,
Abstract
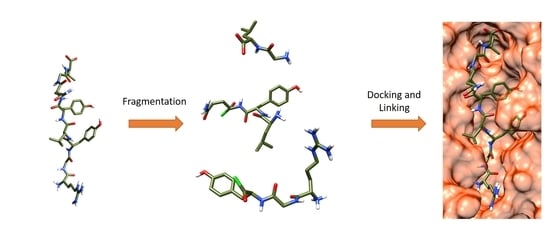
:
1. Introduction
2. Results and Discussion
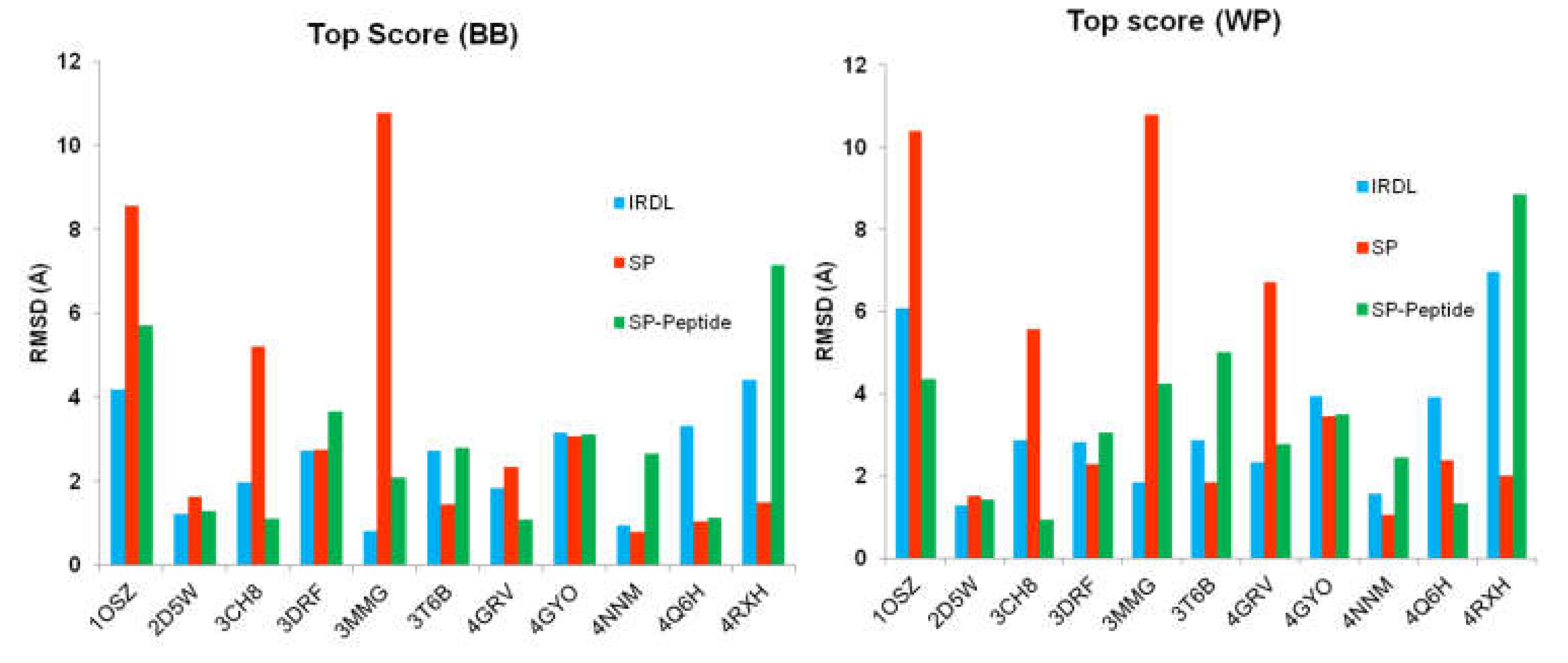
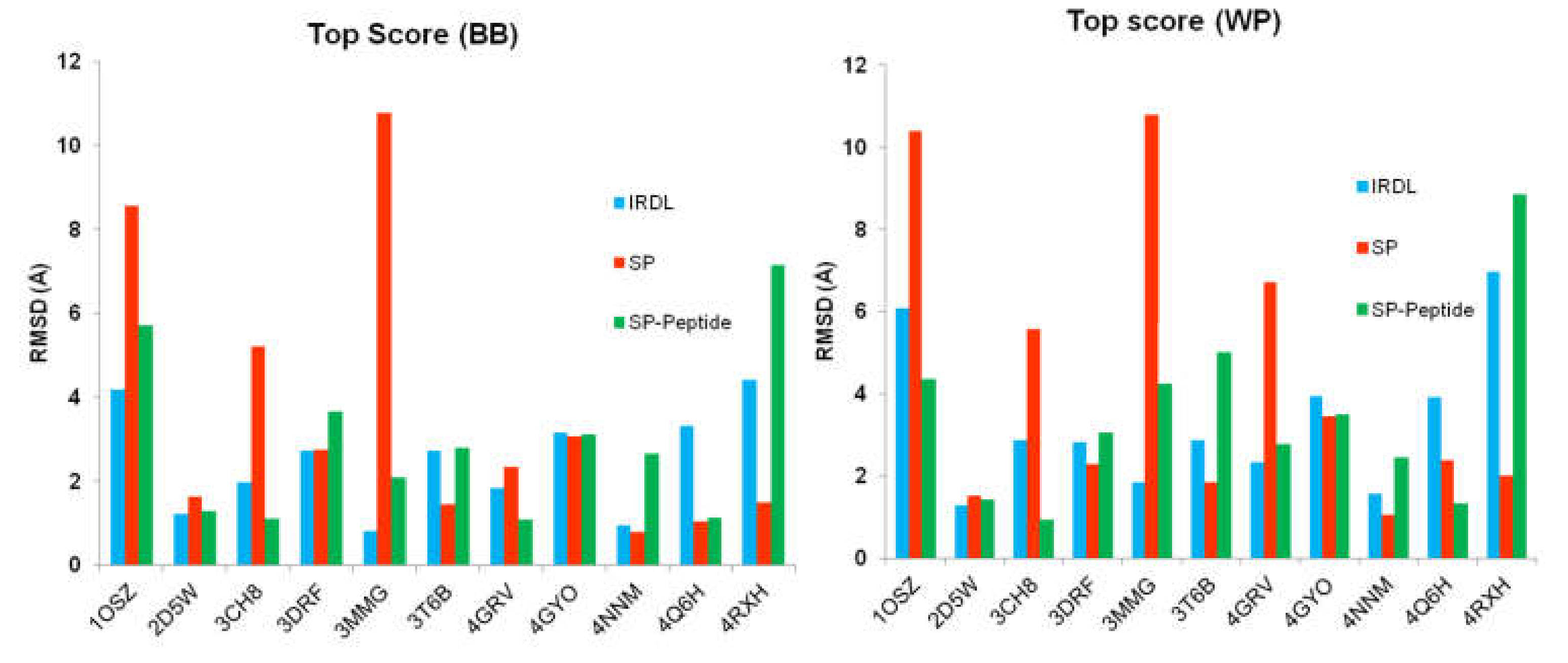
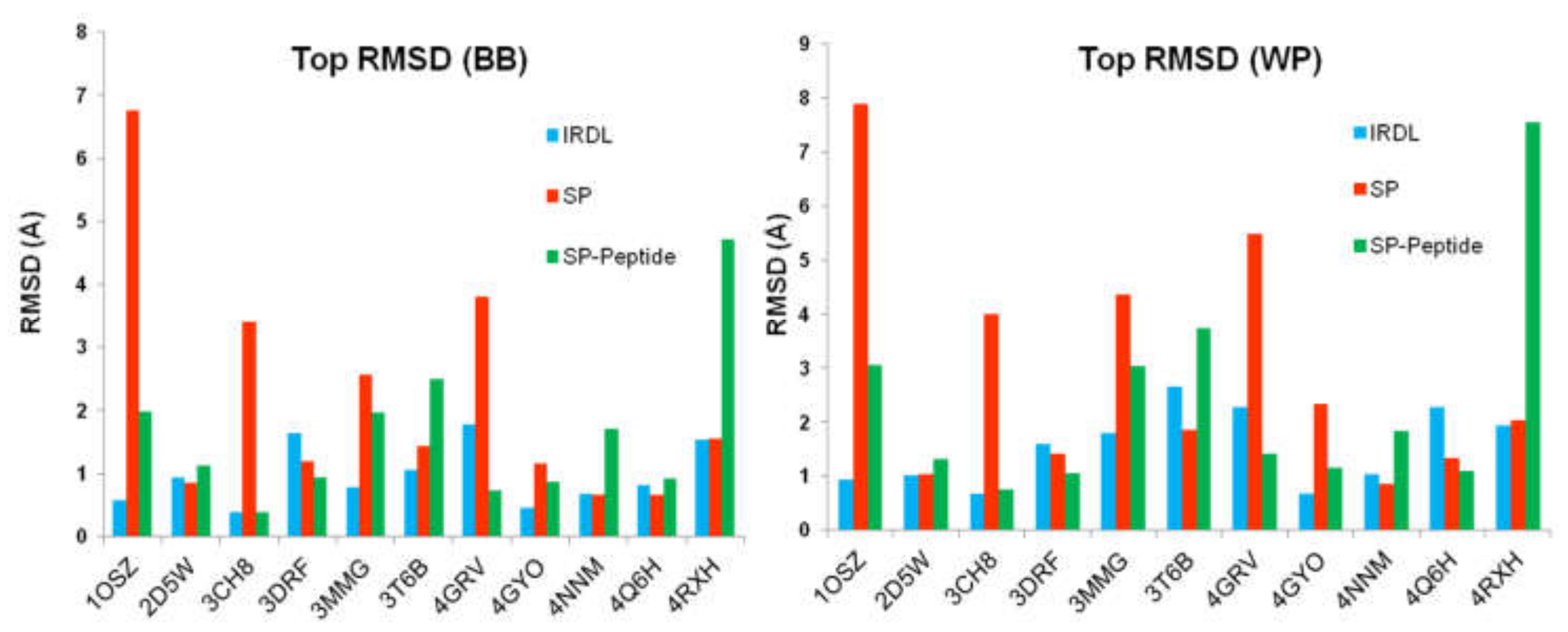
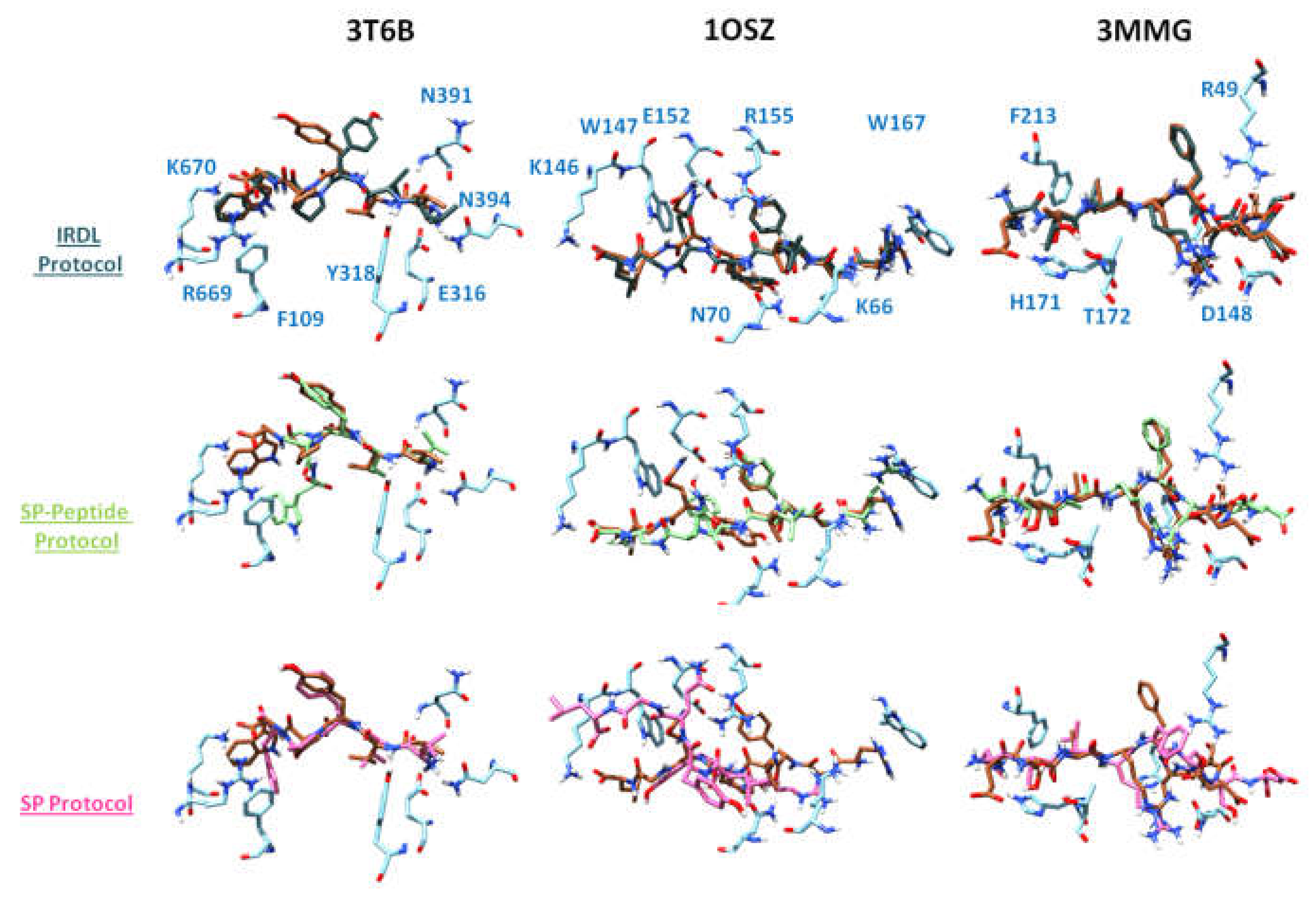
2.1. Highest Scoring Pose
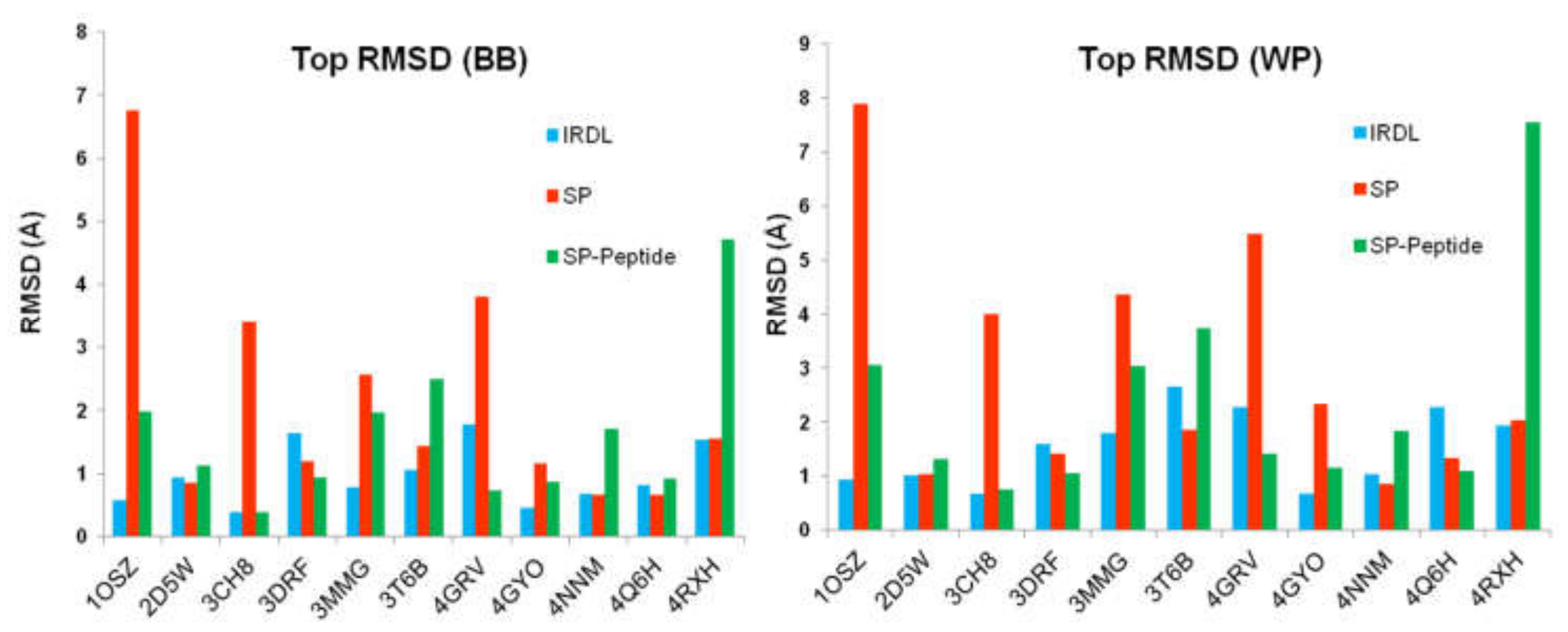
2.2. Poses with the Lowest RMSD to the Crystal Structure
3. Materials and Methods
3.1. Datasets
3.2. Receptor Preparation
3.3. SP and SP-Peptide Docking
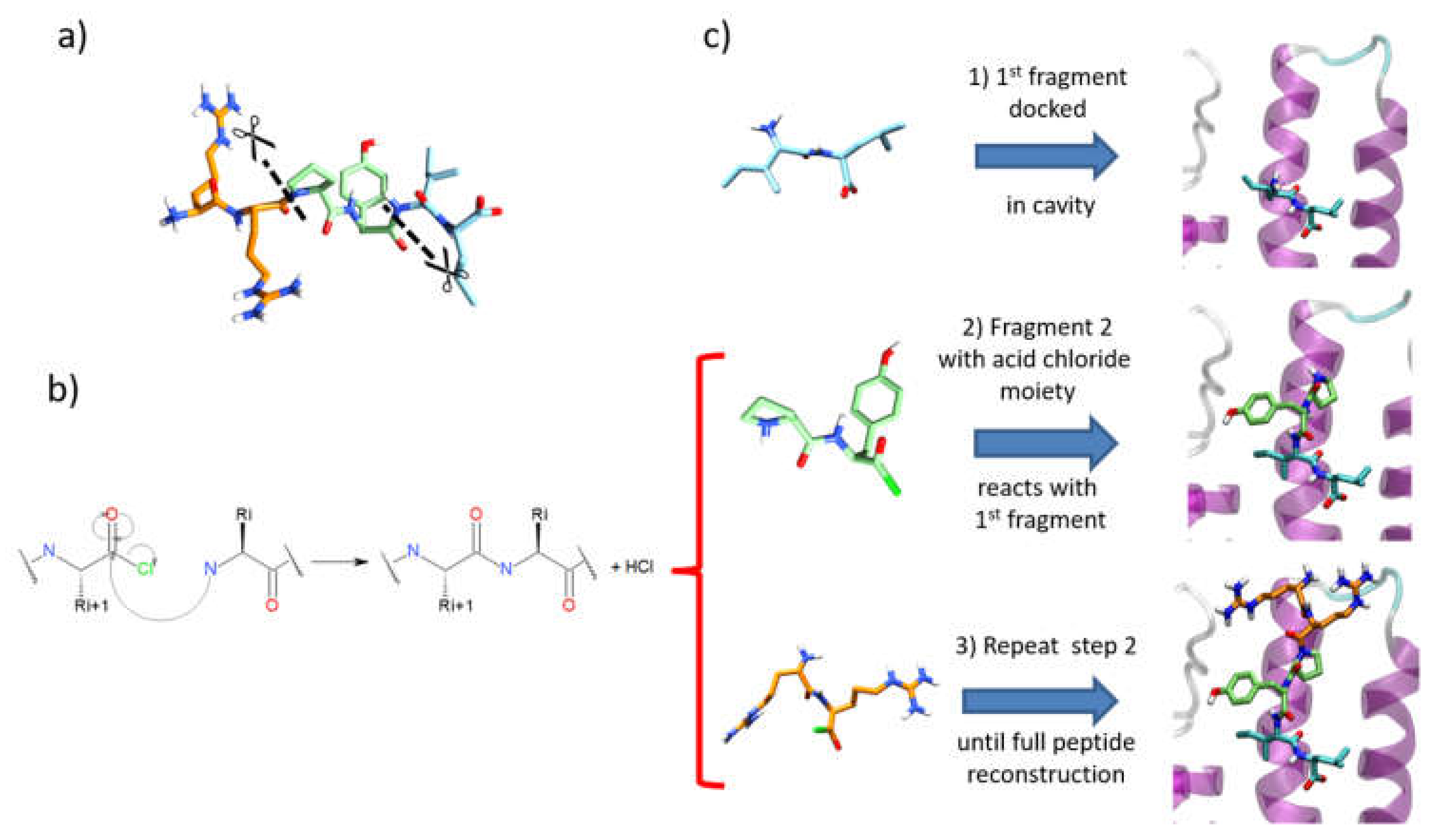
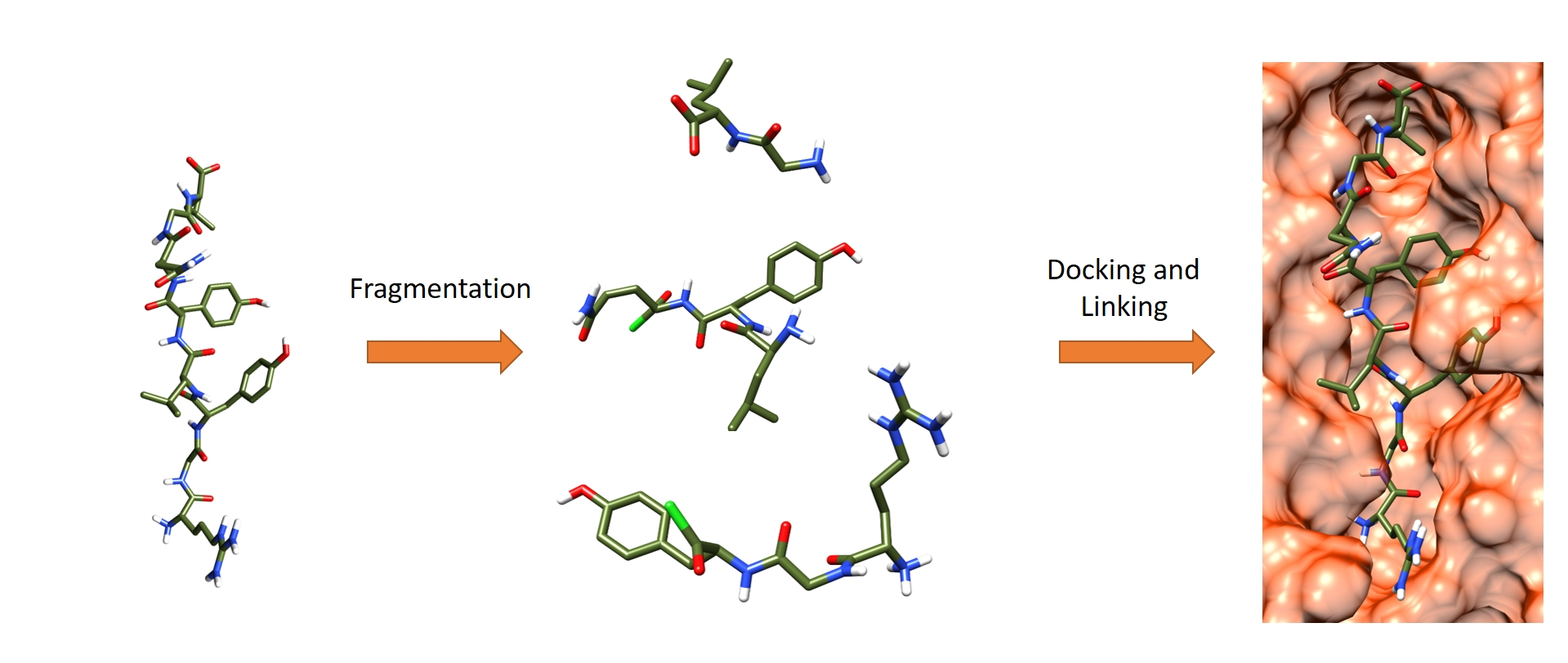
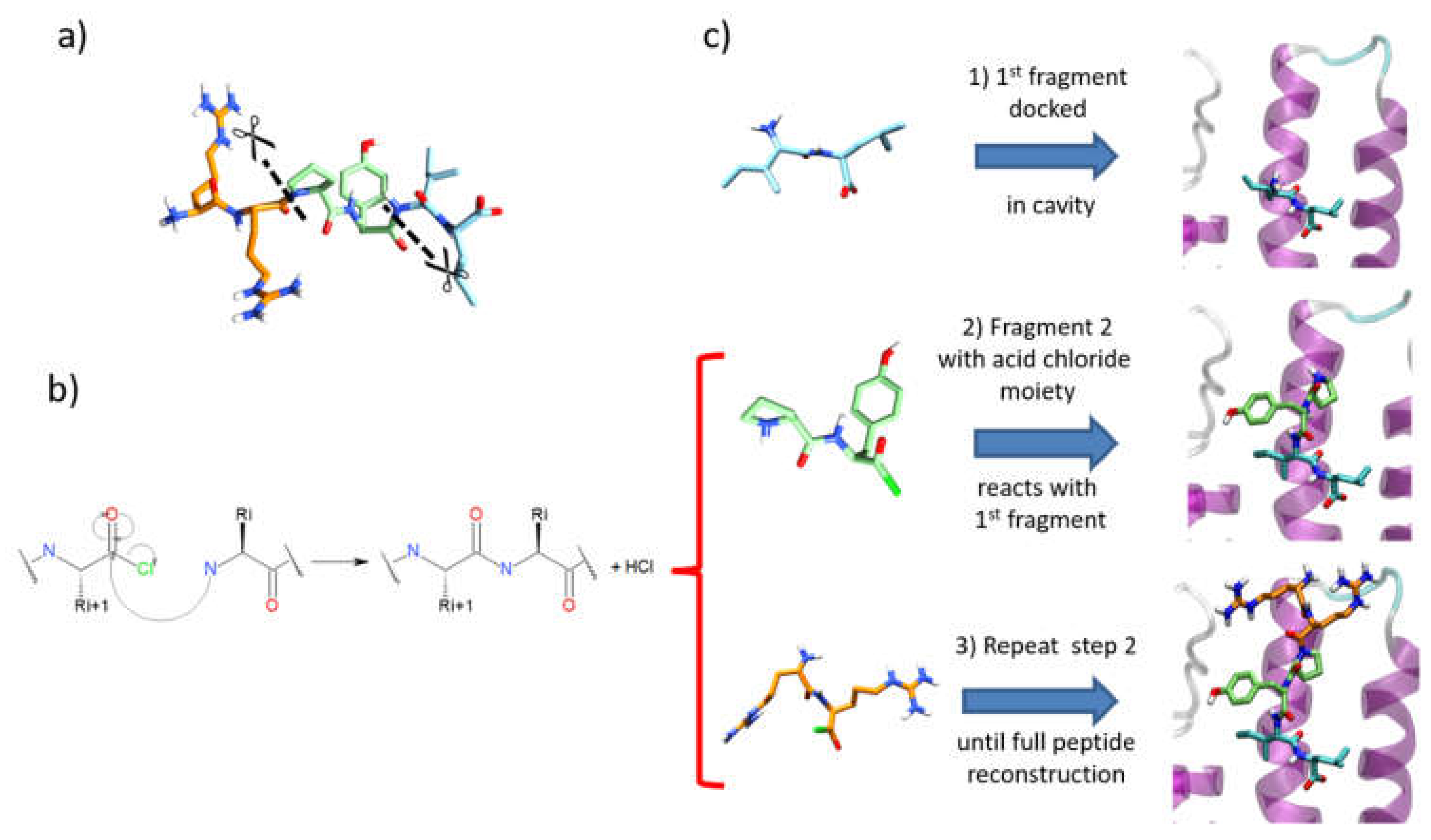
3.4. Iterative Residue Docking and Linking Method
3.4.1. File Preparation
- (a)
- Segments must have 10 or less rotatable bonds. This criterion allows simplification of each calculation by reducing flexibility, therefore improving the conformational search during docking.
- (b)
- To avoid unfeasible covalent docking solutions, we considered that the N-terminal residue should not contain any nucleophilic sidechains. Thus, residues such as Tyr, Cys, Ser, Thr, Asn, and Gln were excluded as N-terminal residues in a fragment, since the covalent docking module of Glide considers those amino acids as nucleophilic moieties. Otherwise, when the molecular topology of the fragment is not conserved after intramolecular covalent docking, the solutions are removed during the post-process analysis (see below).
- (1)
- After a conformational search with MacroModel of the first fragment, as performed for SP and SP-Peptide docking, all conformers were docked in the binding site using the SP-Peptide module of Glide with default parameters. Then, we kept all docking poses up to the pose with the lowest RMSD to crystal structure. In a more realistic project, one would keep all docking poses, which will increase computational time but could improve the results.
- (2)
- The second segment is docked using the covalent docking module of Glide (version 1.3) on each of the selected scoring poses of the first fragment. To allow the chemical reaction to occur, an acid chloride moiety is used on the C-terminal residue of the second fragment. During docking, this acid chloride moiety is automatically placed nearby the primary amine backbone of the first docked fragment. Glide will then optimize the interactions of the molecule in the binding site, keeping the distance between the acid chloride and the primary amine backbone. Then, in order to create the peptide bond, the chemical reaction between the N-terminal part of the first segment and the acid chloride moiety is made. Finally, both residues at the vicinity of the newly formed bond are optimized to remove strains and the receptor stays rigid. Then, the resulting poses are clustered into 15 clusters and a representative pose is proposed for each cluster. All cluster centers are ranked by the scoring function of the Glide covalent docking module. This scoring function averages the Glide docking score values of the two individual noncovalent poses and the resulting covalent poses involved in the peptide bond. Finally, all solutions are rescored with SP scoring function. At this step, we also evaluated the extra precision (XP) scoring function for those intermediate rescoring steps, but the results were less satisfactory (data not shown). As in step one, we decided to keep the highest scoring poses up to the pose with the lowest RMSD to the X-ray structure.
- (3)
- Step 2 is repeated until the whole peptide is entirely created.
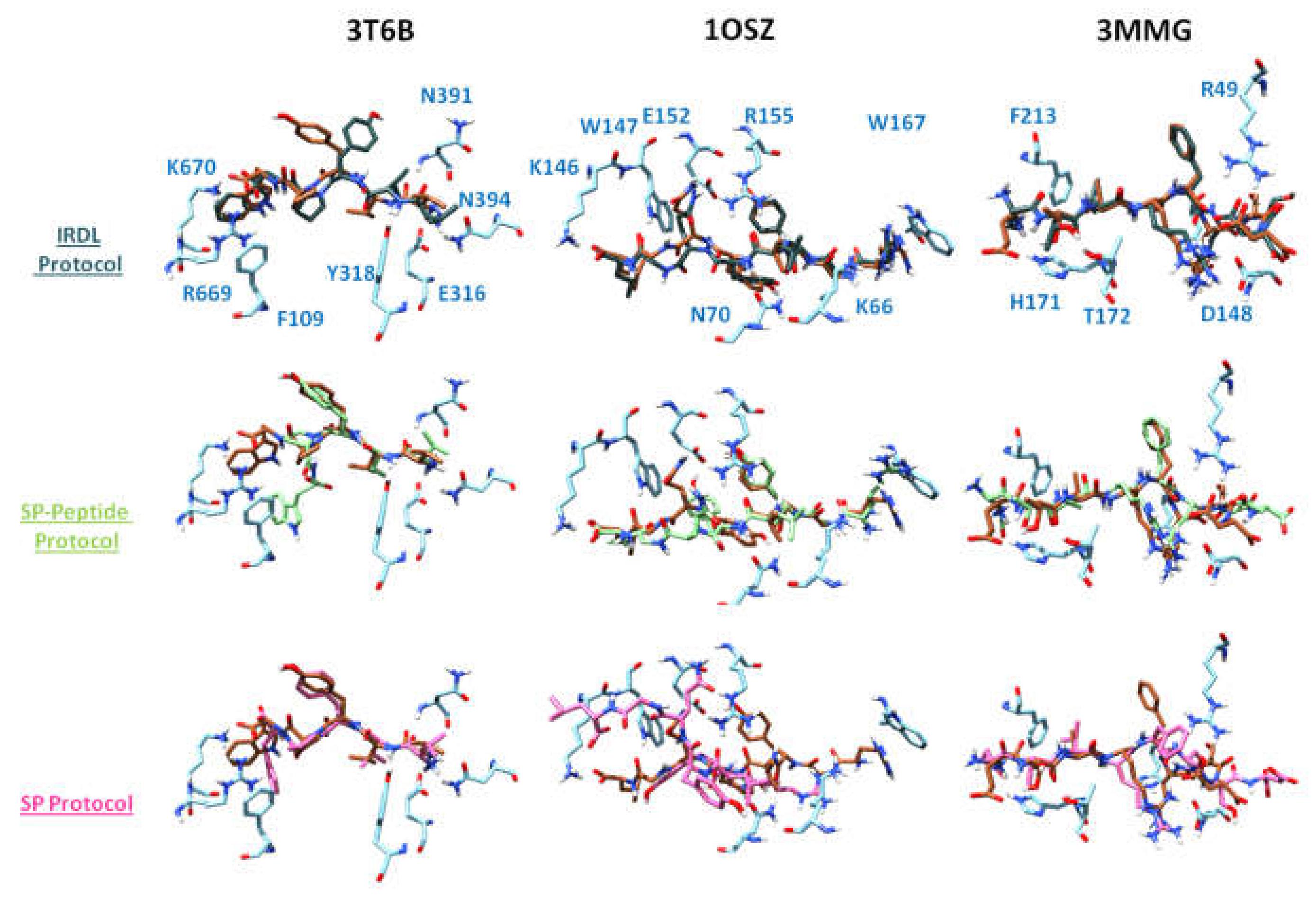
3.4.2. Post-Process Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pawson, T.; Nash, P. Assembly of Cell Regulatory Systems through Protein Interaction Domains. Science 2003, 300, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Blobel, G.; Sabatini, D.D. Ribosome-Membrane Interaction in Eukaryotic Cells. In Biomembranes: Volume 2; Manson, L.A., Ed.; Springer US: Boston, MA, USA, 1971; pp. 193–195. ISBN 978-1-4684-3330-2. [Google Scholar]
- Petsalaki, E.; Russell, R.B. Peptide-mediated interactions in biological systems: New discoveries and applications. Curr. Opin. Biotech. 2008, 19, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Tovar, C.; Rosinski, J.; Filipovic, Z.; Higgins, B.; Kolinsky, K.; Hilton, H.; Zhao, X.; Vu, B.T.; Qing, W.; Packman, K.; et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 1888–1893. [Google Scholar] [CrossRef]
- Perez, M.; Cuadros, R.; Benitez, M.J.; Jimenez, J.S. Interaction of Alzheimer’s disease amyloid beta peptide fragment 25-35 with tau protein, and with a tau peptide containing the microtubule binding domain. J. Alzheimers Dis. 2004, 6, 461–467. [Google Scholar] [CrossRef]
- Penna, G.; Amuchastegui, S.; Cossetti, C.; Aquilano, F.; Mariani, R.; Giarratana, N.; De Carli, E.; Fibbi, B.; Adorini, L. Spontaneous and prostatic steroid binding protein peptide-induced autoimmune prostatitis in the nonobese diabetic mouse. J. Immunol. 2007, 179, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Sievers, S.A.; Karanicolas, J.; Chang, H.W.; Zhao, A.; Jiang, L.; Zirafi, O.; Stevens, J.T.; Munch, J.; Baker, D.; Eisenberg, D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 2011, 475, 96–100. [Google Scholar] [CrossRef]
- Illana, G.; Inna, D. NAP, A Neuroprotective Drug Candidate in Clinical Trials, Stimulates Microtubule Assembly in the Living Cell. Curr. Alzheimer Res. 2007, 4, 507–509. [Google Scholar]
- Rahul, J.; Suresh, C. Advancements in the Anti-Diabetes Chemotherapeutics Based on Amino Acids, Peptides, and Peptidomimetics. Mini RevMed. Chem. 2005, 5, 469–477. [Google Scholar]
- London, N.; Raveh, B.; Movshovitz-Attias, D.; Schueler-Furman, O. Can self-inhibitory peptides be derived from the interfaces of globular protein–protein interactions? Proteins Struct. Funct. Bioinform. 2010, 78, 3140–3149. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.K.; Gellman, S.H. Targeting protein–protein interactions: Lessons from p53/MDM2. Pept. Sci 2007, 88, 657–686. [Google Scholar] [CrossRef] [PubMed]
- Minkovsky, N.; Berezov, A. BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Curr. Opin. Investig. Drugs 2008, 9, 1336–1346. [Google Scholar] [PubMed]
- Filippo, M.; Yi, Z. Targeting Rho GTPases by Peptidic Structures. Curr. Pharm. Des. 2009, 15, 2481–2487. [Google Scholar]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Milhas, S.; Raux, B.; Betzi, S.; Derviaux, C.; Roche, P.; Restouin, A.; Basse, M.-J.; Rebuffet, E.; Lugari, A.; Badol, M.; et al. Protein–Protein Interaction Inhibition (2P2I)-Oriented Chemical Library Accelerates Hit Discovery. ACS Chem. Biol. 2016, 11, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Whitty, A. The road less traveled: Modulating signal transduction enzymes by inhibiting their protein-protein interactions. Curr. Opin. Chem. Biol. 2009, 13, 284–290. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Raveh, B.; Schueler-Furman, O. Modeling Peptide-Protein Interactions; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; ISBN 978-1-4939-6798-8. [Google Scholar]
- London, N.; Raveh, B.; Schueler-Furman, O. Peptide docking and structure-based characterization of peptide binding: From knowledge to know-how. Curr. Opin. Struct. Biol. 2013, 23, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Chen, Y.-G.; Ozdamar, B.; Gyuricza, C.A.; Chong, P.A.; Wrana, J.L.; Massagué, J.; Shi, Y. Structural Basis of Smad2 Recognition by the Smad Anchor for Receptor Activation. Science 2000, 287, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.; Niv, M.Y. Peptidic modulators of protein-protein interactions: Progress and challenges in computational design. Biopolymers 2009, 91, 505–513. [Google Scholar] [CrossRef]
- Schindler, C.E.M.; de Vries, S.J.; Zacharias, M. Fully Blind Peptide-Protein Docking with pepATTRACT. Structure 2015, 23, 1507–1515. [Google Scholar] [CrossRef]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nuc. Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef]
- Raveh, B.; London, N.; Zimmerman, L.; Schueler-Furman, O. Rosetta FlexPepDock ab-initio: Simultaneous Folding, Docking and Refinement of Peptides onto Their Receptors. PLoS ONE 2011, 6, e18934. [Google Scholar] [CrossRef] [PubMed]
- Trellet, M.; Melquiond, A.S.J.; Bonvin, A.M.J.J. A Unified Conformational Selection and Induced Fit Approach to Protein-Peptide Docking. PLoS ONE 2013, 8, e58769. [Google Scholar] [CrossRef] [PubMed]
- Tubert-Brohman, I.; Sherman, W.; Repasky, M.; Beuming, T. Improved Docking of Polypeptides with Glide. J. Chem. Inf. Model 2013, 53, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Antes, I. DynaDock: A new molecular dynamics-based algorithm for protein–peptide docking including receptor flexibility. Proteins Struct. Funct. Bioinform. 2010, 78, 1084–1104. [Google Scholar] [CrossRef] [PubMed]
- Blaszczyk, M.; Kurcinski, M.; Kouza, M.; Wieteska, L.; Debinski, A.; Kolinski, A.; Kmiecik, S. Modeling of protein–peptide interactions using the CABS-dock web server for binding site search and flexible docking. Methods 2016, 93, 72–83. [Google Scholar] [CrossRef]
- London, N.; Movshovitz-Attias, D.; Schueler-Furman, O. The Structural Basis of Peptide-Protein Binding Strategies. Structure 2010, 18, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H.-J. A novel computational tool for automated structure-based drug design. J. Mol. Recognit. 1993, 6, 131–137. [Google Scholar] [CrossRef]
- Miranker, A.; Karplus, M. An automated method for dynamic ligand design. Proteins Struct. Funct. Bioinform. 1995, 23, 472–490. [Google Scholar] [CrossRef]
- Durrant, J.D.; Amaro, R.E.; McCammon, J.A. AutoGrow: A Novel Algorithm for Protein Inhibitor Design. Chem. Biol. Drug Des. 2009, 73, 168–178. [Google Scholar] [CrossRef]
- Durrant, J.D.; Lindert, S.; McCammon, J.A. AutoGrow 3.0: An improved algorithm for chemically tractable, semi-automated protein inhibitor design. J. Mol. Graph. Model. 2013, 44, 104–112. [Google Scholar] [CrossRef]
- Welch, W.; Ruppert, J.; Jain, A.N. Hammerhead: Fast, fully automated docking of flexible ligands to protein binding sites. Chem. Biol. 1996, 3, 449–462. [Google Scholar] [CrossRef]
- Kramer, B.; Rarey, M.; Lengauer, T. Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins 1999, 37, 228–241. [Google Scholar] [CrossRef]
- Moon, J.B.; Howe, W.J. Computer design of bioactive molecules: A method for receptor-based de novo ligand design. Proteins Struct. Funct. Bioinform. 1991, 11, 314–328. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, T.K.; Fisher, J.F.; Hester, J.B.; Smith, C.W.; Tomasselli, A.G.; Tarpley, W.G.; Burton, P.S.; Hui, J.O.; McQuade, T.J.; Conradi, R.A.; et al. Peptidomimetic inhibitors of human immunodeficiency virus protease (HIV-PR): Design, enzyme binding and selectivity, antiviral efficacy, and cell permeability properties. Bioorg. Med. Chem. Lett. 1993, 3, 819–824. [Google Scholar] [CrossRef]
- Thanki, N.; Rao, J.K.M.; Foundling, S.I.; Wlodawer, A.; Howe, W.J.; Moon, J.B.; Hui, J.O.; Tomasselli, A.G.; Heinrikson, R.L.; Thaisrivongs, S. Crystal structure of a complex of HIV-1 protease with a dihydroxyethylene-containing inhibitor: Comparisons with molecular modeling. Protein Sci. 1992, 1, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Thaisrivongs, S.; Tomasselli, A.G.; Moon, J.B.; Hui, J.; McQuade, T.J.; Turner, S.R.; Strohbach, J.W.; Howe, W.J.; Tarpley, W.G.; Heinrikson, R.L. Inhibitors of the protease from human immunodeficiency virus: Design and modeling of a compound containing a dihydroxyethylene isostere insert with high binding affinity and effective antiviral activity. J. Med. Chem. 1991, 34, 2344–2356. [Google Scholar] [CrossRef]
- Glide; Schrödinger, LLC: New York, NY, USA, 2014.
- Bissantz, C.; Folkers, G.; Rognan, D. Protein-Based Virtual Screening of Chemical Databases. 1. Evaluation of Different Docking/Scoring Combinations. J. Med. Chem. 2000, 43, 4759–4767. [Google Scholar] [CrossRef]
- Kellenberger, E.; Rodrigo, J.; Muller, P.; Rognan, D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins Struct. Funct. Bioinform. 2004, 57, 225–242. [Google Scholar] [CrossRef]
- Cummings, M.D.; DesJarlais, R.L.; Gibbs, A.C.; Mohan, V.; Jaeger, E.P. Comparison of Automated Docking Programs as Virtual Screening Tools. J. Med. Chem. 2005, 48, 962–976. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nuc. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Maestro; Schrödinger, LLC: New York, NY, USA, 2014.
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- MacroModel; Schrödinger, LLC: New York, NY, USA, 2014.
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach To Pose Prediction and Scoring. J. Chem. Inf. Model 2014, 54, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.M. A Ranking-Based Scoring Function for Peptide–Spectrum Matches. J. Proteome Res. 2009, 8, 2241–2252. [Google Scholar] [CrossRef] [PubMed]
- Spiliotopoulos, D.; Kastritis, P.L.; Melquiond, A.S.J.; Bonvin, A.M.J.J.; Musco, G.; Rocchia, W.; Spitaleri, A. dMM-PBSA: A New HADDOCK Scoring Function for Protein-Peptide Docking. Front. Mol. Biosci. 2016, 3. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Code | IRDL | SP | SP-Peptide | ||||
|---|---|---|---|---|---|---|---|
| BB | WP | H-Bond Number Ratio a | BB | WP | BB | WP | |
| 1OSZ | 2 | 2 | 1 | - | - | 12 | 2 |
| 2D5W | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 3CH8 | 1 | 1 | 1 | - | - | 1 | 1 |
| 3DRF | 6 | 1 | 0.9 | 11 | 1 | 32 | 4 |
| 3MMG | 1 | 1 | 1 | - | - | 3 | 2 |
| 3T6B | 67 | 1 | 0.75 | 1 | 1 | - | - |
| 4GRV | 1 | 1 | 0.5 | - | - | 1 | 1 |
| 4GYO | 2 | 2 | 0.86 | 9 | 9 | 9 | 9 |
| 4NNM | 1 | 1 | 0.82 | 1 | 1 | 27 | 1 |
| 4Q6H | 2 | 2 | 0.88 | 1 | 1 | 1 | 1 |
| 4RXH | 36 | 36 | 0.8 | 1 | 1 | - | - |
| PDB Code | Classification | Target | Resolution (Å) | Organism | Peptide Sequence Length | Torsion Angles |
|---|---|---|---|---|---|---|
| 1OSZ | Complex (MHC-1) | H-2K1 | 2.1 | Mus Musculus | 8 | 33 |
| 2D5W | Protein Binding | Oligopeptide binding protein | 1.3 | Thermus thermophilus | 5 | 18 |
| 3CH8 | Protein Binding | PDZ-Fibronectin | 1.9 | Homo Sapiens | 8 | 25 |
| 3DRF | Protein Binding | OppA | 1.3 | Lactococcus lactis | 8 | 25 |
| 3MMG | Hydrolase | Tobacco Vein Mottling Virus protease | 1.7 | Tobacco Vein Mottling Virus | 8 | 35 |
| 3T6B | Hydrolase | DPP III | 2.4 | Homo Sapiens | 5 | 15 |
| 4GRV | Signaling protein | NTS1 receptor | 2.8 | Rattus Norvegicus | 6 | 27 |
| 4GYO | Hydrolase | Rap-J | 2.2 | Homo Sapiens | 5 | 22 |
| 4NNM | Protein Binding | TIP-1 | 1.6 | Homo Sapiens | 6 | 21 |
| 4Q6H | Transport Protein | CAL | 1.9 | Homo Sapiens | 6 | 26 |
| 4RXH | Transport Protein | Importin-α | 1.8 | Neurospora Crassa | 6 | 43 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diharce, J.; Cueto, M.; Beltramo, M.; Aucagne, V.; Bonnet, P. In Silico Peptide Ligation: Iterative Residue Docking and Linking as a New Approach to Predict Protein-Peptide Interactions. Molecules 2019, 24, 1351. https://doi.org/10.3390/molecules24071351
Diharce J, Cueto M, Beltramo M, Aucagne V, Bonnet P. In Silico Peptide Ligation: Iterative Residue Docking and Linking as a New Approach to Predict Protein-Peptide Interactions. Molecules. 2019; 24(7):1351. https://doi.org/10.3390/molecules24071351
Chicago/Turabian StyleDiharce, Julien, Mickaël Cueto, Massimiliano Beltramo, Vincent Aucagne, and Pascal Bonnet. 2019. "In Silico Peptide Ligation: Iterative Residue Docking and Linking as a New Approach to Predict Protein-Peptide Interactions" Molecules 24, no. 7: 1351. https://doi.org/10.3390/molecules24071351
APA StyleDiharce, J., Cueto, M., Beltramo, M., Aucagne, V., & Bonnet, P. (2019). In Silico Peptide Ligation: Iterative Residue Docking and Linking as a New Approach to Predict Protein-Peptide Interactions. Molecules, 24(7), 1351. https://doi.org/10.3390/molecules24071351