
Effect of the Immobilization Strategy on the Efficiency and Recyclability of the Versatile Lipase from Ophiostoma piceae


 ,
,
Abstract
1. Introduction
2. Results
2.1. Immobilization of Enzyme Crudes Containing the Lipase OPEr
2.2. Activity of the Nanobiocatalysts in Synthesis of Butyl Esters of Volatile Fatty Acids
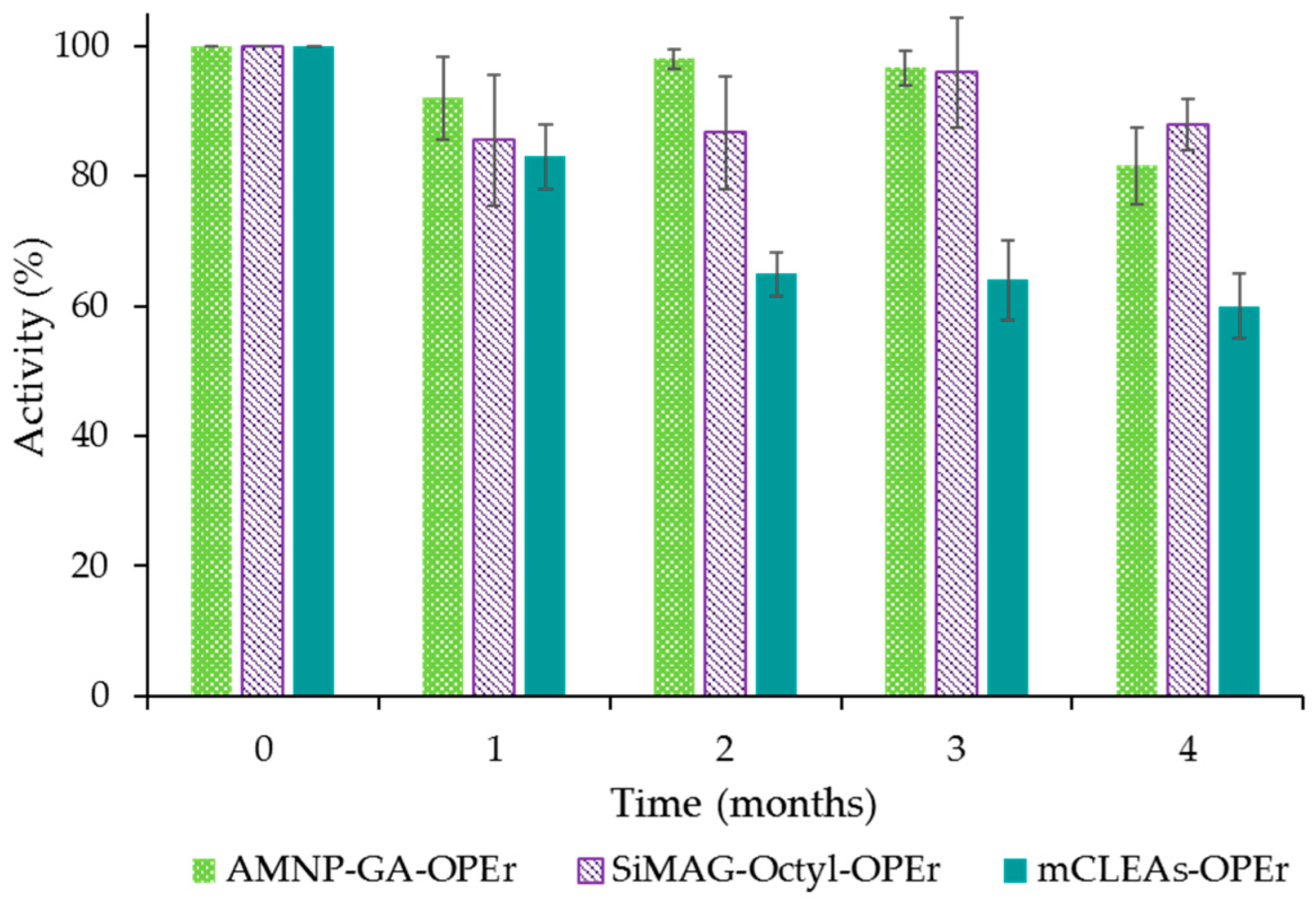
2.3. Storage Stability
2.4. Effect of Several Reaction Parameters in the Synthesis of Butyl Esters of Volatile Fatty Acids Catalyzed by AMNP-GA-OPEr
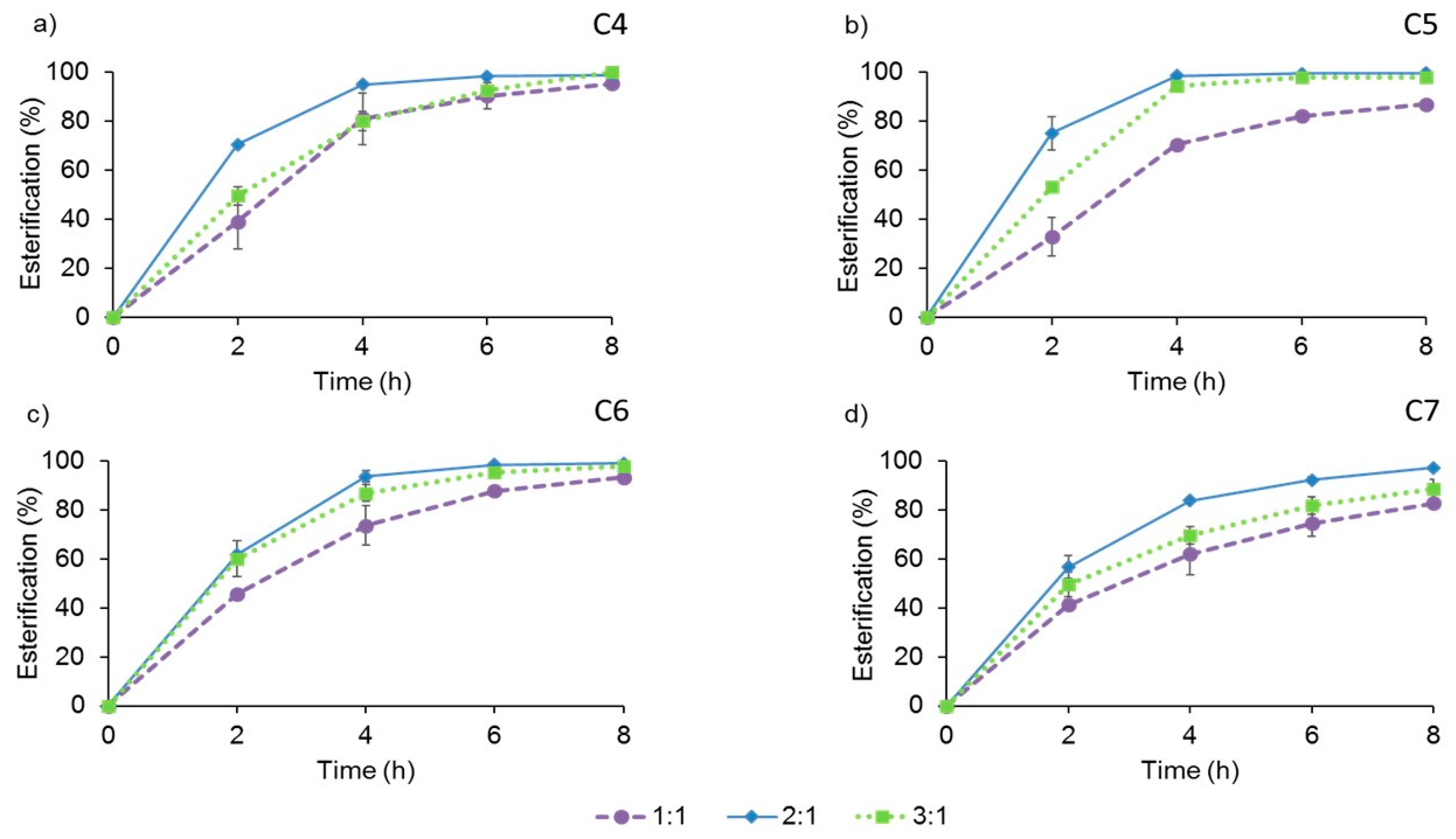
2.4.1. Molar Ratio of Substrates
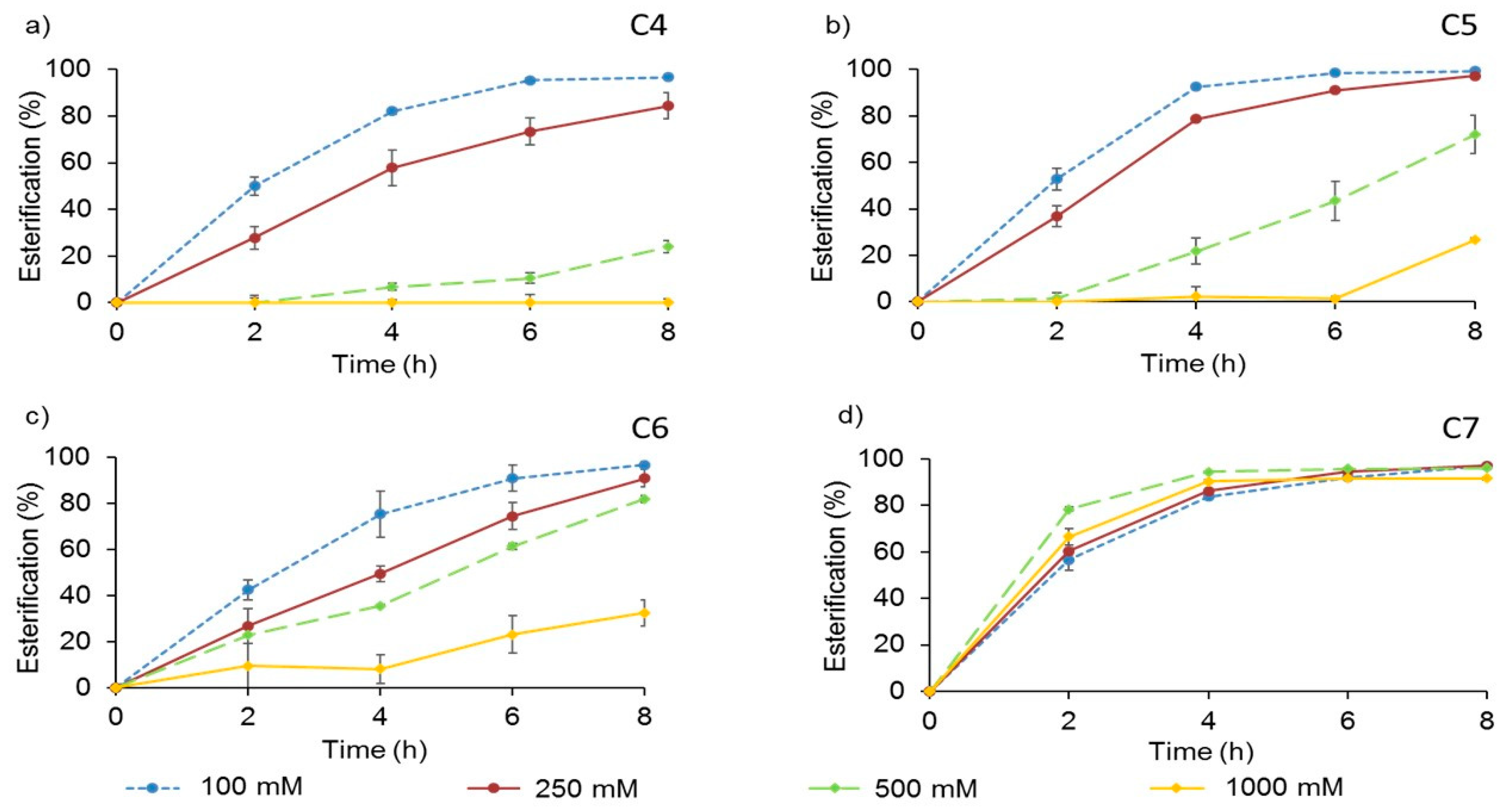
2.4.2. Substrates Concentration
2.4.3. Branching of the Acyl Donor
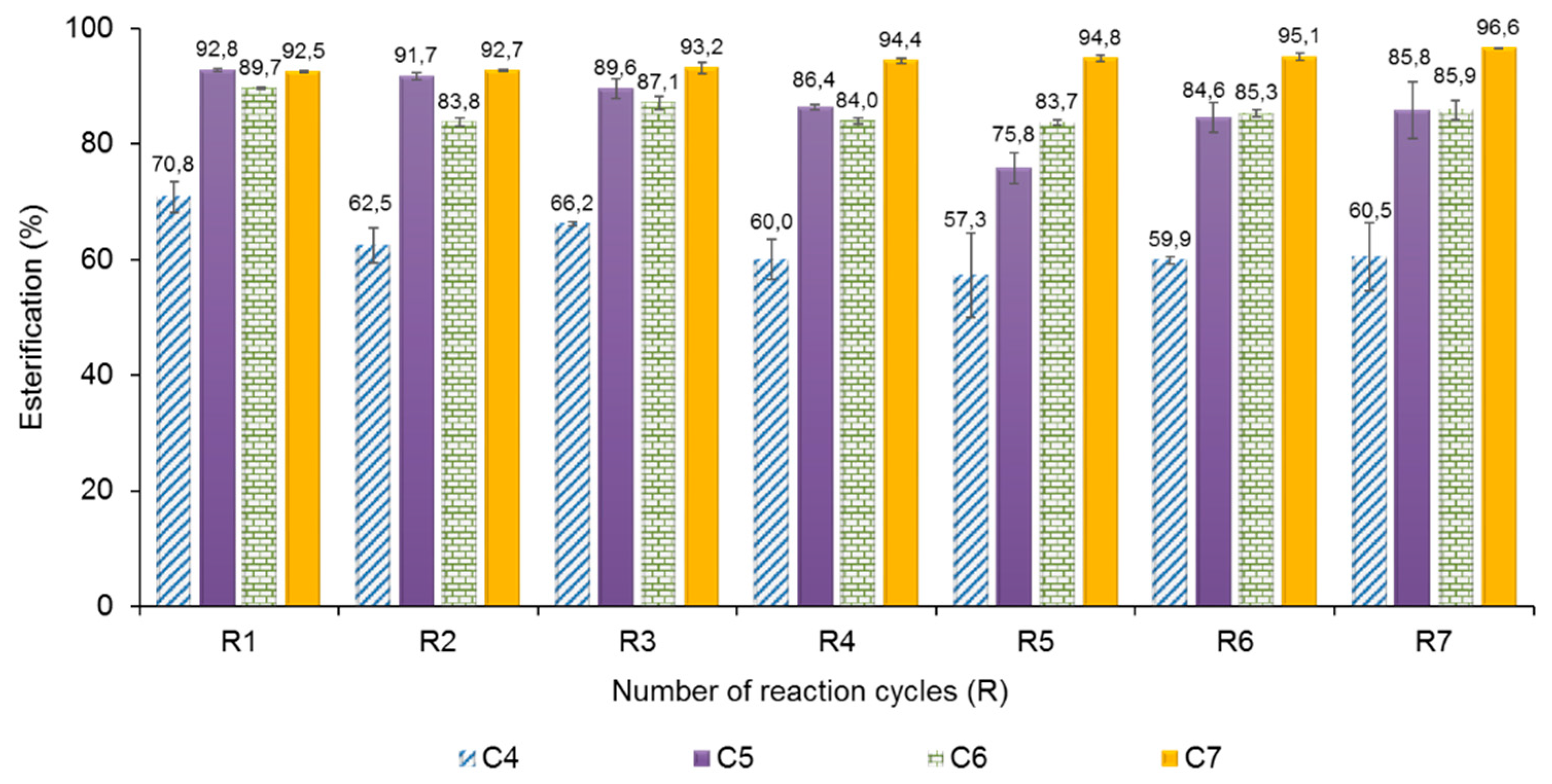
2.5. Operational Stability of AMNP-GA-OPEr
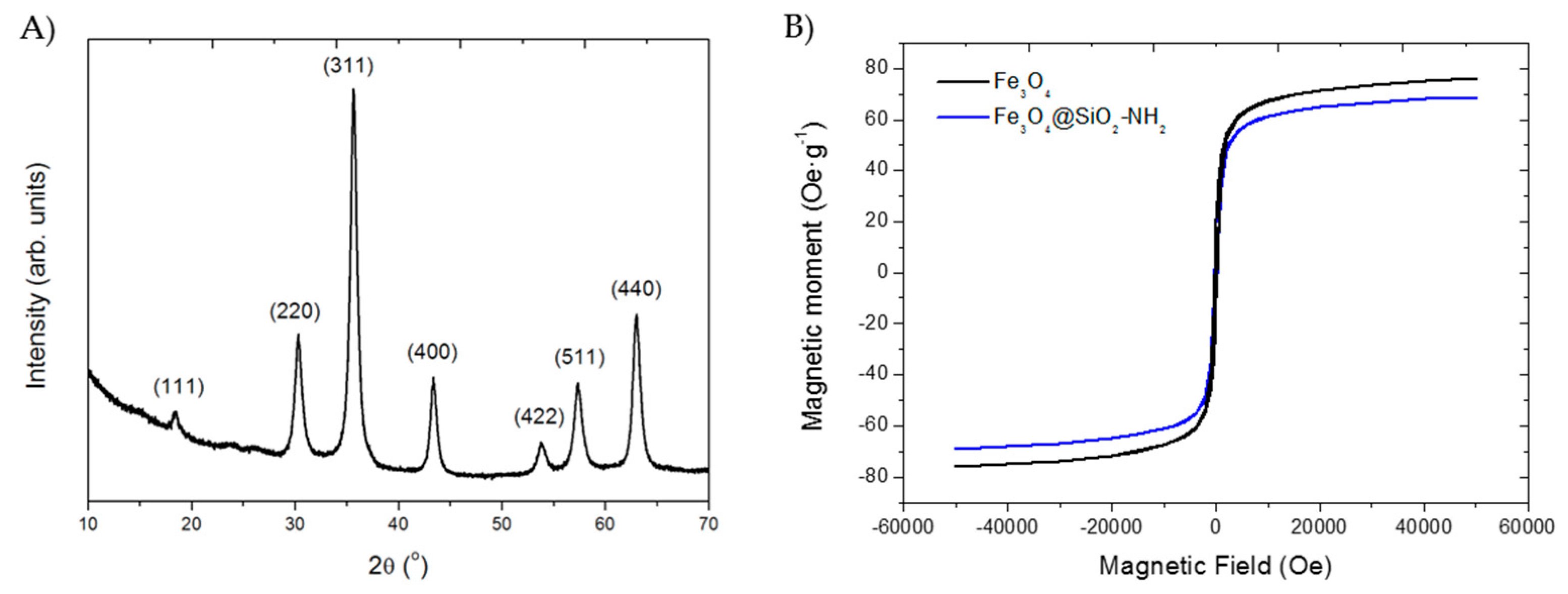
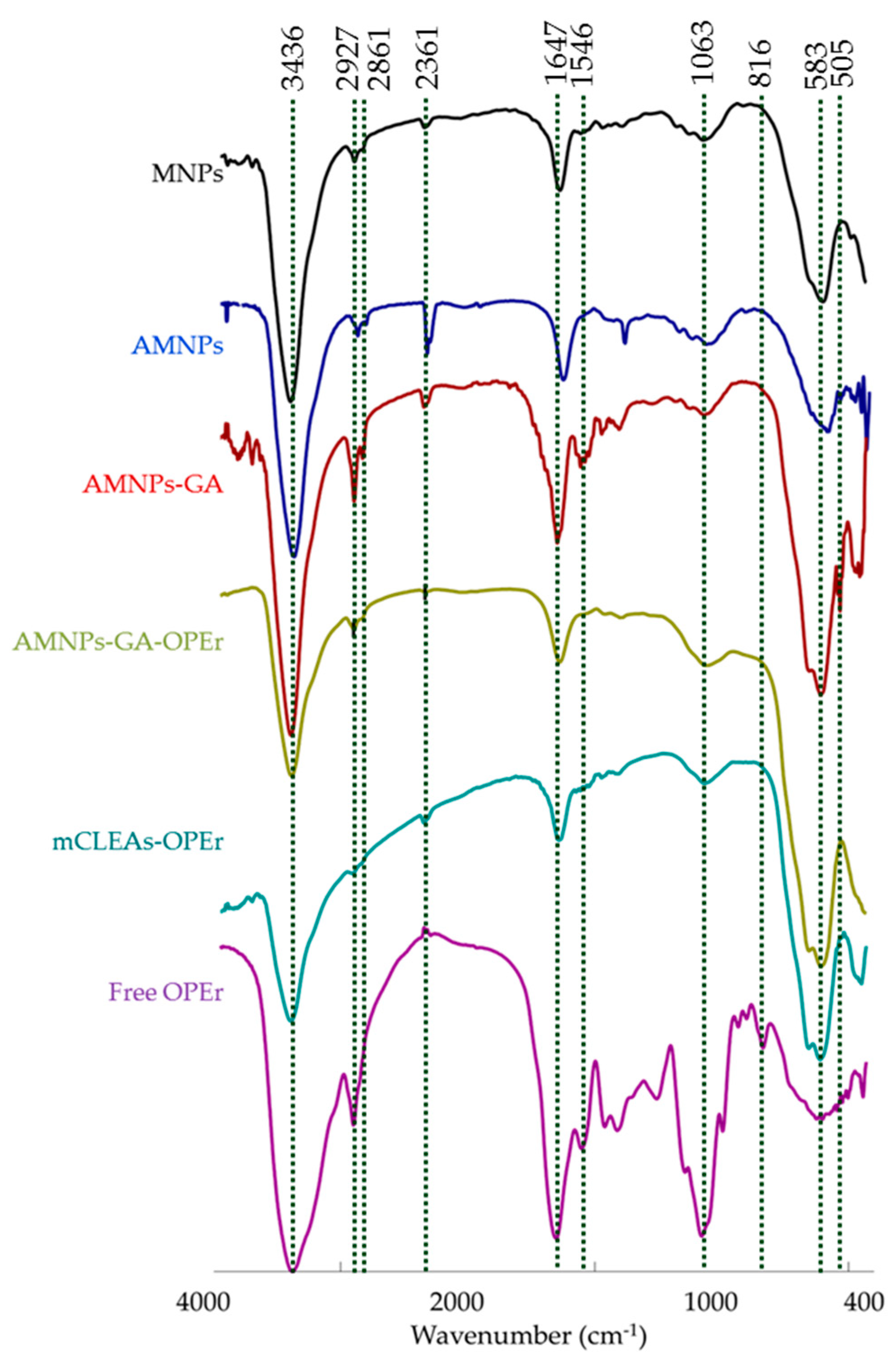
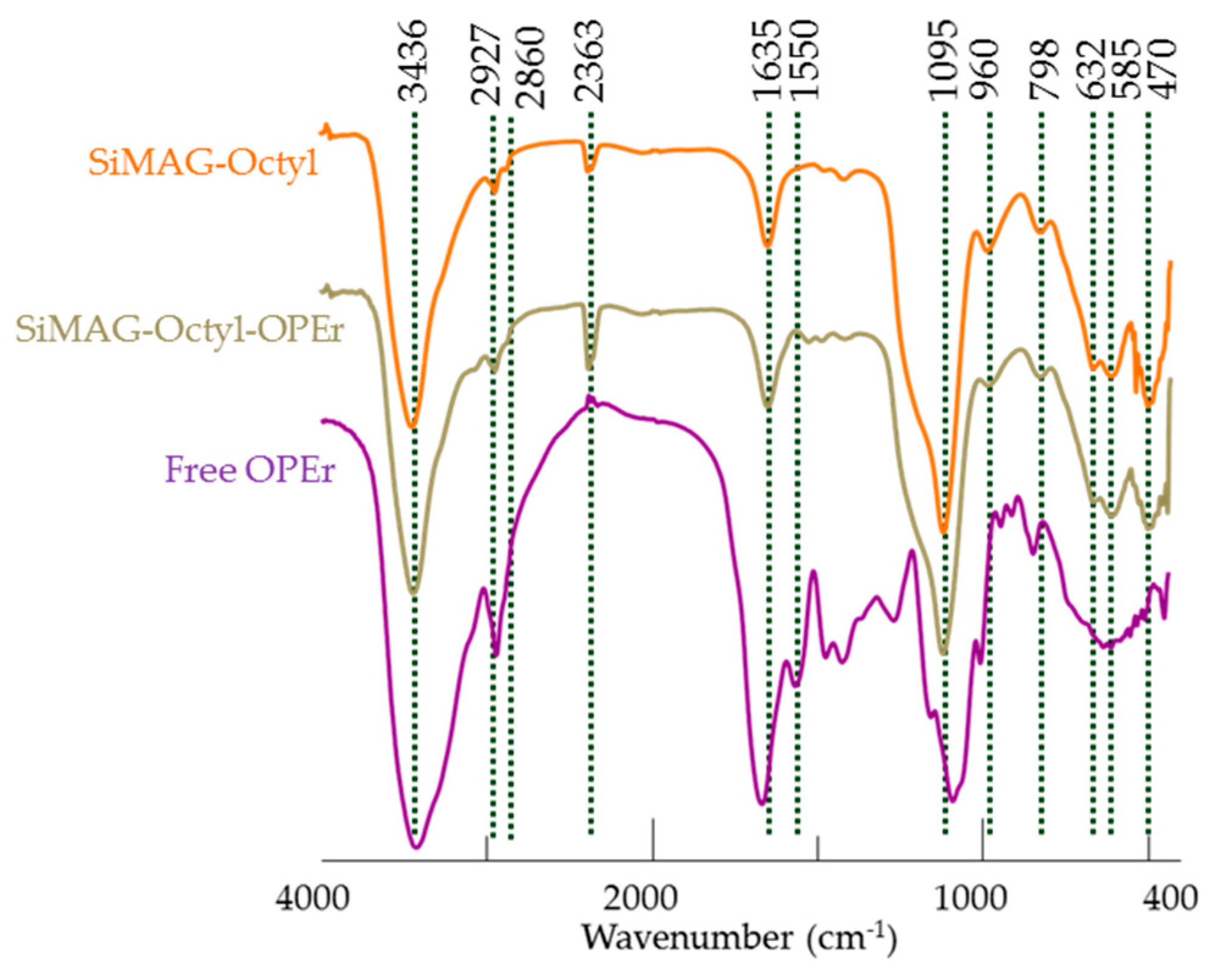
2.6. Characterization of the Nanoparticles and Nanobiocatalysts
3. Discussion
3.1. Synthesis of the Magnetic Nanobiocatalysts with OPEr and Comparison of Their Activity in Hydrolysis of pNPB
3.2. The OPEr Magnetic Nanobiocatalysts Catalyze the Synthesis of the Butyl Esters of Straight-Chain Fatty Acids C4-C7 with Different Efficiency
3.3. Effect of Other Variables in the Enzymatic Synthesis of Butyl Esters of VFA Catalyzed by AMNP-GA-OPEr
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Strains, Culture Conditions, and Preparation of Enzyme Crudes
4.3. Evaluation of Enzyme Activity and Protein Content
4.4. Functionalization of Nude Magnetic Nanoparticles
4.5. Characterization of the Nanoparticles
4.6. Immobilization of Crudes with the Recombinant Versatile Lipase from O. piceae
4.6.1. Covalent Immobilization on Amino-Functionalized Magnetic Nanoparticles Activated with Glutaraldehyde
4.6.2. Immobilization as Magnetic CLEAS
4.6.3. Immobilization by Adsorption on Commercial Magnetic Nanoparticles Functionalized with Hydrophobic Octyl Groups
4.7. Activity of Immobilized Enzymes
4.8. Esterification of Volatile Fatty Acids in Isooctane
4.9. Monitoring Reactions by Gas Chromatography
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ansorge-Schumacher, M.B.; Thum, O. Immobilised lipases in the cosmetics industry. Chem. Soc. Rev. 2013, 42, 6475–6490. [Google Scholar] [CrossRef] [PubMed]
- Ivić, J.T.; Veličković, D.; Dimitrijević, A.; Bezbradica, D.; Dragačević, V.; Jankulović, M.G.; Milosavić, N. Design of biocompatible immobilized Candida rugosa lipase with potential application in food industry. J. Sci. Food Agric. 2016, 96, 4281–4287. [Google Scholar] [CrossRef] [PubMed]
- Adlercreutz, P. Immobilisation and application of lipases in organic media. Chem. Soc. Rev. 2013, 42, 6406–6436. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Dhar, K.; Kanwar, S.; Arora, P. Lipase catalysis in organic solvents: Advantages and applications. Biol. Proced. Online 2016, 18, 2. [Google Scholar] [CrossRef]
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed]
- Vaghari, H.; Jafarizadeh-Malmiri, H.; Mohammadlou, M.; Berenjian, A.; Anarjan, N.; Jafari, N.; Nasiri, S. Application of magnetic nanoparticles in smart enzyme immobilization. Biotechnol. Lett. 2016, 38, 223–233. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Woodley, J.M. Role of biocatalysis in sustainable chemistry. Chem. Rev. 2018, 118, 801–838. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, N.R.; Marzuki, N.H.C.; Buang, N.A.; Huyop, F.; Wahab, R.A. An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechnol. Biotechnol. Equip. 2015, 29, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Shuai, W.; Das, R.K.; Naghdi, M.; Brar, S.K.; Verma, M. A review on the important aspects of lipase immobilization on nanomaterials. Biotechnol. Appl. Biochem. 2017, 64, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Meng, G.; Tao, K.; Feng, M.; Zhao, X.; Li, Z.; Xu, H.; Xia, D.; Lu, J.R. Immobilization of lipases on alkyl silane modified magnetic nanoparticles: Effect of alkyl chain length on enzyme activity. PLoS ONE 2012, 7, e43478. [Google Scholar] [CrossRef]
- Netto, C.G.C.M.; Toma, H.E.; Andrade, L.H. Superparamagnetic nanoparticles as versatile carriers and supporting materials for enzymes. J. Mol. Catal. B Enzym. 2013, 85–86, 71–92. [Google Scholar] [CrossRef]
- Kim, M.; Park, J.M.; Um, H.J.; Lee, D.H.; Lee, K.H.; Kobayashi, F.; Iwasaka, Y.; Hong, C.S.; Min, J.; Kim, Y.H. Immobilization of cross-linked lipase aggregates onto magnetic beads for enzymatic degradation of polycaprolactone. J. Basic Microbiol. 2010, 50, 218–226. [Google Scholar] [CrossRef]
- Tudorache, M.; Gheorghe, A.; Viana, A.S.; Parvulescu, V.I. Biocatalytic epoxidation of α-pinene to oxy-derivatives over cross-linked lipase aggregates. J. Mol. Catal. B Enzym. 2016, 134, 9–15. [Google Scholar] [CrossRef]
- Cruz-Izquierdo, Á.; Picó, E.A.; López, C.; Serra, J.L.; Llama, M.J. Magnetic Cross-Linked Enzyme Aggregates (mCLEAs) of Candida antarctica lipase: An efficient and stable biocatalyst for biodiesel synthesis. PLoS ONE 2014, 9, e115202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, H.; Liu, W.; Wang, N.; Yu, X. Improved performance of magnetic cross-linked lipase aggregates by interfacial activation: A robust and magnetically recyclable biocatalyst for transesterification of jatropha oil. Molecules 2017, 22, 2157. [Google Scholar] [CrossRef] [PubMed]
- Turcheniuk, K.; Tarasevych, A.; Kukhar, V.; Boukherroub, R.; Szunerits, S. Recent advances in surface chemistry strategies for the fabrication of functional iron oxide based magnetic nanoparticles. Nanoscale 2013, 5, 10729–10752. [Google Scholar] [CrossRef]
- Viñambres, M.; Filice, M.; Marciello, M. Modulation of the catalytic properties of lipase B from Candida antarctica by immobilization on tailor-made magnetic iron oxide nanoparticles: The key role of nanocarrier surface engineering. Polymers 2018, 8, 615. [Google Scholar] [CrossRef]
- Barriuso, J.; Vaquero, M.E.; Prieto, A.; Martínez, M.J. Structural traits and catalytic versatility of the lipases from the Candida rugosa-like family: A review. Biotechnol. Adv. 2016, 34, 874–885. [Google Scholar] [CrossRef]
- Cedillo, V.B.; Prieto, A.; María, M.J. Potential of Ophiostoma piceae sterol esterase for biotechnologically relevant hydrolysis reactions. Bioengineered 2013, 4, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Barba, V.; Plou, F.J.; Martínez, M.J. Recombinat sterol esterase from Ophiostoma piceae: An improvement biocatalyst expressed in Pichia pastoris. Microb. Cell Fact. 2012, 11, 1–14. [Google Scholar]
- Molina-Gutiérrez, M.; Hakalin, N.L.S.; Rodríguez-Sanchez, L.; Prieto, A.; Martínez, M.J. Green synthesis of β-sitostanol esters catalyzed by the versatile lipase/sterol esterase from Ophiostoma piceae. Food Chem. 2017, 221, 1458–1465. [Google Scholar] [CrossRef]
- Hakalin, N.L.S.; Molina-Gutiérrez, M.; Prieto, A.; Martínez, M.J. Optimization of lipase-catalyzed synthesis of β-sitostanol esters by response surface methodology. Food Chem. 2018, 261, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, M.E.; Barriuso, J.; Medrano, F.J.; Prieto, A.; Martínez, M.J. Heterologous expression of a fungal sterol esterase/lipase in different hosts: Effect on solubility, glycosylation and production. J. Biosci. Bioeng. 2015, 120, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Calero-Rueda, O.; Barba, V.; Rodríguez, E.; Plou, F.; Martínez, Á.T.; Martínez, M.J. Study of a sterol esterase secreted by Ophiostoma piceae: Sequence, model and biochemical properties. Biochim. Biophys. Acta-Proteins Proteomics 2009, 1794, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Sá, A.G.A.; de Meneses, A.C.; de Araújo, P.H.H.; de Oliveira, D. A review on enzymatic synthesis of aromatic esters used as flavor ingredients for food, cosmetics and pharmaceuticals industries. Trends Food Sci. Technol. 2017, 69, 95–105. [Google Scholar] [CrossRef]
- Khan, N.R.; Rathod, V.K. Enzyme catalyzed synthesis of cosmetic esters and its intensification: A review. Process Biochem. 2015, 50, 1793–1806. [Google Scholar] [CrossRef]
- Brault, G.; Shareck, F.; Hurtubise, Y.; Lépine, F.; Doucet, N. Short-chain flavor ester synthesis in organic media by an E. coli whole-cell biocatalyst expressing a newly characterized heterologous lipase. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.A.; Sanromán, M.A. Production of food aroma compounds: Microbial and enzymatic methodologies. Food Technol. Biotechnol. 2006, 44, 335–353. [Google Scholar]
- Martins, A.B.; Friedrich, J.L.R.; Cavalheiro, J.C.; Garcia-Galan, C.; Barbosa, O.; Ayub, M.A.Z.; Fernandez-Lafuente, R.; Rodrigues, R.C. Improved production of butyl butyrate with lipase from Thermomyces lanuginosus immobilized on styrene-divinylbenzene beads. Bioresour. Technol. 2013, 134, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.B.; Da Silva, A.M.; Schein, M.F.; Garcia-Galan, C.; Záchia Ayub, M.A.; Fernandez-Lafuente, R.; Rodrigues, R.C. Comparison of the performance of commercial immobilized lipases in the synthesis of different flavor esters. J. Mol. Catal. B Enzym. 2014, 105, 18–25. [Google Scholar] [CrossRef]
- Dutta, K.; Dasu, V.V. Synthesis of short chain alkyl esters using cutinase from Burkholderia cepacia NRRL B2320. J. Mol. Catal. B Enzym. 2011, 72, 150–156. [Google Scholar] [CrossRef]
- Su, L.; Hong, R.; Guo, X.; Wu, J.; Xia, Y. Short-chain aliphatic ester synthesis using Thermobifida fusca cutinase. Food Chem. 2016, 206, 131–136. [Google Scholar] [CrossRef]
- Nikolaivits, E.; Makris, G.; Topakas, E. Immobilization of a cutinase from Fusarium oxysporum and application in pineapple flavor synthesis. J. Agric. Food Chem. 2017, 65, 3505–3511. [Google Scholar] [CrossRef] [PubMed]
- Lemos, F.; Fonseca, L.P.; Cabral, J.M.S.; de Barros, D.P.C.; Fonseca, L.P.; Cabral, J.M.S. Kinetic cutinase-catalyzed esterification of caproic acid in organic solvent system. J. Mol. Catal. 2010, 66, 285–293. [Google Scholar]
- Azevedo, A.M.; Cabral, J.M.S.; Fonseca, L.P.; De Barros, D.P.C.; Azevedo, A.N.A.M.; Cabral, J.M.S.; Fonseca, L.P. Optimization of flavor esters synthesis by Fusarium solani pisi cutinase. J. Food Biochem. 2012, 36, 275–284. [Google Scholar]
- Patil, R.M.; Shete, P.B.; Thorat, N.D.; Otari, S.V.; Barick, K.C.; Prasad, A.; Ningthoujam, R.S.; Tiwale, B.M.; Pawar, S.H. Superparamagnetic iron oxide/chitosan core/shells for hyperthermia application: Improved colloidal stability and biocompatibility. J. Magn. Magn. Mater. 2014, 355, 22–30. [Google Scholar] [CrossRef]
- Shi, S.; Yang, J.; Liang, S.; Li, M.; Gan, Q.; Xiao, K.; Hu, J. Enhanced Cr(VI) removal from acidic solutions using biochar modified by Fe3O4@SiO2-NH2 particles. Sci. Total Environ. 2018, 628–629, 499–508. [Google Scholar] [CrossRef]
- Alcaraz, L.; Isasi, J.; Caballero, A.C.; Izquierdo, J.G.; Bañares, L. Nanopowders Y1−yNdyV1−xCrxO4 with y=0 and 1; x=0, 0.1, 0.2 and 0.5 synthesized by a sol–gel process. Relationship between morphological characteristics and optical properties. J. Lumin. 2015, 161, 110–116. [Google Scholar] [CrossRef]
- Wu, W.; Wu, Z.; Yu, T.; Jiang, C.; Kim, W.S. Recent progress on magnetic iron oxide nanoparticles: Synthesis, surface functional strategies and biomedical applications. Sci. Technol. Adv. Mater. 2015, 16, 23501. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; He, J.; Deng, L.; Gao, X. Fabrication of cyclodextrin-functionalized superparamagnetic Fe3O4/amino-silane core-shell nanoparticles via layer-by-layer method. Appl. Surf. Sci. 2009, 255, 7974–7980. [Google Scholar] [CrossRef]
- Vadivel, M.; Babu, R.R.; Arivanandhan, M.; Ramamurthi, K.; Hayakawa, Y. Role of SDS surfactant concentrations on the structural, morphological, dielectric and magnetic properties of CoFe2O4 nanoparticles. RSC Adv. 2015, 5, 27060–27068. [Google Scholar] [CrossRef]
- Zhao, J.; Niu, Y.; Ren, B.; Chen, H.; Zhang, S.; Jin, J.; Zhang, Y. Synthesis of Schiff base functionalized superparamagnetic Fe3O4 composites for effective removal of Pb(II) and Cd(II) from aqueous solution. Chem. Eng. J. 2018, 347, 574–584. [Google Scholar] [CrossRef]
- Griffiths, P.R. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules; Elsevier: Amsterdam, The Netherlands, 1992; Volume 4, ISBN 9780124511606. [Google Scholar]
- Petrovský, E.; Herrero-Bervera, E.; Harinarayana, T.; Ivers, D. The Earth’s Magnetic Interior; Petrovský, E., Ivers, D., Harinarayana, T., Herrero-Bervera, E., Eds.; Springer: Dordrecht, The Netherlands, 2011; ISBN 978-94-007-0322-3. [Google Scholar]
- Tural, B.; Özkan, N.; Volkan, M. Preparation and characterization of polymer coated superparamagnetic magnetite nanoparticle agglomerates. J. Phys. Chem. Solids 2009, 70, 860–866. [Google Scholar] [CrossRef]
- Barbosa, O.; Torres, R.; Ortiz, C.; Fernandez-Lafuente, R. Versatility of glutaraldehyde to immobilize lipases: Effect of the immobilization protocol on the properties of lipase B from Candida antarctica. Process Biochem. 2012, 47, 1220–1227. [Google Scholar] [CrossRef]
- Betancor, L.; López-Gallego, F.; Hidalgo, A.; Alonso-Morales, N.; Mateo, G.D.-O.C.; Fernández-Lafuente, R.; Guisán, J.M. Different mechanisms of protein immobilization on glutaraldehyde activated supports: Effect of support activation and immobilization conditions. Enzyme Microb. Technol. 2006, 39, 877–882. [Google Scholar] [CrossRef]
- Zhang, Y.; Ge, J.; Liu, Z. Enhanced activity of immobilized or chemically modified enzymes. ACS Catal. 2015, 5, 4503–4513. [Google Scholar] [CrossRef]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Glutaraldehyde in bio-catalysts design: A useful crosslinker and a versatile tool in enzyme immobilization. RSC Adv. 2014, 4, 1583–1600. [Google Scholar] [CrossRef]
- Xie, W.; Ma, N. Immobilized lipase on Fe3O4 nanoparticles as biocatalyst for biodiesel production. Energy Fuels 2009, 23, 1347–1353. [Google Scholar]
- Wang, X.; Dou, P.; Zhao, P.; Zhao, C.; Ding, Y.; Xu, P. Immobilization of lipases onto magnetic Fe3O4 nanoparticles for application in biodiesel production. Chem. Sus. Chem. 2009, 2, 947–950. [Google Scholar] [CrossRef] [PubMed]
- López, C.; Cruz-Izquierdo, Á.; Picó, E.A.; García-Bárcena, T.; Villarroel, N.; Llama, M.J.; Serra, J.L. Magnetic biocatalysts and their uses to obtain biodiesel and biosurfactants. Front. Chem. 2014, 2, 1–11. [Google Scholar]
- Tükel, S.S.; Hürrem, F.; Yildirim, D.; Alptekin, Ö. Preparation of crosslinked enzyme aggregates (CLEA) of catalase and its characterization. J. Mol. Catal. B Enzym. 2013, 97, 252–257. [Google Scholar] [CrossRef]
- Fernandez-Lorente, G.; Cabrera, Z.; Godoy, C.; Fernandez-Lafuente, R.; Palomo, J.M.; Guisan, J.M. Interfacially activated lipases against hydrophobic supports: Effect of the support nature on the biocatalytic properties. Process Biochem. 2008, 43, 1061–1067. [Google Scholar] [CrossRef]
- Fernandez-Lafuente, R.; Armisén, P.; Sabuquillo, P.; Fernández-Lorente, G.; Guisán, J.M. Immobilization of lipases by selective adsorption on hydrophobic supports. Chem. Phys. Lipids 1998, 93, 185–197. [Google Scholar] [CrossRef]
- Tacias-Pascacio, V.G.; Peirce, S.; Torrestiana-Sanchez, B.; Yates, M.; Rosales-Quintero, A.; Virgen-Ortíz, J.J.; Fernandez-Lafuente, R. Evaluation of different commercial hydrophobic supports for the immobilization of lipases: Tuning their stability, activity and specificity. RSC Adv. 2016, 6, 100281–100294. [Google Scholar] [CrossRef]
- Virgen-Ortíz, J.J.; Tacias-Pascacio, V.G.; Hirata, D.B.; Torrestiana-Sanchez, B.; Rosales-Quintero, A.; Fernandez-Lafuente, R. Relevance of substrates and products on the desorption of lipases physically adsorbed on hydrophobic supports. Enzym. Microb. Technol. 2017, 96, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Zaak, H.; Siar, E.H.; Kornecki, J.F.; Fernandez-Lopez, L.; Pedrero, S.G.; Virgen-Ortíz, J.J.; Fernandez-Lafuente, R. Effect of immobilization rate and enzyme crowding on enzyme stability under different conditions. The case of lipase from Thermomyces lanuginosus immobilized on octyl agarose beads. Process Biochem. 2017, 56, 117–123. [Google Scholar] [CrossRef]
- Sarno, M.; Iuliano, M.; Polichetti, M.; Ciambelli, P. High activity and selectivity immobilized lipase on Fe3O4 nanoparticles for banana flavour synthesis. Process Biochem. 2017, 56, 98–108. [Google Scholar] [CrossRef]
- Ondul, E.; Dizge, N.; Albayrak, N. Immobilization of Candida antarctica A and Thermomyces lanuginosus lipases on cotton terry cloth fibrils using polyethyleneimine. Colloids Surf. B Biointerfaces 2012, 95, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Abbas, H.; Comeau, L. Aroma synthesis by immobilized lipase from Mucor sp. Enzym. Microb. Technol. 2003, 32, 589–595. [Google Scholar] [CrossRef]
- Romero, M.D.; Calvo, L.; Alba, C.; Daneshfar, A.; Ghaziaskar, H.S. Enzymatic synthesis of isoamyl acetate with immobilized Candida antarctica lipase in n-hexane. Enzym. Microb. Technol. 2005, 37, 42–48. [Google Scholar] [CrossRef]
- Pires-Cabral, P.; da Fonseca, M.M.R.; Ferreira-Dias, S. Synthesis of ethyl butyrate in organic media catalyzed by Candida rugosa lipase immobilized in polyurethane foams: A kinetic study. Biochem. Eng. J. 2009, 43, 327–332. [Google Scholar] [CrossRef]
- Friedrich, J.L.R.; Peña, F.P.; Garcia-Galan, C.; Fernandez-Lafuente, R.; Ayub, M.A.Z.; Rodrigues, R.C. Effect of immobilization protocol on optimal conditions of ethyl butyrate synthesis catalyzed by lipase B from Candida antarctica. J. Chem. Technol. Biotechnol. 2013, 88, 1089–1095. [Google Scholar] [CrossRef]
- Martins, A.B.; Schein, M.F.; Friedrich, J.L.R.; Fernandez-Lafuente, R.; Ayub, M.A.Z.; Rodrigues, R.C. Ultrasound-assisted butyl acetate synthesis catalyzed by Novozym 435: Enhanced activity and operational stability. Ultrason. Sonochem. 2013, 20, 1155–1160. [Google Scholar] [CrossRef]
- Calero-Rueda, O.; Plou, F.J.; Ballesteros, A.; Martínez, A.T.; Martínez, M.J. Production, isolation and characterization of a sterol esterase from Ophiostoma piceae. Biochim. Biophys. Acta Proteins Proteomics 2002, 1599, 28–35. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Tsai, M.G.; Chi, M.C.; Wang, T.F.; Lin, L.L. Covalent immobilization of Bacillus licheniformis gamma-glutamyl transpeptidase on aldehyde-functionalized magnetic nanoparticles. Int. J. Mol. Sci. 2013, 14, 4613–4628. [Google Scholar] [CrossRef]
- del Campo, A.; Sen, T.; Lellouche, J.-P.; Bruce, I.J. Multifunctional magnetite and silica–magnetite nanoparticles: Synthesis, surface activation and applications in life sciences. J. Magn. Magn. Mater. 2005, 293, 33–40. [Google Scholar] [CrossRef]
- Villars, P.; Cenzual, K. Pearson’s Crystal Data Crystal Structure Database for Inorganic Compounds; ASM International: Materials Park, OH, USA, 2009. [Google Scholar]
- Mendelson, M.I. Average grain size in polycrystalline ceramics. J. Am. Ceram. Soc. 1969, 52, 443–446. [Google Scholar] [CrossRef]
Sample Availability: Not available. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biocatalyst | Immobilization Type | Offered Protein (mg/mg Carrier) | Offered Activity (mU/mg Carrier) | Yield (%) | Specific Activity (mU/mg Carrier) |
|---|---|---|---|---|---|
| SiMAG-Octyl-OPEr | Hydrophobicity | 0.07 | 1200 | 99 | 430 ± 60 |
| AMNP-GA-OPEr | Covalent | 0.07 | 1200 | 53 | 440 ± 20 |
| mCLEAS-OPEr | Covalent | 0.05 | 990 | 97 | 728 ± 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molina-Gutiérrez, M.; Hakalin, N.L.S.; Rodríguez-Sánchez, L.; Alcaraz, L.; López, F.A.; Martínez, M.J.; Prieto, A. Effect of the Immobilization Strategy on the Efficiency and Recyclability of the Versatile Lipase from Ophiostoma piceae. Molecules 2019, 24, 1313. https://doi.org/10.3390/molecules24071313
Molina-Gutiérrez M, Hakalin NLS, Rodríguez-Sánchez L, Alcaraz L, López FA, Martínez MJ, Prieto A. Effect of the Immobilization Strategy on the Efficiency and Recyclability of the Versatile Lipase from Ophiostoma piceae. Molecules. 2019; 24(7):1313. https://doi.org/10.3390/molecules24071313
Chicago/Turabian StyleMolina-Gutiérrez, María, Neumara L. S. Hakalin, Leonor Rodríguez-Sánchez, Lorena Alcaraz, Félix A. López, María Jesús Martínez, and Alicia Prieto. 2019. "Effect of the Immobilization Strategy on the Efficiency and Recyclability of the Versatile Lipase from Ophiostoma piceae" Molecules 24, no. 7: 1313. https://doi.org/10.3390/molecules24071313
APA StyleMolina-Gutiérrez, M., Hakalin, N. L. S., Rodríguez-Sánchez, L., Alcaraz, L., López, F. A., Martínez, M. J., & Prieto, A. (2019). Effect of the Immobilization Strategy on the Efficiency and Recyclability of the Versatile Lipase from Ophiostoma piceae. Molecules, 24(7), 1313. https://doi.org/10.3390/molecules24071313