
Formation, Photophysics, and Photochemistry of Anionic Lanthanide(III) Mono- and Bisporphyrins

Abstract
1. Introduction
2. Results and Discussion
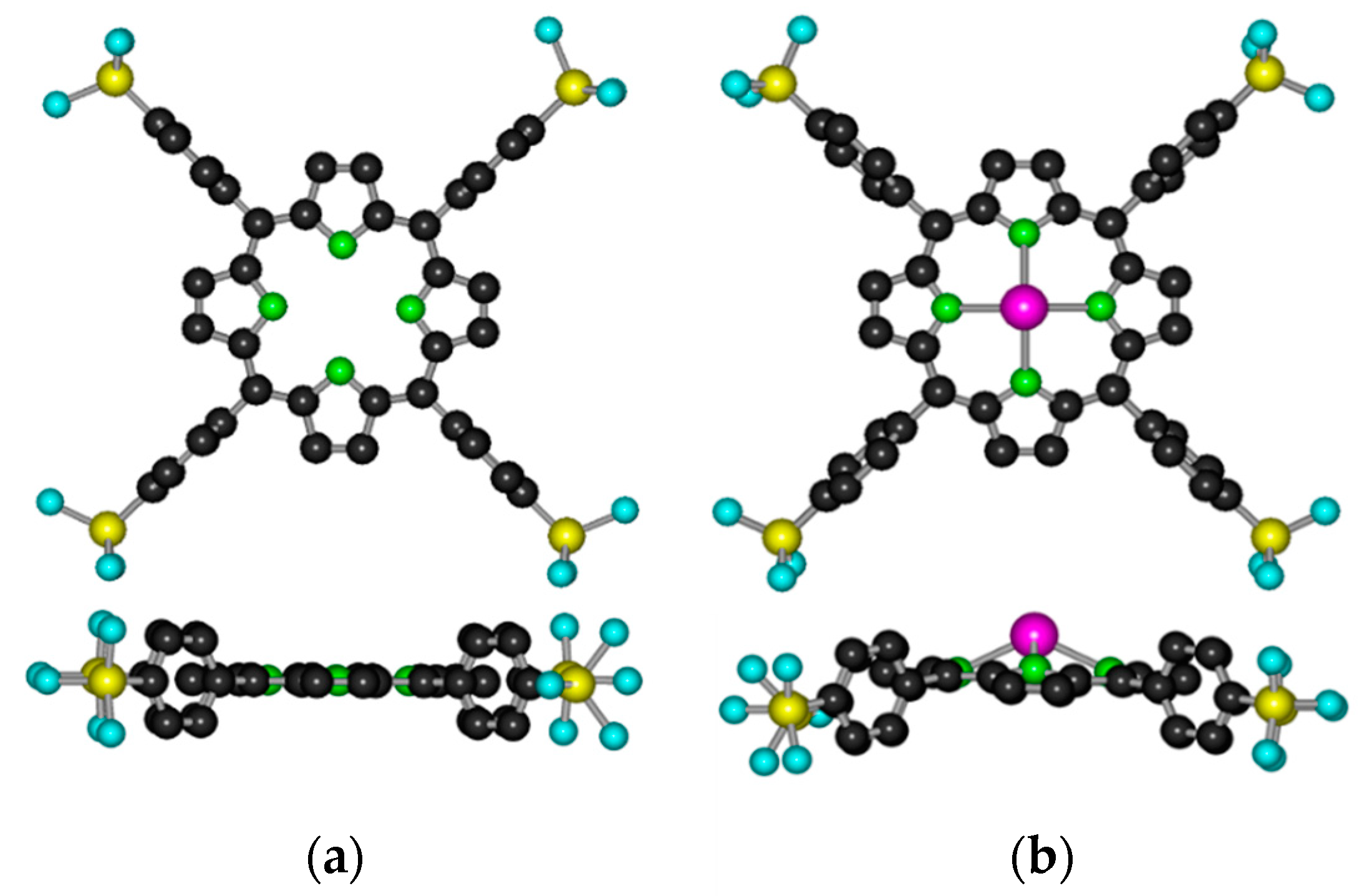
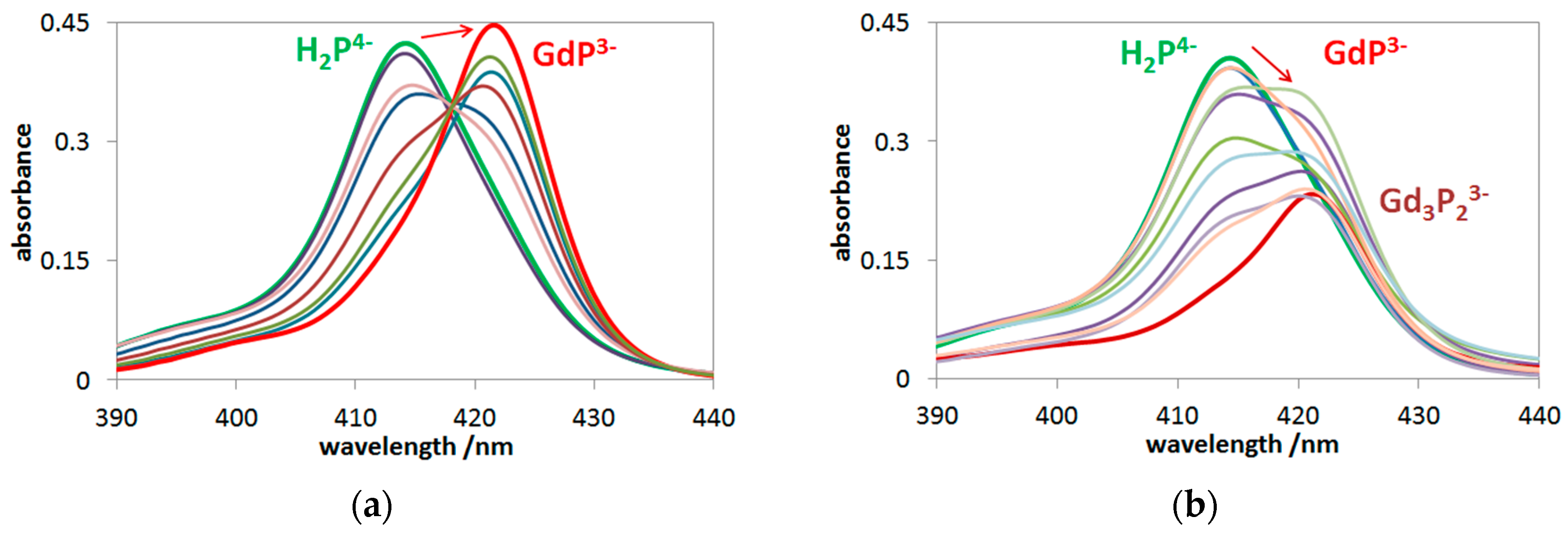
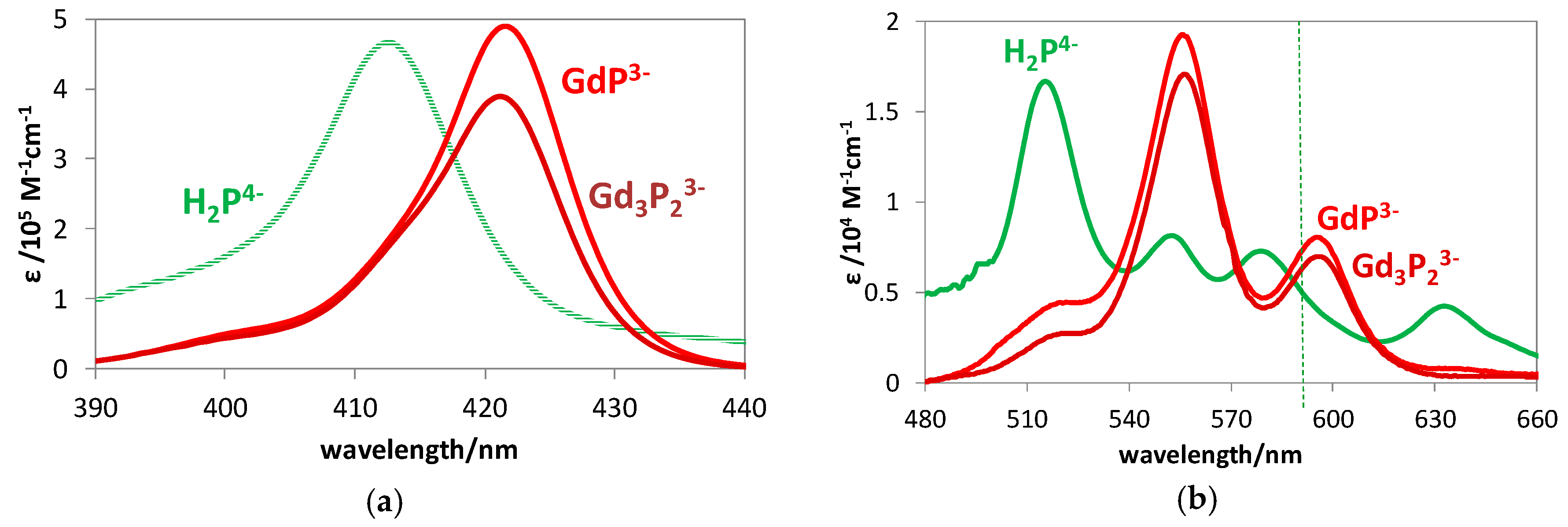
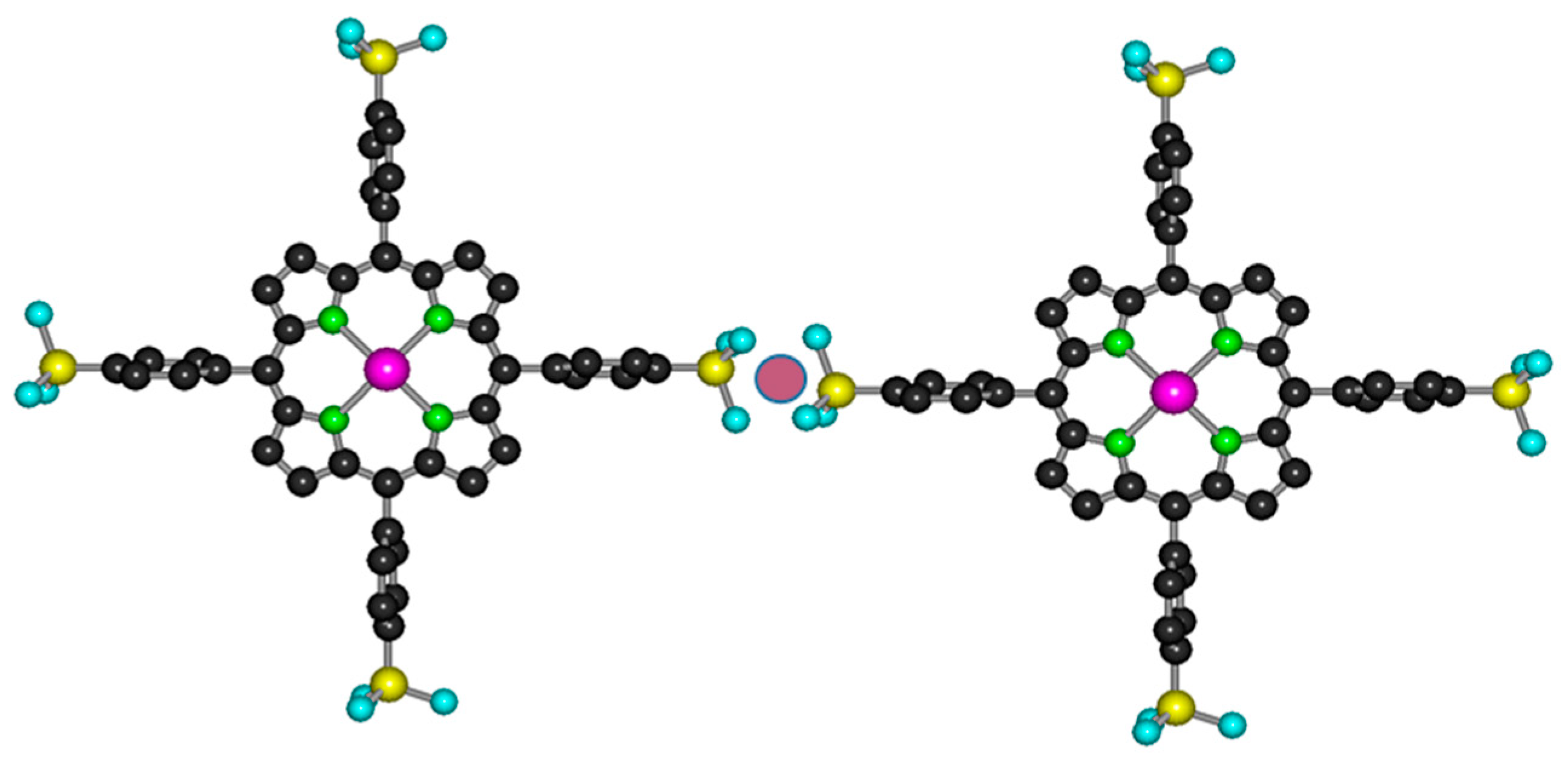
2.1. Formation, Composition, and Absorption Spectra
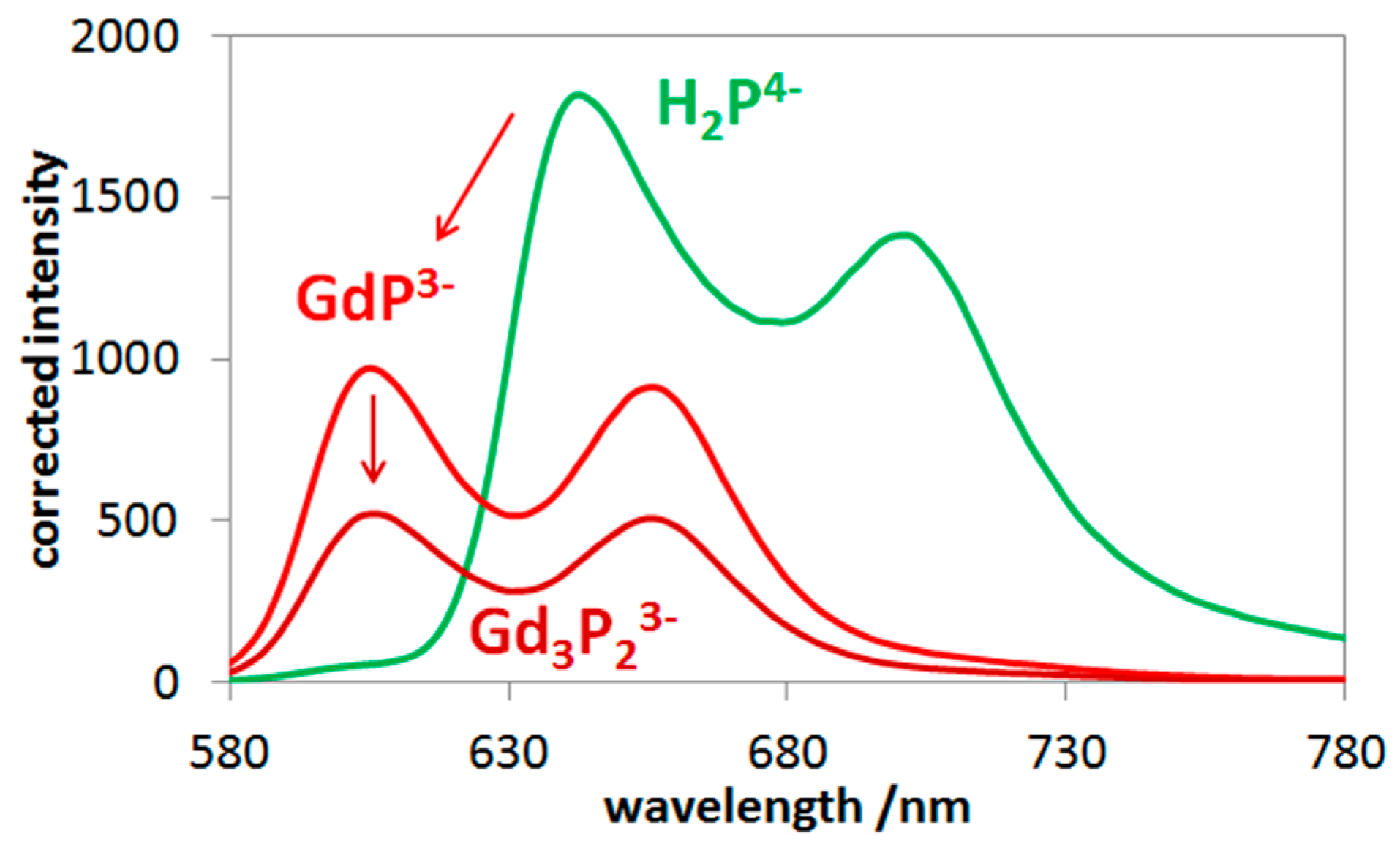
2.2. Photophysics of Mono- and Bisporphyrins
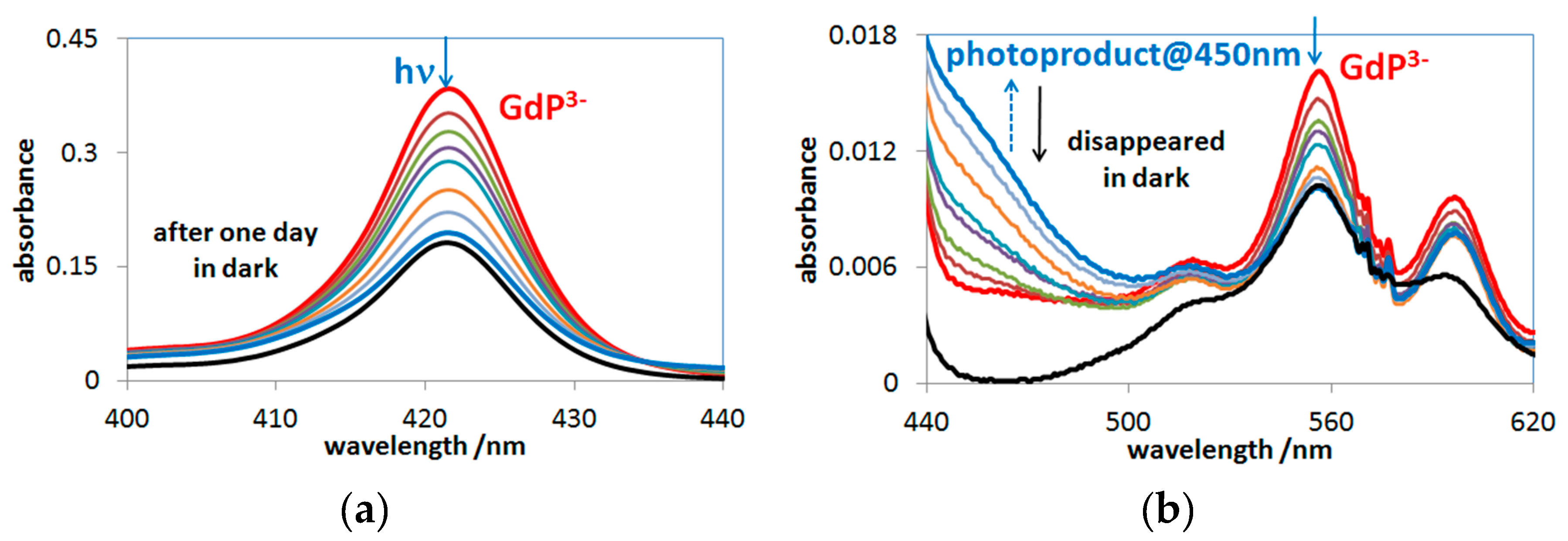
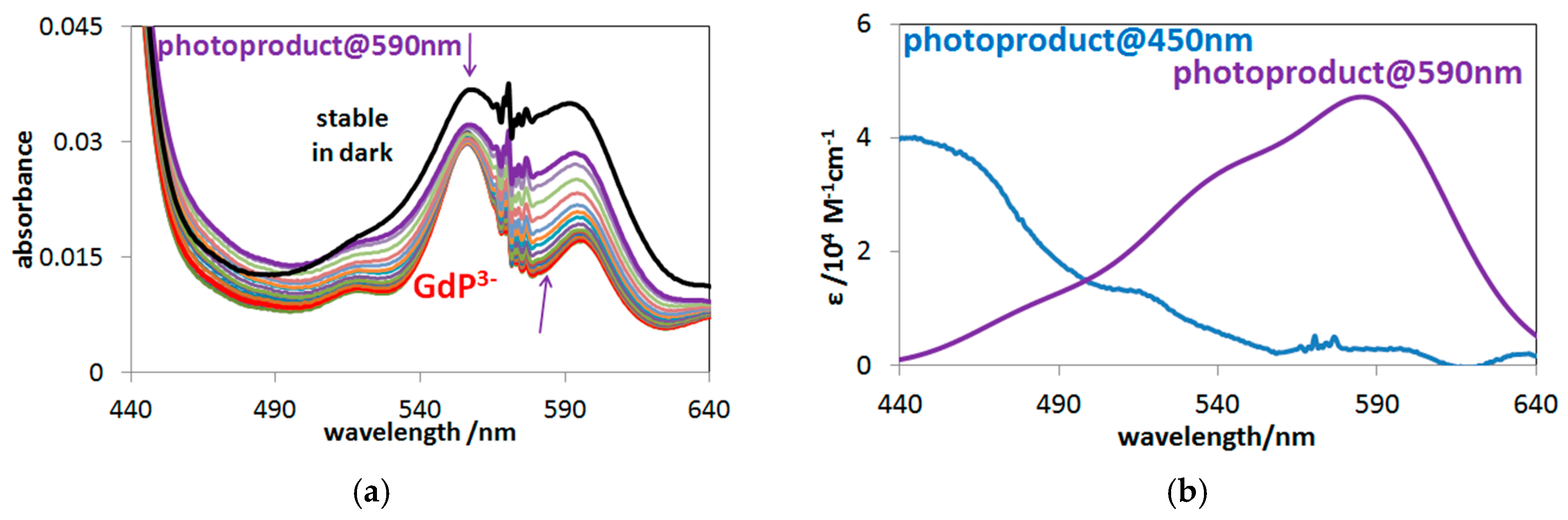
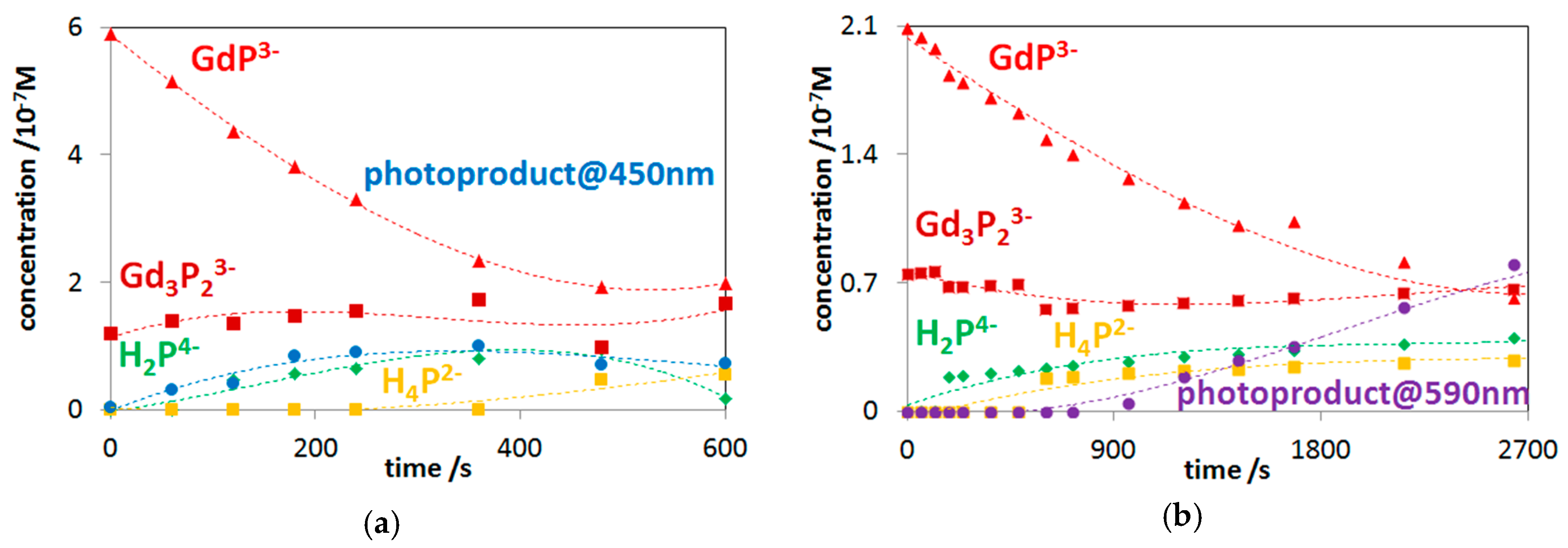
2.3. Photochemistry of Mono- and Bisporphyrins
3. Materials and Methods
3.1. Materials
3.2. Methods
3.3. Evaluations
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stochel, G.; Brindell, M.; Wojciech Macyk, W.; Zofia Stasicka, Z.; Szaciłowski, K. Bioinorganic Photochemistry; Wiley: Chichester, UK, 2009; pp. 169–187. ISBN 9781405161725. [Google Scholar]
- Garrett, R.H.; Grisham, C.M. Biochemistry, 4th ed.; Brooks/Cole: Boston, MA, USA, 2010; pp. 467–478. ISBN 978-0-495-10935-8. [Google Scholar]
- Shriver, D.; Weller, M.; Overton, T.; Rourke, J.; Armstrong, F. Inorganic Chemistry, 6th ed.; W. H. Freeman and Co.: New York, NY, USA, 2014; pp. 783–795. ISBN 978-1-4292-9906-0. [Google Scholar]
- Dąbrowski, J.M.; Pucelik, B.; Regiel-Futyra, A.; Brindell, M.; Mazuryk, O.; Kyzioł, A.; Stochel, G.; Macyk, W.; Arnaut, L.G. Engineering of relevant photodynamic processes through structural modifications of metallotetrapyrrolic photosensitizers. Coord. Chem. Rev. 2016, 325, 67–101. [Google Scholar] [CrossRef]
- Ladomenou, K.; Natali, M.; Iengo, E.; Charalampidis, G.; Scandola, F.; Coutsolelos, A.G. Photochemical hydrogen generation with porphyrin-based systems. Coord. Chem. Rev. 2015, 304–305, 38–54. [Google Scholar] [CrossRef]
- Ertl, M.; Wöβ, E.; Knör, G. Antimony porphyrins as red-light powered photocatalysts for solar fuel production from halide solutions in the presence of air. Photochem. Photobiol. Sci. 2015, 14, 1826–1830. [Google Scholar] [CrossRef] [PubMed]
- Knör, G. Recent progress in homogeneous multielectron transfer photocatalysis and artificial photosynthetic solar energy conversion. Coord. Chem. Rev. 2015, 304–305, 102–108. [Google Scholar] [CrossRef]
- Salzl, S.; Ertl, M.; Knör, G. Evidence for photosensitised hydrogen production from water in the absence of precious metals, redox-mediators and co-catalysts. Phys. Chem. Chem. Phys. 2017, 19, 8141–8147. [Google Scholar] [CrossRef]
- Radford, R.J.; Lim, M.D.; Da Silva, R.S.; Ford, P.C. Photochemical cleavage of nitrate ion coordinated to a Cr(III) porphyrin. J. Coord. Chem. 2010, 63, 2743–2749. [Google Scholar] [CrossRef]
- Kurtikyan, T.S.; Hovhannisyan, A.A.; Gulyan, G.M.; Ford, P.C. Interaction of Nitrogen Bases with Iron−Porphyrin Nitrito Complexes Fe(Por)(ONO) in Sublimed Solids. Inorg. Chem. 2007, 46, 7024–7031. [Google Scholar] [CrossRef] [PubMed]
- Kurtikyan, T.S.; Ford, P.C. FTIR and optical spectroscopic studies of the reactions of heme models with nitric oxide and other NOx in porous layered solids. Coord. Chem. Rev. 2008, 252, 1486–1496. [Google Scholar] [CrossRef]
- Fodor, M.A.; Horváth, O.; Fodor, L.; Grampp, G.; Wankmüller, A. Photophysical and photocatalytic behavior of cobalt(III) 5,10,15,20-tetrakis(1-methylpyridinium-4-yl)porphyrin. Inorg. Chem. Commun. 2014, 50, 110–112. [Google Scholar] [CrossRef]
- Fodor, M.A.; Horváth, O.; Fodor, L.; Vazdar, K.; Grampp, G.; Wankmüller, A. Photophysical and photochemical properties of manganese complexes with cationic porphyrin ligands: Effects of alkyl substituents and micellar environment. J. Photochem. Photobiol. A Chem. 2016, 328, 233–239. [Google Scholar] [CrossRef]
- Major, M.M.; Horváth, O.; Fodor, M.A.; Fodor, L.; Valicsek, Z.; Grampp, G.; Wankmüller, A. Photophysical and photocatalytic behavior of nickel(II) 5,10,15,20-tetrakis(1-methylpyridinium-4-yl)porphyrin. Inorg. Chem. Commun. 2016, 73, 1–3. [Google Scholar] [CrossRef]
- Horváth, O.; Valicsek, Z.; Fodor, M.A.; Major, M.M.; Imran, M.; Grampp, G.; Wankmüller, A. Visible light-driven photophysics and photochemistry of water-soluble metalloporphyrins. Coord. Chem. Rev. 2016, 325, 59–66. [Google Scholar] [CrossRef]
- Miskolczy, Z.; Biczók, L. Photochromism of a Merocyanine Dye Bound to Sulfonatocalixarenes: Effect of pH and the Size of Macrocycle on the Kinetics. J. Phys. Chem. B 2013, 117, 648–653. [Google Scholar] [CrossRef]
- Takahashi, Y.; Fujihara, T.; Kobayashi, N.; Nakabayashi, S.; Miskolczy, Z.; Biczók, L. Electron transfer kinetics of methylviologen included in 4-sulfonatocalix[n]arenes at glassy carbon electrode; adiabaticity and activation energy. Chem. Phys. Lett. 2018, 708, 222–227. [Google Scholar] [CrossRef]
- Valicsek, Z.; Horváth, O.; Lendvay, G.; Kikaš, I.; Škorić, I. Formation, photophysics, and photochemistry of cadmium(II) complexes with 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrin and its octabromo derivative: The effects of bromination and the axial hydroxo ligand. J. Photochem. Photobiol. A Chem. 2011, 218, 143–155. [Google Scholar] [CrossRef]
- Valicsek, Z.; Horváth, O.; Patonay, K. Formation, photophysical and photochemical properties of water-soluble bismuth(III) porphyrins: The role of the charge and structure. J. Photochem. Photobiol. A Chem. 2011, 226, 23–35. [Google Scholar] [CrossRef]
- Horváth, O.; Valicsek, Z.; Harrach, G.; Lendvay, G.; Fodor, M.A. Spectroscopic and photochemical properties of water-soluble metalloporphyrins of distorted structure. Coord. Chem. Rev. 2012, 256, 1531–1545. [Google Scholar] [CrossRef]
- Valicsek, Z.; Horváth, O. Application of the electronic spectra of porphyrins for analytical purposes: The effects of metal ions and structural distortions. Microchem. J. 2013, 107, 47–62. [Google Scholar] [CrossRef]
- Cotton, S. Lanthanide and Actinide Chemistry; Wiley: Chichester, UK, 2006; ISBN 0-470-01005-3. [Google Scholar]
- Bünzli, J.-C.G. Rising Stars in Science and Technology: Luminescent Lanthanide Materials. Eur. J. Inorg. Chem. 2017, 2017, 5058–5063. [Google Scholar] [CrossRef]
- de Bettencourt-Dias, A.; Barber, P.S.; Bauer, S. A Water-Soluble Pybox Derivative and Its Highly Luminescent Lanthanide Ion Complexes. J. Am. Chem. Soc. 2012, 134, 6987–6994. [Google Scholar] [CrossRef]
- Guenet, A.; Eckes, F.; Bulach, V.; Strassert, C.A.; De Cola, L.; Hosseini, M.W. Sensitisation of the Near-Infrared Emission of NdIII from the Singlet State of Porphyrins Bearing Four 8-Hydroxyquinolinylamide Chelates. ChemPhysChem 2012, 13, 3163–3171. [Google Scholar] [CrossRef] [PubMed]
- Semenishyn, N.N.; Smola, S.S.; Efryushina, N.P.; Rusakova, N.V. Spectral and Luminescence Properties of Lanthanide(III) Complexes with Porphyrins and Corroles with Varied Structure. Theor. Exp. Chem. 2015, 51, 224–229. [Google Scholar] [CrossRef]
- Faulkner, S.; Matthews, J.L. Fluorescent Complexes for Biomedical Applications. In Comprehensive Coordination Chemistry II; Elsevier: Amsterdam, The Netherlands, 2003; pp. 913–944. ISBN 9780080437484. [Google Scholar]
- Bünzli, J.-C.G. Luminescence Bioimaging with Lanthanide Complexes. In Luminescence of Lanthanide Ions in Coordination Compounds and Nanomaterials; John Wiley & Sons Ltd.: Chichester, UK, 2014; pp. 125–196. ISBN 9781118682760. [Google Scholar]
- Bünzli, J.-C.G. Lanthanide light for biology and medical diagnosis. J. Lumin. 2016, 170, 866–878. [Google Scholar] [CrossRef]
- Imran, M.; Szentgyörgyi, C.; Eller, G.; Valicsek, Z.; Horváth, O. Peculiar photoinduced properties of water-soluble, early lanthanide(III) porphyrins. Inorg. Chem. Commun. 2015, 52, 60–63. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, J. N-Based Rare Earth Complexes. In Rare Earth Coordination Chemistry: Fundamentals and Applications; Huang, C., Ed.; John Wiley & Sons (Asia) Pte Ltd.: Singapore, 2010; pp. 137–192. ISBN 978-0-470-82485-6. [Google Scholar]
- Bouvet, M.; Gaudillat, P.; Suisse, J.-M. Lanthanide macrocyclic complexes: From molecules to materials and from materials to devices. J. Porphyr. Phthalocyanines 2013, 17, 628–635. [Google Scholar] [CrossRef]
- Kiss, M.P.; Valicsek, Z.; Horváth, O. Effects of the axial ligands on the formation kinetics of water-soluble cerium(III) porphyrins. Molecules 2019. under review. [Google Scholar]
- Spyroulias, G.A.; Despotopoulos, A.P.; Raptopoulou, C.P.; Terzis, A.; de Montauzon, D.; Poilblanc, R.; Coutsolelos, A.G. Comparative Study of Structure−Properties Relationship for Novel β-Halogenated Lanthanide Porphyrins and Their Nickel and Free Bases Precursors, as a Function of Number and Nature of Halogens Atoms. Inorg. Chem. 2002, 41, 2648–2659. [Google Scholar] [CrossRef]
- Liao, M.-S.; Watts, J.D.; Huang, M.-J. DFT/TDDFT Study of Lanthanide III Mono- and Bisporphyrin Complexes. J. Phys. Chem. A 2006, 110, 13089–13098. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-J.; Zhang, T.; Zhao, S.; Wong, W.-K.; Wong, W.-Y. Synthesis, Structure, and Photophysical Properties of Some Gadolinium(III) Porphyrinate Complexes. Eur. J. Inorg. Chem. 2011, 2011, 3314–3320. [Google Scholar] [CrossRef]
- Valicsek, Z.; Eller, G.; Horváth, O. Equilibrium, photophysical and photochemical examination of anionic lanthanum(III) mono- and bisporphyrins: The effects of the out-of-plane structure. Dalton Trans. 2012, 41, 13120–13131. [Google Scholar] [CrossRef] [PubMed]
- Kiss, M.P.; Imran, M.; Szentgyörgyi, C.; Valicsek, Z.; Horváth, O. Peculiarities of the reactions between early lanthanide(III) ions and an anionic porphyrin. Inorg. Chem. Commun. 2014, 48, 22–25. [Google Scholar] [CrossRef]
- Wittmer, L.L.; Holten, D. Photophysics of Lanthanide Triple Decker Porphyrin Sandwich Complexes. Phys. J. Chem. 1996, 100, 860–868. [Google Scholar] [CrossRef]
- Valicsek, Z.; Lendvay, G.; Horváth, O. Equilibrium, Photophysical, Photochemical, and Quantum Chemical Examination of Anionic Mercury(II) Mono- and Bisporphyrins. J. Phys. Chem. B 2008, 112, 14509–14524. [Google Scholar] [CrossRef] [PubMed]
- Bünzli, J.-C.G. Lanthanide Luminescence for Biomedical Analyses and Imaging. Chem. Rev. 2010, 110, 2729–2755. [Google Scholar] [CrossRef] [PubMed]
- Khalil, G.E.; Thompson, E.K.; Gouterman, M.; Callis, J.B.; Dalton, L.R.; Turro, N.J.; Jockusch, S. NIR luminescence of gadolinium porphyrin complexes. Chem. Phys. Lett. 2007, 435, 45–49. [Google Scholar] [CrossRef]
- Kalota, B.; Tsvirko, M. Fluorescence and phosphorescence of lutetium(III) and gadolinium(III) porphyrins for the intraratiometric oxygen sensing. Chem. Phys. Lett. 2015, 634, 188–193. [Google Scholar] [CrossRef]
- Bulach, V.; Sguerra, F.; Hosseini, M.W. Porphyrin lanthanide complexes for NIR emission. Coord. Chem. Rev. 2012, 256, 1468–1478. [Google Scholar] [CrossRef]
- Hu, J.Y.; Ning, Y.; Meng, Y.S.; Zhang, J.; Wu, Z.Y.; Gao, S.; Zhang, J.L. Highly near-IR emissive ytterbium(III) complexes with unprecedented quantum yields. Chem. Sci. 2017, 8, 2702–2709. [Google Scholar] [CrossRef]
- He, H. Near-infrared emitting lanthanide complexes of porphyrin and BODIPY dyes. Coord. Chem. Rev. 2014, 273–274, 87–99. [Google Scholar] [CrossRef]
- Wei, C.; Ma, L.; Wei, H.B.; Liu, Z.W.; Bian, Z.Q.; Huang, C.H. Advances in luminescent lanthanide complexes and applications. Sci. China Technol. Sci. 2018, 61, 1265–1285. [Google Scholar] [CrossRef]
- Lipstman, S.; Muniappan, S.; George, S.; Goldberg, I. Framework coordination polymers of tetra(4-carboxyphenyl)porphyrin and lanthanide ions in crystalline solids. Dalton Trans. 2007, 30, 3273–3281. [Google Scholar] [CrossRef]
- Horváth, O.; Valicsek, Z.; Vogler, A. Unique photoreactivity of mercury(II) 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrin. Inorg. Chem. Commun. 2004, 7, 854–857. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Imran, M. Formation, Photophysics and Photochemistry of Water-Soluble Lanthanide(III) Porphyrins. Ph.D. Thesis, University of Pannonia, Veszprém, Hungary, 2016. [Google Scholar]
- Valicsek, Z.; Horváth, O. Formation, photophysics and photochemistry of thallium(III) 5,10,15,20-tetrakis(4-sulphonatophenyl)porphyrin: New supports of typical sitting-atop features. J. Photochem. Photobiol. A Chem. 2007, 186, 1–7. [Google Scholar] [CrossRef]
- Horváth, O.; Huszánk, R.; Valicsek, Z.; Lendvay, G. Photophysics and photochemistry of kinetically labile, water-soluble porphyrin complexes. Coord. Chem. Rev. 2006, 250, 1792–1803. [Google Scholar] [CrossRef]
- Davila, J.; Harriman, A.; Richoux, M.-C.; Milgrom, L.R. Sterically-hindered zinc porphyrins for solar-energy conversion. J. Chem. Soc. Chem. Commun. 1987, 7, 525–527. [Google Scholar] [CrossRef]
- Huszánk, R.; Lendvay, G.; Horváth, O. Air-stable, heme-like water-soluble iron(II) porphyrin: In situ preparation and characterization. J. Biol. Inorg. Chem. 2007, 12, 681–690. [Google Scholar] [CrossRef]
Sample Availability: Not available. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | SmP3− | Sm3P23− | EuP3− | Eu3P23− | GdP3− | Gd3P23− |
|---|---|---|---|---|---|---|
| Ln3+ radius /pm * | 107.9 | 106.6 | 105.3 | |||
| lgβ1:1 and lgβ3:2 | 4.64 | 17.47 | 4.80 | 17.51 | 4.94 | 17.53 |
| lgβ1:1(Ac−) | 4.89 | 5.01 | 5.11 | |||
| Complex | H2P4− | SmP3− | Sm3P23− | EuP3− | Eu3P23− | GdP3− | Gd3P23− | |
|---|---|---|---|---|---|---|---|---|
| Ln3+ radius/pm * | - | 107.9 | 106.6 | 105.3 | ||||
| λ B(0,0)/nm | 413 | 421 | 423 | 421 | 423 | 421 | 422 | |
| ε B(0,0) /105 M−1cm−1 | 4.66 | 4.92 | 4.20 | 4.92 | 4.10 | 4.90 | 3.98 | |
| λ Q(1,0)/nm | y 516 | x 579 | 555 | 557 | 555 | 557 | 555 | 557 |
| ε Q(1,0) /104 M−1cm−1 | y 1.67 | x 0.67 | 1.96 | 1.74 | 1.95 | 1.73 | 1.92 | 1.70 |
| ϕ(S1-fluo@Q) /10−2 ** | 7.53 | 4.39 | 3.09 | 4.47 | 2.88 | 4.15 | 2.37 | |
| Ф(S1-fluo@B) /10−2 ** | 5.62 | 3.03 | 2.20 | 2.86 | 1.92 | 2.91 | 1.73 | |
| ϕ(IC S2–S1) | 0.746 | 0.69 | 0.71 | 0.64 | 0.67 | 0.70 | 0.73 | |
| τ(S1) /ns | 10.0 | 1.93 | 1.94 | 1.94 | 1.95 | 1.93 | 1.94 | |
| kr(S1) /106 s−1 | 7.5 | 22.8 | 16.0 | 23.1 | 14.8 | 21.5 | 12.3 | |
| knr(S1) /107 s−1 | 9.2 | 49.6 | 50.0 | 49.2 | 49.8 | 49.7 | 50.4 | |
| Complex | H2P4− | SmP3− | Sm3P23− | EuP3− | Eu3P23− | GdP3− | Gd3P23− |
|---|---|---|---|---|---|---|---|
| Ln3+ radius /pm * | - | 107.9 | 106.6 | 105.3 | |||
| Ф (S2-photochem) /10−3 | 0.006 | 0.78 | 1.67 | 1.20 | 1.31 | 0.86 | 1.01 |
| Redox % | 100% | 86% | 96% | 86% | 91% | 72% | 95% |
| Dissociation % | - | 11% | 3% | 7% | 4% | 15% | 4% |
| Transformation % | 0% ** | 3% | 1% | 7% | 5% | 13% | 1% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imran, M.; Kiss, M.P.; Valicsek, Z.; Horváth, O. Formation, Photophysics, and Photochemistry of Anionic Lanthanide(III) Mono- and Bisporphyrins. Molecules 2019, 24, 1309. https://doi.org/10.3390/molecules24071309
Imran M, Kiss MP, Valicsek Z, Horváth O. Formation, Photophysics, and Photochemistry of Anionic Lanthanide(III) Mono- and Bisporphyrins. Molecules. 2019; 24(7):1309. https://doi.org/10.3390/molecules24071309
Chicago/Turabian StyleImran, Muhammad, Melitta P. Kiss, Zsolt Valicsek, and Ottó Horváth. 2019. "Formation, Photophysics, and Photochemistry of Anionic Lanthanide(III) Mono- and Bisporphyrins" Molecules 24, no. 7: 1309. https://doi.org/10.3390/molecules24071309
APA StyleImran, M., Kiss, M. P., Valicsek, Z., & Horváth, O. (2019). Formation, Photophysics, and Photochemistry of Anionic Lanthanide(III) Mono- and Bisporphyrins. Molecules, 24(7), 1309. https://doi.org/10.3390/molecules24071309