HPLC-DAD Determination of Iodide in Mineral Waters on Phosphatidylcholine Column

 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
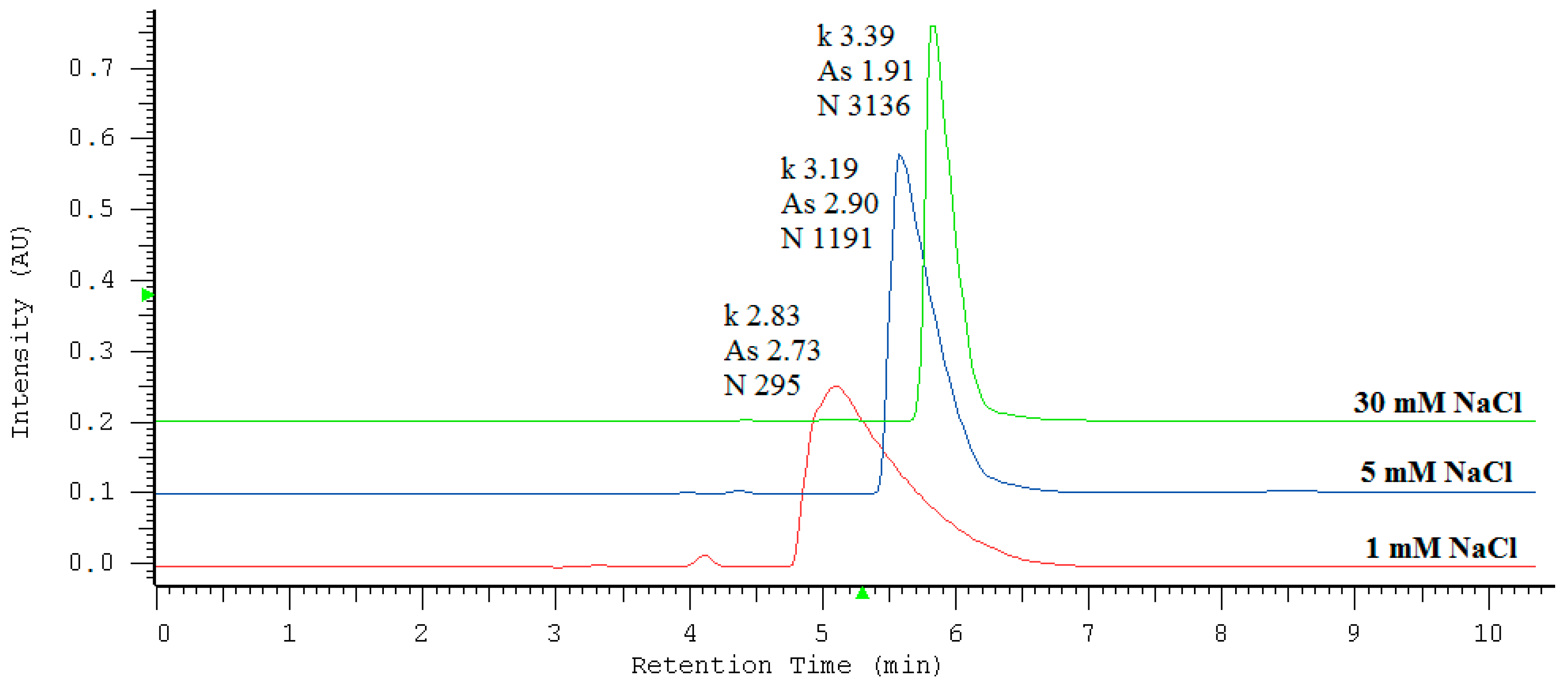
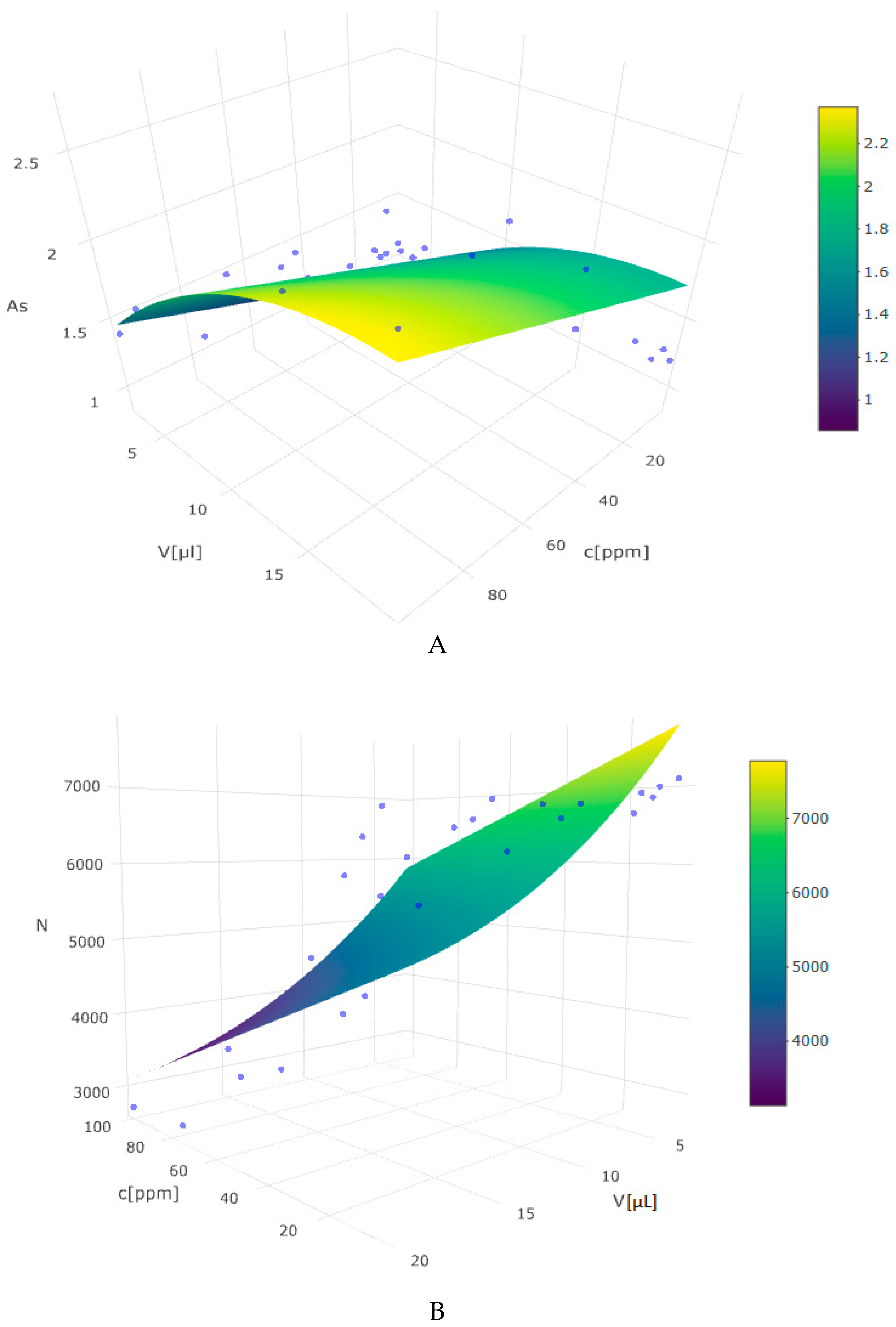
2.1. Effect of Experimental Variables
2.2. Analytical Quality Control
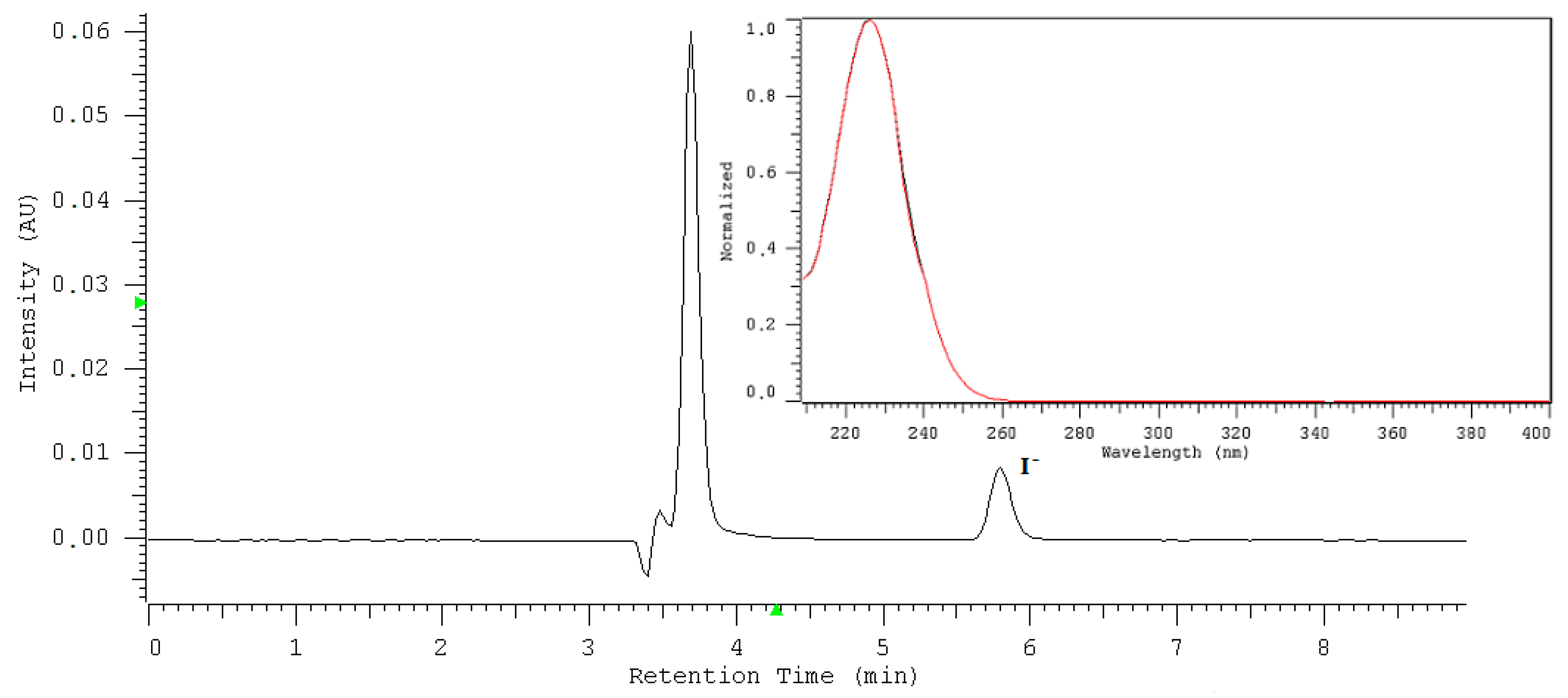
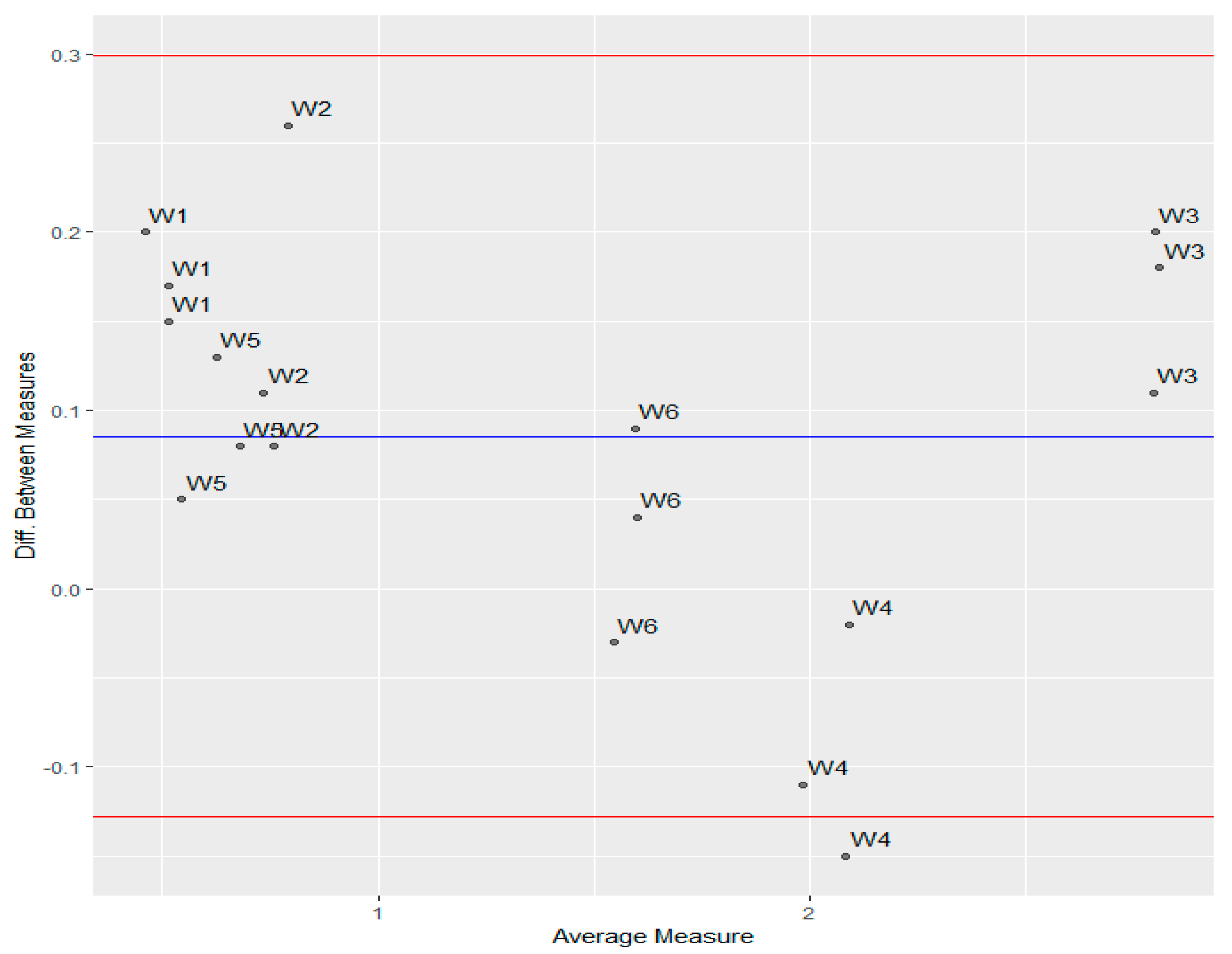
2.3. The Mineral Water Sample Analysis
2.4. Interferences of Coexisting Ions
3. Experimental
3.1. Materials and Sample
3.2. HPLC Conditions
3.3. ICP-MS
3.4. Method Optimization and Validation
3.5. Data Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zimmermann, M.B.; Jooste, P.L.; Pandav, C.S. Iodine-deficiency disorders. Lancet 2008, 372, 1251–1262. [Google Scholar] [CrossRef]
- World Health Organization/United Nations Children’s Fund/International Council for Control of Iodine Deficiency Disorders. Assessment of Iodine Deficiency Disorders and Monitoring Their Elimination: A Guide for Programme Managers, 3rd ed.; World Health Organization: Geneva, Switzerland, 2007. [Google Scholar]
- Zimmermann, M.B. Iodine deficiency. Endocr. Rev. 2009, 30, 376–408. [Google Scholar] [CrossRef]
- Delange, F.; Bürgi, H.; Chen, Z.P.; Dunn, J.T. World status of monitoring of iodine deficiency disorders control programs. Thyroid 2002, 12, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-B.; Chen, F.; Xu, X.-R. Determination of iodide in seawater and urine by size exclusion chromatography with iodine-starch complex. J. Chromatogr. A 2001, 918, 335–339. [Google Scholar] [CrossRef]
- Wisnu, C. Determination of iodine species content in iodized salt and foodstuff during cooking. Int. Food Res. J. 2008, 15, 325–330. [Google Scholar]
- Merian, E.; Anke, M.; Ihnat, M.; Stoeppler, M. Elements and Their Compounds in the Environment, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2004; pp. 1457–1485. [Google Scholar]
- Food and Nutrition Board, Institute of Medicine. Dietary Reference Intakes; National Academy Press: Washington, WA, USA, 2001.
- Hansen, V.; Yi, P.; Hou, X.; Aldahan, A.; Roos, P.; Possnert, G. Iodide and iodate in surface water of the Baltic Sea, Kattegat and Skagerrak. Sci. Total Environ. 2011, 412–413, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Gilfedder, B.S.; Petri, M.; Biester, H. Iodine speciation and cycling in fresh waters: A case study from a humic rich headwater lake (Mummelsee). J. Limnol. 2009, 68, 396–408. [Google Scholar] [CrossRef]
- Schwehr, K.A.; Santschi, P.H. Sensitive determination of iodine species, including organo-iodine, for freshwater and seawater samples using high performance liquid chromatography and spectrophotometric detection. Anal. Chim. Acta 2003, 482, 59–71. [Google Scholar] [CrossRef]
- Zhang, S.; Schwehr, K.A.; Ho, Y.F.; Xu, C.; Roberts, K.A.; Kaplan, D.I.; Brinkmeyer, R.; Yeager, C.M.; Santschi, P.H. A novel approach for the simultaneous determination of iodide, iodate and organo-Iodide for 127I and 129I in environmental samples using gas chromatography-mass spectrometry. Environ. Sci. Technol. 2010, 44, 9042–9048. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.D.; Maiti, B.; Mathur, P.K. Determination of iodate and sulphate in iodized common salt by ion chromatography with conductivity detection. Talanta 2001, 53, 701–705. [Google Scholar] [CrossRef]
- Rebary, B.; Paul, P.; Ghosh, P.K. Determination of iodide and iodate in edible salt by ion chromatography with integrated amperometric detection. Food Chem. 2010, 123, 529–534. [Google Scholar] [CrossRef]
- Sandell, E.B.; Kolthoff, I.M. Chronometric catalytic method for the determination of micro quantities of iodine. J. Am. Chem. Soc. 1934, 56, 1426. [Google Scholar] [CrossRef]
- Association of Officiating Analytical Chemists. Official Method of Analysis: Method 935.14, 18th ed.; AOAC International: Washington, DC, USA, 2005. [Google Scholar]
- Association of Officiating Analytical Chemists. Official method of Analysis: Method 992.24, 18th ed.; AOAC International: Washington, DC, USA, 2005. [Google Scholar]
- Kosminsky, L.; Bertotti, M. Determination of iodate in salt samples with amperometric detection at a molybdenum oxide modified electrode. Electroanalysis 1999, 11, 623–626. [Google Scholar] [CrossRef]
- Shelor, C.P.; Dasgupta, P.K. Review of analytical methods for the quantification of iodine in complex matrices. Anal. Chim. Acta 2011, 702, 16–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Mnatsakanov, A.; Hower, R.; Cantor, H.; Wang, Y. Urinary iodine assays and ionophore based potentiometric iodide sensors. Front. Biosci. 2005, 10, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, T.R.I.; Rubino, A.; Laviola, M.C.; Ciriello, R. Comparison of silver, gold and modified platinum electrodes for the electrochemical detection of iodide in urine samples following ion chromatography. J. Chromatogr. B 2005, 827, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Judprasong, K.; Jongjaithet, N.; Chavasit, V. Comparison of methods for iodine analysis in foods. Food Chem. 2016, 193, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Association of Officiating Analytical Chemists. Official Method of Analysis: Method 992.22, 18th ed.; AOAC International: Washington, DC, USA, 2007. [Google Scholar]
- Association of Officiating Analytical Chemists. Official Method of Analysis: Method 2012.14 and 2012.15, 18th ed.; AOAC International: Washington, DC, USA, 2012. [Google Scholar]
- Wang, T.; Zhao, S.; Shen, C.; Tang, J.; Wang, D. Determination of iodate in table salt by transient isotachophoresis-capillary zone electrophoresis. Food Chem. 2009, 112, 215–220. [Google Scholar] [CrossRef]
- Huang, X.; Li, Y.; Chen, Y.; Wang, L. Electrochemical determination of nitrite and iodate by use of gold nanoparticles/poly(3-methylthiophene) composites coated glassy carbon electrode. Sens. Actuators B Chem. 2008, 134, 780–786. [Google Scholar] [CrossRef]
- Dionex Corporation. Determination of Iodide and Iodate in Seawater and Iodized Table Salt by HPLC With UV Detection (Application Note 236). Available online: http://www.cromlab.es/Articulos/Columnas/HPLC/Thermo/Acclaim/WAX-1/81306-AN236-LC-Iodide-Seawater-15Sept2009-LPN2312.pdf (accessed on 28 March 2019).
- Hurum, D.; Rohrer, J. Determination of Iodide in Seawater and Other Saline Matrices Using a Reagent-Free Ion Chromatography System with Suppressed Conductivity and UV Detections (Application Note 239). Available online: https://assets.thermofisher.com/TFS-Assets/CMD/Application-Notes/AN-239-IC-Iodide-Seawater-AN71348-EN.pdf (accessed on 28 March 2019).
- Yamada, H.; Kajiyama, S.; Yonebayashi, K. Determination of trace amounts of iodine in soils by HPLC with fluorescence detection. Bunseki Kagaku 1995, 44, 1027–1032. [Google Scholar] [CrossRef]
- Yamada, H.; Kiriyama, T.; Onagawa, Y.; Hisamori, I.; Miyazaki, C.; Yonebayashi, K. Speciation of Iodine in Soils. Soil Sci. Plant Nutr. 1999, 45, 563–568. [Google Scholar] [CrossRef]
- Hu, W.; Haddad, P.R.; Tanaka, K.; Mori, M.; Tekura, K.; Hase, K.; Ohno, M.; Kamo, N. Creation and characteristics of phosphatidylcholine stationary phases for the chromatographic separation of inorganic anions. J. Chromatogr. A 2003, 997, 237–242. [Google Scholar] [CrossRef]
- Wiedmer, S.K.; Jussila, M.S.; Riekkola, M.L. Phospholipids and liposomes in liquid chromatographic and capillary electromigration techniques. Trends Anal. Chem. 2004, 23, 562–582. [Google Scholar] [CrossRef]
- Giaginis, C.; Tsantili-Kakoulidou, A. Alternative measures of lipophilicity from octanol-water partitioning to IAM retention. J. Pharm. Sci. 2008, 97, 2984–3004. [Google Scholar] [CrossRef] [PubMed]
- Barbato, F.; Di Martino, G.; Grumetto, L.; La Rotonda, M.I. Retention of quinolones on human serum albumin and alpha1-acid glycoprotein HPLC columns: Relationships with different scales of lipophilicity. Eur. J. Pharm. Sci. 2007, 30, 211–219. [Google Scholar] [CrossRef]
- Lázaro, E.; Castillo, J.A.; Ráfols, C.; Rosés, M.; Clapés, P.; Torres, J.L. Immunomodulatory Activity of a New Family of Antioxidants Obtained from Grape Polyphenols. J. Agric. Food Chem. 2007, 55, 2901–2905. [Google Scholar] [CrossRef]
- Zhang, W.N.; Hu, Z.X.; Liu, Y.; Feng, Y.Q.; Da, S.L. Study on interaction between drug and membrane by using liposome coated zirconia–magnesia chromatography. Talanta 2005, 67, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Barbato, F.; di Martino, G.; Grumetto, L.; La Rotonda, M. Prediction of drug-membrane interactions by IAM-HPLC: Effects of different phospholipid stationary phases on the partition of bases. Eur. J. Pharm. Sci. 2004, 22, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Ollila, F.; Halling, K.; Vuorela, P.; Vuorela, H.; Slotte, J.P. Characterization of flavonoid--biomembrane interactions. Arch. Biochem. Biophys. 2002, 399, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Sarr, F.S.; André, C.; Guillaume, Y.C. Statins (HMG-coenzyme A reductase inhibitors)-biomimetic membrane binding mechanism investigated by molecular chromatography. J. Chromatogr. B 2008, 868, 20–27. [Google Scholar] [CrossRef]
- Mei, J.; Xu, J.R.; Xiao, Y.X.; Zhang, Q.R.; Feng, Y.Q. Immobilized phospholipid capillary electrophoresis for study of drug–membrane interactions and prediction of drug activity. Talanta 2008, 75, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirokawa, T. Enhanced detection of iodide in seawater by ion chromatography using an ODS column coated with cetyltrimethyl ammonium. Anal. Sci. 2001, 17, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Nomura, R.; Fujii, T.; Tanaka, M.; Tsumura, T.; Shibata, H.; Hirokawa, T. Determination of nitrite, nitrate, bromide, and iodide in seawater by ion chromatography with UV detection using dilauryl dimethyl ammonium-coated monolithic ODS columns and sodium chloride as an eluent. Anal. Bioanal. Chem. 2012, 404, 2513–2517. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.S.; Setiawan, A.N.; Pope, S.; Haddad, P.R.; Nesterenko, P.N.; Paull, B. A simple and sensitive method for the determination of iodide and iodate in raw, ultraviolet- and ozone-treated aqua cultural seawater samples using ion chromatography coupled to an ultraviolet detector. Anal. Methods 2016, 8, 5587–5595. [Google Scholar] [CrossRef]
- Giavarina, D. Understanding Bland Altman Analysis. Biochem. Med. 2015, 25, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Bland, J.M.; Altman, D.G. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet 1986, 1, 307–310. [Google Scholar] [CrossRef]
- Altman, D.G.; Bland, J.M. Measurement in Medicine: The Analysis of Method Comparison Studies. J. R. Stat. Soc. Ser. D 1983, 32, 307–317. [Google Scholar]
- Bland, J.M.; Altman, D.G. Statistical Methods for Assessing Agreement Between Two Methods of Clinical Measurement. Int. J. Nurs. Stud. 2010, 47, 931–936. [Google Scholar] [CrossRef]
- R Core Team R: A language and environment for statistical computing. R Foundation for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 8 February 2008).
- International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use, Validation of analytical procedures: Text and Methodology; ICH-Q2B: Geneva, Switzerland, 1996.
- Nitscke, U.; Stengel, D.B. A new HPLC method for the detection of iodine applied to natural samples of edible seaweeds and commercial seaweed food products. Food Chem. 2015, 172, 326–334. [Google Scholar] [CrossRef]
Sample Availability: Samples of the investigated water are available from the authors. |
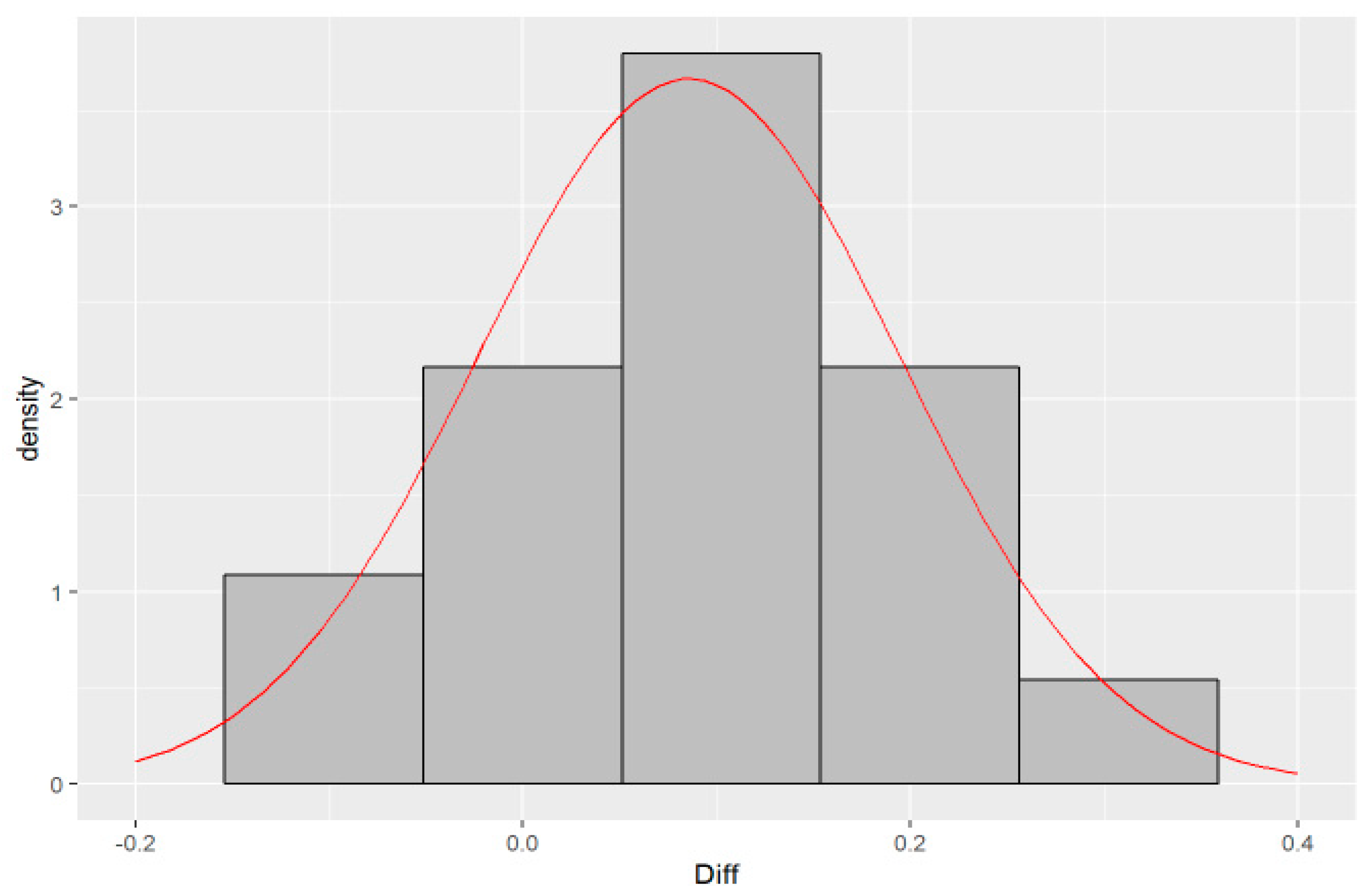

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NaCl Concentration [mM] | Retention Factor k | Symmetry Factor As * | Theoretical Plates Number N (EUP) ** |
|---|---|---|---|
| 1 | 2.83 (± 0.08) | 2.73 (± 0.05) | 295 (± 97) |
| 5 | 3.19 (± 0.07) | 2.90 (± 0.04) | 1191 (± 147) |
| 10 | 3.57 (± 0.06) | 2.97 (± 0.07) | 1973 (± 198) |
| 15 | 3.59 (± 0.10) | 2.74 (± 0.08) | 2352 (± 116) |
| 20 | 3.59 (± 0.07) | 2.57 (± 0.06) | 2794 (± 174) |
| 25 | 3.50 (± 0.08) | 2.32 (± 0.07) | 3082 (± 196) |
| 30 | 3.39 (± 0.06) | 1.91 (± 0.06) | 3136 (± 177) |
| Parameters | As | N |
|---|---|---|
| C | 0.021 (0.005) | −49.876 (11.734) |
| I(C2) | −0.0001 (0.00004) | 0.204 (0.112) |
| V | 0.037 (0.006) | −113.931 (14.506) |
| constant | 0.646 (0.100) | 8360.409 (257.425) |
| observations | 35 | 35 |
| R2 | 0.812 | 0.853 |
| Adjusted R2 | 0.794 | 0.839 |
| Residual Std Error | 0.209 (df = 31) | 538.961 (df = 31) |
| F statistic | 44.771 (df = 3;31) | 60.201 (df = 3;31) |
| LOD * [mg L−1] | LOQ * [mg L−1] | Linear Range * [mg L−1] | Regression Equation ** (y = ax + b) | ||||
|---|---|---|---|---|---|---|---|
| a ± Sa | b ± Sb | r | Se | F | |||
| 0.02284 | 0.06852 | 0.5–10.0 | 16156.1 (± 175.33) | −2264.6 (± 902.5) | 0.9996 | 1361.5 | 8491.4 |
| Sample | Added [mg L−1] | Measured Mean ± SD (n = 3) [mg L−1] | Percentage of Recovery | RSD % |
|---|---|---|---|---|
| Canal water | 0 | 0 | - | - |
| 2 | 1.97 (± 0.07) | 98.5 | 3.55 | |
| 5 | 4.96 (± 0.22) | 99.2 | 4.43 | |
| 10 | 10.02 (± 0.46) | 100.2 | 4.59 |
| Sample | Contents Declared [mg L−1] | IAM.PC.DD2 Regis HPLC Agilent Technologies [mg L−1] Mean ± SD | Relative Bias * (%) | ICP-MS [mg L−1] Mean ± SD | Relative Bias ** (%) |
|---|---|---|---|---|---|
| 1 | 0.52 | 0.58 (± 0.02) | 11.5 | 0.41 (± 0.04) | −21.1 |
| 2 | 2.49 | 0.84 (± 0.07) | −66.2 | 0.69 (± 0.03) | −72.2 |
| 3 | 2.50 | 2.88 (± 0.03) | 15.2 | 2.72 (± 0.02) | 8.8 |
| 4 | 2.36 | 2.01 (± 0.07) | −14.8 | 2.10 (± 0.06) | −11.0 |
| 5 | 0.90 | 0.66 (± 0.08) | −26.6 | 0.57 (± 0.06) | −36.6 |
| 6 | 4.43 | 1.60 (± 0.06) | −63.8 | 1.56 (± 0.02) | −64.8 |
| Statistical Evaluation of the Iodide Peak Height [I−] = 4 ppm | H2O | HCO3− [1000 ppm] | Cl− [1000 ppm] | SO42− [100 ppm] | F− [10 ppm] | Br− [10 ppm] |
|---|---|---|---|---|---|---|
| Average peak height | 6188.50 | 6209.67 | 6263.67 | 6138.67 | 6140.33 | 5890.33 |
| SD | 254.26 | 100.01 | 59.70 | 274.97 | 109.70 | 157.02 |
| CV | 4.11 | 1.61 | 0.95 | 4.48 | 1.79 | 2.67 |
| Tolerance limit [%] | - | 0.3 | 1.2 | 0.8 | 0.7 | 4.8 |
| Statistical Evaluation of the Iodide Peak Area [I−] = 2 ppm | H2O | HCO3− [1000 ppm] | Cl− [1000 ppm] | SO42− [100 ppm] | F− [10 ppm] | Br− [10 ppm] |
|---|---|---|---|---|---|---|
| Average peak area | 232,474.7 | 231,092.7 | 233,938.9 | 230,291.6 | 230,719.1 | 225,425.5 |
| SD | 3185.09 | 3122.82 | 3182.58 | 2255.27 | 1715.77 | 4106.21 |
| CV | 1.37 | 1.35 | 1.36 | 0.98 | 0.74 | 1.82 |
| Tolerance limit [%] | 0.6 | 0.6 | 0.9 | 0.8 | 3.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tatarczak-Michalewska, M.; Flieger, J.; Kawka, J.; Flieger, W.; Blicharska, E. HPLC-DAD Determination of Iodide in Mineral Waters on Phosphatidylcholine Column. Molecules 2019, 24, 1243. https://doi.org/10.3390/molecules24071243
Tatarczak-Michalewska M, Flieger J, Kawka J, Flieger W, Blicharska E. HPLC-DAD Determination of Iodide in Mineral Waters on Phosphatidylcholine Column. Molecules. 2019; 24(7):1243. https://doi.org/10.3390/molecules24071243
Chicago/Turabian StyleTatarczak-Michalewska, Małgorzata, Jolanta Flieger, Justyna Kawka, Wojciech Flieger, and Eliza Blicharska. 2019. "HPLC-DAD Determination of Iodide in Mineral Waters on Phosphatidylcholine Column" Molecules 24, no. 7: 1243. https://doi.org/10.3390/molecules24071243
APA StyleTatarczak-Michalewska, M., Flieger, J., Kawka, J., Flieger, W., & Blicharska, E. (2019). HPLC-DAD Determination of Iodide in Mineral Waters on Phosphatidylcholine Column. Molecules, 24(7), 1243. https://doi.org/10.3390/molecules24071243