Abstract
Background: Selectively targeting dopamine receptors (DRs) has been a persistent challenge in the last years for the development of new treatments to combat the large variety of diseases involving these receptors. Although, several drugs have been successfully brought to market, the subtype-specific binding mode on a molecular basis has not been fully elucidated. Methods: Homology modeling and molecular dynamics were applied to construct robust conformational models of all dopamine receptor subtypes (D1-like and D2-like). Fifteen structurally diverse ligands were docked. Contacts at the binding pocket were fully described in order to reveal new structural findings responsible for selective binding to DR subtypes. Results: Residues of the aromatic microdomain were shown to be responsible for the majority of ligand interactions established to all DRs. Hydrophobic contacts involved a huge network of conserved and non-conserved residues between three transmembrane domains (TMs), TM2-TM3-TM7. Hydrogen bonds were mostly mediated by the serine microdomain. TM1 and TM2 residues were main contributors for the coupling of large ligands. Some amino acid groups form electrostatic interactions of particular importance for D1R-like selective ligands binding. Conclusions: This in silico approach was successful in showing known receptor-ligand interactions as well as in determining unique combinations of interactions, which will support mutagenesis studies to improve the design of subtype-specific ligands.
1. Introduction
1.1. Dopamine Receptors
The dopaminergic system has been intensively studied over the past 75 years due to the (patho)physiological role in modulating cognitive and motor behaviour [1,2]. The importance of dopamine has dramatically emerged from being just an intermediate in the formation of noradrenaline to having a celebrity status as the most important mammalian neurotransmitter [3]. Dopamine binds to five distinct dopamine receptors (DRs; D1–5 Receptor), grouped into two classes —D1-like and D2-like receptors— that differ in their physiological effects and signal transduction. The D1-like receptors, D1R and D5R, are principally coupled to Gs proteins and enhance the activity of adenylyl-cyclase, whereas D2-like receptors, D2–4R, are primarily coupled to inhibitory Gi proteins and suppress the activity of adenylyl cyclases [1,4]. The DRs belong to the G Protein-Coupled Receptor family (GPCRs), the largest and most diverse protein family in humans with approximately 800 members [5,6]. GPCRs share a conserved overall fold of seven transmembrane helices (TMs) linked by three intracellular loops (ICLs) and three extracellular loops (ECLs). Around 30–40% of all available pharmacotherapeutics target this protein family [7].
Many severe neuropsychiatric and neurodegenerative disorders such as Tourette’s syndrome, schizophrenia, Parkinson’s disease and Huntington’s disease are believed to occur as a result of imbalances and alterations in dopamine signaling [8,9,10]. Moreover, also a wide array of psychiatric disorders such as hallucinations, paranoia, bipolar disorder, gambling, alcoholism, mania, depression, eating disorders, movement and hyperactivity disorders are linked to malfunctioning dopaminergic transmission [3,11,12,13]. The discovery of chlorpromazine, the first antipsychotic drug, in the 1950s, was the hallmark of the development of a collection of antipsychotics [14] which were later reported to commonly bind to the D2R subtype (with different affinity) [4,11]. The “first-generation/classical” antipsychotics came along with significant motor side effects such as tardive dyskinesia, extrapyramidal symptoms and related conditions. These problems were not eliminated in “second-generation/atypical” antipsychotics, and others such as weight gain and the “metabolic syndrome” also appeared [11,15,16,17]. It was then later discovered that these multiple clinical and adverse effects of several antipsychotics depended on the combination of occupied receptors from other systems such as cholinergic, histaminergic and serotoninergic receptors (but always including the D2R) resulting in non-selective profiles and therefore in an insufficient explanation of the mechanism of action [11,15]. Until today, no drug has yet been identified with antipsychotic action without a significant affinity for the D2R [15]. However, antipsychotics remain a necessary first-line treatment for the management of a variety of the already mentioned psychiatric disorders (Figure 1). In fact, it is difficult to directly target one of these diseases with one specific antipsychotic, since there are also numerous cases of non-responding patients to first-line or any antipsychotic treatment or which become resistant to treatment over time [11,18].
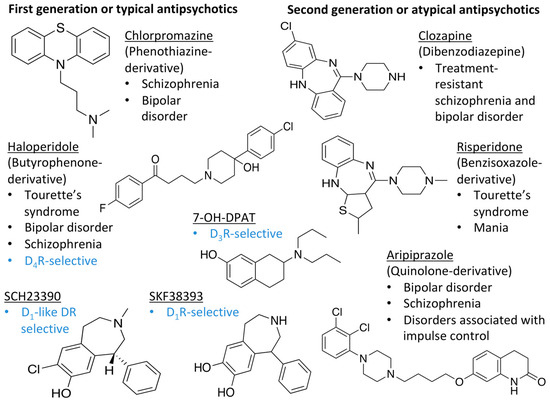
Figure 1.
Examples for first-line treatments of neurological diseases and selective dopamine receptor ligands. Drugs are classified in typical and atypical antipsychotics [19]. The targets of the selective ligands haloperidole, 7-OH-DPAT, SCH23390 and SKF38393 were colored in blue.
The search for DR subtype selective (foremost D2R-selective) therapeutics is an ongoing field of research [15] as current drugs have D2R/D3R-affinity or affinity for all DRs [19,20]. It has been proposed that substituted 4-phenylpiperazine compounds distinguish between D3R and D2R selectivity [21,22]. In addition, the aminotetraline derivative 7-OH-DPAT was identified as a selective D3R agonist [23,24], whereas it was shown that most D4R available therapeutics are not selective [22], with only one exception, haloperidole [25] (Figure 1). Also, finding D1-like DR targeting ligands, a more challenging aim [26,27], may improve antipsychotic treatment, as D1R was also shown to be relevant for modulating behaviour in health and disease (reviewed in O’Sullivan et al. [28]). So far, SKF38393 was the only selective agonist attained for the D1R, while D5R completely lacks a selective ligand [29,30]. SCH23390 was proposed as the only D1-like DR selective antagonist [31] (Figure 1).
1.2. Computer-Aided Drug Design
The strive for finding new and effective therapeutics led to a growing interest in the use of Computer-Aided Drug Design (CADD). Originally developed for high-throughput screening (HTS) of compound libraries, the use of CADD nowadays plays an important role in drug discovery [32]. The CADD pipeline can be classified into two general categories: structure-based and ligand-based, dependent on the available information about the topic of investigation [33]. A structure-based CADD is used when the target, e.g., a receptor, is known and compound libraries can be screened to find suitable structures for the target. In contrast, ligand-based CADD relies on known active and inactive compounds with their affinities in order to construct quantitative pharmacophore models and to perform virtual screening that is carried out target-independently [32]. Both CADD approaches are only fruitful if sufficient information is present. Structure-based approaches rely on the availability of the target crystal structure or homologs proteins whereas pharmacophore and other ligand-based methodologies rely on the existence of a sufficient number of ligands. For example, for GPCRs potentially involved in Parkinson’s disease, a variety of molecular docking studies were carried out using resolved crystal structures to which self-synthesized ligands were docked (reviewed in Lemos et al., [34]). Vice versa, inspection of known ligands was used to build pharmacophores or Quantitative Structure-Activity-Relationships (QSAR) to screen for new bioactive molecules (reviewed in Lemos et al., [34]). All in all, CADD is capable of addressing many challenges in hit-to-lead-development and is currently widely used in the pharmaceutical industry [34,35].
1.3. Aim
Modeling GPCRs remains problematic due to the complex structure of these membrane proteins and the lack of structural information about the desired receptor to target. However, the recent boom of resolved X-ray crystallography structures leads to a more promising application of CADD approaches to this receptor. Herein, we used tools of structure-based CADD to investigate the receptor-ligand properties of all DR-subtypes with marketed DR therapeutics. In particular, we: (i) applied homology modeling by using the resolved X-ray crystallography structures of the dopamine receptors D2R, D3R and D4R (D2R bound to the atypical antipsychotic risperidone, PDBid: 6CM4 [36]; D3R bound to D2R/D3R-selective antagonist eticlopride, PDBid: 3PBL [37]; and D4R in complex with D2R/D3R-selective antagonist nemonapride, PDBid: 5WIU [38]) in order to provide models with structural ligand-free properties; (ii) performed Molecular Dynamics (MD) of the five model structures, and (iii) performed molecular docking studies of 15 ligands targeting different conformational rearrangements’ of DR subtypes. The binding energies, number of conformations as well as the distances between ligands and receptor interacting residues of the binding pocket were calculated for all complexes. The interaction between ligands and receptors were further analyzed using an in-house pipeline that takes advantage of BINding ANAlyzer (BINANA), a Python- implemented algorithm for analyzing ligand binding [39]. BINANA was shown to successfully atomically characterize key interactions between protein amino acids and ligand atoms, and as such it is a promising approach to map such interactions in GPCRs [39]. The main goal was to reveal new structural findings to help explain the mode of binding of the selected ligands and their selectivity for certain DR-subtypes.
2. Results
2.1. Homology Models of Dopamine Receptors D1R-D5R Are Stable
The ligand-free D2-like homology models were generated using the resolved ligand-bound crystal structures of the D2R (PDBid: 6CM4) [36], D3R (PDBid: 3PBL) [37] and D4R (PDBid: 5WIU) [38] (over 90% identity). The 3D crystal structures of DRs are typically incomplete, lacking key regions for intracellular partner coupling such as intracellular loop 3 (ICL3). In contrast, D1-like DRs lack their own templates and therefore, the most suitable template to each DR was selected according to the percentage of similarity obtained upon sequence alignment by BLAST [40] in combination with ClustalOmega [41]. In fact, the D3R crystal structure was chosen as template for D1R (35.0% identity with BLAST and 39.5% with ClustalOmega), and the D4R crystal structure was chosen for D5R models (total similarity of 35.0% BLAST/39.1% ClustalOmega, check Materials and Methods section). We also calculated the similarity of the TMs in relation to the respective template and the results are summarized in Table S1. All TMs of the D2-like subtypes showed almost 100% identity with their crystal structure templates, which is also in line with the total similarity. For D1R an average TM similarity with its template was 41.0%, compared to a total similarity of 39.5%, while for D5R 36.0% TM identity was calculated compared to the total similarity of 39.1%. For the D1-like subtypes no differences between the TM similarity and the total similarity with their template were obtained. Furthermore, for D1R the highest similarity between the model and its template was observed for TM1-3, whereas for D5R it was achieved for TM2, TM3 and TM7. Consequently, TM2 and TM3 seem to be very conserved among all DR subtypes.
The combination of different metrics and scores were used to choose the most accurate models in order to perform MD and molecular docking: (i) the Discrete Optimized Protein Energy (DOPE) [42] score, MODELLER’s standard metrics, (ii) Protein Structure Analysis (ProSA-web) [43,44] and (iii) Protein Quality (ProQ)-LGscore and MaxSub score [45,46]. All final DR models (check Materials and Methods section) achieved LGscores > 4 and MaxSub scores > 0.5. The highest z-score was obtained for D4R model, whereas the lowest were counted for D1-like DR models.
MD simulations of 100 ns were briefly run for each ligand-free modelled receptor and analyzed to confirm their stability. Root-Mean-Square-Deviations (RMSD) of Cα atom mean values ranged from 0.3 nm and 0.5 nm (Figure S1). Each DR model showed good overall stability. However, D1-like models showed slightly higher RMSD values than D2-like models: D1R (0.48 ± 0.07 nm) and D5R (0.49 ± 0.06 nm) vs D2R (0.35 ± 0.09 nm), D3R (0.34 ± 0.04 nm) and D4R (0.36 ± 0.09 nm). This behavior is justified by the higher homology scores attained for the D2-like subfamily. Additionally, RMSD of Cα carbons for individual TM was computed and their average values were listed in Table S2. The low values obtained for TM’s RMSD further supports the good overall stability of the model structures.
2.2. Dopamine Receptor Binding Pocket Definition
In this work, we used the comprehensive review of Floresca and Schnetz [47], highly cited [48,49,50], as a base for the definition of the binding pocket of all DRs (Figure 2). Furthermore, by applying Ballesteros and Weinstein numbering (B&W) [51], the position of considered critical residues was more easily comparable between all receptors. This nomenclature is based on the presence of a highly conserved residue in each TMs [51], the so called X.50, in which X varies between 1 and 7 depending on the TM helix. The remaining residues were numbered according to their position relative to the most conserved one.
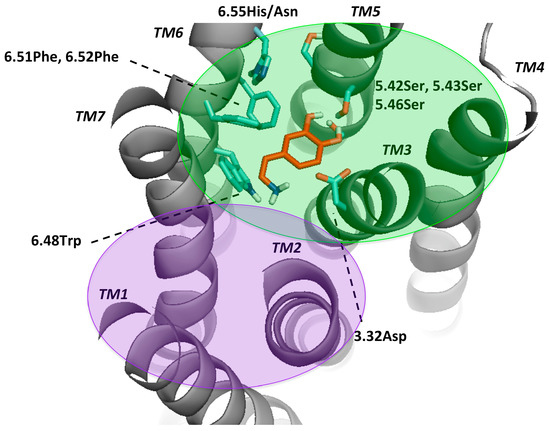
Figure 2.
Key residues for molecular docking of Dopamine to DR models. Residues were designated in bold and colored in cyan, TMs in bold and italic. This set-up was kept for Figures S3–S7 and highly corresponds to the definition of the binding pocket of Floresca and Schnetz [47]. The area of the orthosteric and secondary binding pockets were colored and violet, respectively.
Mutagenesis studies induced the believe that for dopamine binding, the endogenous agonist of the DR, a negatively charged aspartate (3.32Asp) forms an ionic bond interaction with the protonable amine of dopamine [2,50,52]. Moreover, it was shown that this effect was crucial for ligand binding and that this amino-acid was not only conserved among the DR, but also in all biogenic amine GPCRs [53,54]. Also, a serine microdomain in TM5 (5.42Ser, 5.43Ser, 5.46Ser) was considered as an important feature for dopaminergic binding in all DRs as it is believed that the serines form hydrogen bonds with the catechol hydroxyls of dopamine, increasing the binding affinity and orienting ligands in the orthosteric binding pocket [47,52,55,56,57]. While 5.42Ser seems to be critical, 5.43Ser plays a less important role [47]. A further microdomain, the aromatic microdomain, consisting of 6.48Trp, 6.51Phe, 6.52Phe and 6.55His/Asn was reported to trigger the activation of the dopamine receptor. All amino-acids in this microdomain share the same hydrophobic face in the water-assessable binding-site crevice, indicating that any reorientation of these residues by binding to a ligand would cause steric clashes and would therefore force the residues to reorient themselves in a domino-like fashion, which lastly leads to the so-called “rotamer toggle switch” [47,50,53,58]. In addition, 6.48Trp was reported together with 6.55His to stabilize the position of the ligand in the binding pocket via π-π-stacking [47,58]. Therefore, 6.48Trp and 6.55His as well as one phenylalanine (6.51Phe) were chosen for the docking protocol to mimic the ligand-binding on TM6. Dependent on the ligand properties other residues of TM3 were also considered, such as 3.33Val and 3.36Cys. 3.36Cys is believed to be part of a deeper subpocket below the orthosteric binding pocket (OBP) [36]. Additionally, Ericksen et al. reported that this cysteine was a relevant residue for benzamide binding [49]. Regarding 3.33Val, it was reported to show interaction with N-methylspiperdone by Moreira et al. [53] as well as with the methoxy ring of nemonapride, determined in the crystal structure of the D4R [38]. Different authors hypothesized that DRs have a secondary binding pocket (SBP) next to the OBP, which was confirmed by the resolved crystal structures together with computational analyses [37,38,59]. Crystal structures of D2R (PDBid: 6CM4) [36] and D3R (PDBid: 3PBL) [37] and computational data suggest that 7.43Tyr is also a crucial amino-acid for interaction in the SBP [17,36,37]. 2.57Val was shown to form a hydrophobic pocket for antagonists like clozapine and haloperidole [57]. However, since the OBP is widely explored through experimental, computational and crystal structure data, there could be other residues important in the SBP. Detailed information about the literature, mostly regarding D2-like DR can be found on Table S3. In order to compare all DRs ligand-binding properties and specificity, we focused on the mentioned residues in the OBP. The residues considered flexible in the different dockings were listed in Methods and Materials section.
2.3. Proof-of-Concept of Molecular Docking Success
Ten conformational rearrangements were chosen every 5 ns upon a 50 ns stabilization MD run. These 10 plus the initial model (time 0 ns) were then subjected to molecular docking of 15 different ligands. The results of the molecular docking were evaluated by AutoDock4.2, which ranks the possible binding positions by energy level and clusters these positions by RMSD of 2 Å. In addition, the total number of conformations (NoC) in these clusters were counted. Binding poses with more than five conformations per cluster were considered as a valid ligand position, despite the binding energy (BE) of this pose. All results of the docking can be checked in the Supplementary Information (Tables S4–S8).
As proof of concept, redocking of the co-crystalized ligands to the crystal structure templates of the D2R, D3R and D4R [36,37,38] was conducted (Figure S2, Table S9). Receptors and ligands coordinates were retrieved from PDB files. Top clusters achieved a ligand pose equivalent to the pose in the correspondent crystal, presenting very small RMSD values. Lastly, these results were compared to the docking poses of the corresponding DR-models and ligands at time point 0 ns. The binding energies of the two sets were found to fall within a similar range. This is a further evidence of docking protocol reliability.
For a general overview, dopamine docking was analyzed in detail (Figure 3A) as it is the endogenous ligand of the DRs and its binding mode is well-known compared to the other ligands [47]. However, we have to stress out the lack of a crystal structure with the dopamine-bound DR as the ligand’s structural properties are not suitable for crystallization (too small, not suitable for stabilizing a GPCR). We observed that the binding energy of D2R was the most stable at different analyzed MD conformations, while for the other subtypes it oscillated more frequently. Over time the average binding energy for all DRs was found to be at −9 kcal/mol. The highest NoC during all MD conformations were obtained for D4R and D1R (up to > 80 for D4R at 95 ns), whereas for D2R around 30 conformations were counted for all conformational arrangements (Figure 3B). Lastly, for all DRs complexed with dopamine, the first or the second cluster with the lowest binding energy also contained the highest NoC, indicating that the docking of dopamine was indeed stable and reliable (Tables S4–S8). In summary, the binding energy and 3D positions of dopamine docking may demonstrate the binding mode of dopamine to DRs. According to Floresca and Schnetz, these features are crucial for dopamine’s binding affinity and DR activation but may not necessarily be true for all dopaminergic ligands (selective and non-selective) [47].

Figure 3.
Results of the molecular docking of dopamine to all DR subtypes at all MD time steps. (A) The average binding energy of the three lowest energies of dopamine was calculated. (B) The mean of the number of conformations of the three clusters with the lowest binding energies are shown for each time point and receptor.
The binding position of dopamine to all DR complexes was stable over time namely, the protonable amine was always directed towards the aspartic acid on TM3 (3.32Asp) and the hydroxy groups were facing the serine microdomain (5.42Ser, 4.32Ser and 4.46Ser), in agreement with Floresca and Schetz [47] and Durdagi et al. [60] (Figures S3–S8). As known from the literature dopamine’s interaction with the serine microdomain only typically requires two of the serines binding to the hydroxy groups [47]. At 0 ns dopamine was located planar in the OBP in the position described above. Notably, D2R and D4R hydroxyl groups were more directed towards serine microdomain (Figure S3). At 55 ns torsions were observed for dopamine bounded to all DR, which included a switch of interactions with the serines at TM5 for D3R, since it is known that dopamine is only capable of interacting with two of the three serines [47]. At 60 ns dopamine is shifted more to the serine and aromatic microdomain (TM6) for all DRs in a different manner. However, only at D4R a strong direction of dopamine’s protonable amine towards 3.32Asp was observed. At 65 ns dopamine bounded to all DRs was located again planar in the OBP (Figure S4). Small individual torsions were observed during the period of 70–90 ns (Figures S4 and S5). Interestingly, at 95 ns dopamine was strongly involved in the aromatic microdomain (TM6) at all DR, which was then vanished especially for D3R at 100 ns. The large decrease in D4R binding energy at 90 ns can be explained, by the approximation of dopamine to 3.32Asp distance from the serine microdomain (Figure S5).
2.4. Docking of Various Ligands to DR Models
Since non-selective agonistic activity was already covered by dopamine docking, chlorpromazine was chosen as a non-selective antagonist [61,62]. Herein, we also selected the following ligands: SKF38393 as selective D1R agonist [27,30] and SCH23390 as D1-like DR antagonist [31,63], apomorphine as selective D2R agonist [60], 7-OH-DPAT as selective D3R agonist [23], nemonapride as D2R and D3R selective antagonist [64] and lastly haloperidole, due to its affinity for D4R [25]. This set of ligands was chosen as example of ligands with different DR selectivity (Table 1). The obtained binding energies and NoC in these clusters are summarized in Figure 4 (graphical output of the other ligands can be found in the appendix: Figure S7).

Table 1.
Ligands used for molecular docking and information on their function.


Figure 4.
Results of the molecular docking of 7-OH-DPAT, apomorphine, nemonapride, SCH23390, SKF38393, haloperidole and chlorpromazine for all DR subtypes at time points [ns]. The average of the three lowest binding energies of dopamine were calculated in the left plots. The mean of the number of conformations of the three clusters with the lowest binding energies were plotted for each time point and receptor (right plot).
For 7-OH-DPAT we observed a low and stable binding energy upon binding to all DRs. For apomorphine, a decrease in the binding energy was determined for D2R at 65 ns (−11 kcal/mol), whereas an increase at 85 ns was shown for D4R (−9 kcal/mol). Stable binding energies around −10 kcal/mol were observed for DR:nemonapride complexes, however a massive increase was observed for the D5R at 100 ns. For SCH23390, but not for SKF38393 the binding energy was stable over time at −9 kcal/mol for all DRs. The binding energy of SKF38393 at D2R and D4R increased at 85 ns. Haloperidole displayed the most interesting docking-profile: while the binding energies of DRs were stable at −10 kcal/mol, only for D4R a massive increase was observed at 55 ns and 80–90 ns into the positive range, meaning these binding positions were extremely unfavorable for haloperidole. Lastly, the chlorpromazine binding energy was increased only for D1R at 70 ns up to −3 kcal/mol.
Similar to dopamine binding, the NoC of 7-OH-DPAT decreased at all DRs from 0 to 65 ns. For apomorphine, the lowest binding energies were obtained for D1R and D2R. Lesser NoC were counted for nemonapride in total at all DRs (max. 30 at 85 ns for D2R). The NoC for SKF38393 were the lowest over 70–85 ns period for D1R, D2R and D3R. In contrast to the BE of haloperidole, the NoC was found to be stable over time except for D1R with up to 40 conformations at 60 ns. In addition, most conformations were counted for the D4R especially at 0–70 ns for haloperidole.
We also calculated the distance between the center of mass of the ligand and the alpha carbon of the binding pocket residues. Overall results of all ligand-residue measurements (Figure 5) showed that 3.32Asp was the closest residue for the majority of ligands. The ligand center of mass-residue alpha carbon distance was lower than 7–8 Å, particularly for D1R (<6 Å). We noted an increase in the distance between 3.32Asp and several ligands for D4R. The distances between 3.32Asp and both SKF38393 and SCH23390 ligands were larger at for D3R, D4R and D5R, but also D2R. This effect might occur due to the fact, that SCH23390 and SKF38393 are reported to be D1R-selective [30,63]. Subtype specific tendencies were observed for the serine microdomain. 5.42Ser was shown to be most distant at D1-like receptors and 5.43Ser for D2R and D3R (D2-like). These differences are less accentuated for dopamine, 7-OH-DPAT, apomorphine and bromocriptine.
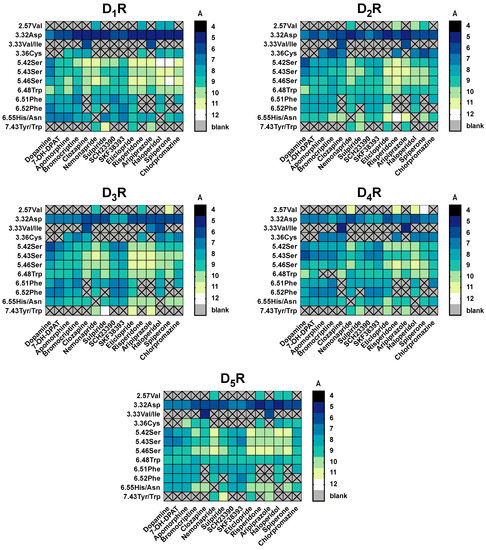
Figure 5.
Summary of the distances between ligands and residues used in molecular docking for all DR subtypes. For each ligand-residue-distance [Å], we calculated the mean of all time points of the conformational models (11) of the three best docked clusters ranked by binding energy [kcal/mol] Noteworthy is that not all ligands were set to interact with all residues shown in the x-axis in the molecular docking. (e.g., only clozapine and aripiprazole were set to interact with 3.33Val). The distances are color coded: while dark colors indicate short distances, light colors indicate wider distances.
For 7-OH-DPAT, a known D3R selective agonist, distances between ligand and the defined pocket are higher for D1-like receptors and distinctive residue between D2-like seems to be 6.52Phe, that is closer to the ligand on D3R. The same pattern was visible with apomorphine, a selective D2R agonist, where distances in D1-like are higher, although distinction within D2-like family is less pronounced. Clozapine, sulpiride and risperidone are known as “dirty drugs” because of their non-selective profile, and for that reason none of these ligands showed distinctive differences between DR subtypes. Likewise, residues 3.32Asp and 3.33Val/Ile were the closest to clozapine in all five subtypes, suggesting that these residues are crucial for this ligand’s binding. Haloperidole, categorized as D2R selective antagonist with some activity on D4R, has distinctive differences between D1-like and D2-like family, being closer to the second (although within D2-like family there is no great differences on distances pattern). Spiperone and chlorpromazine have affinity for all DR subtypes, which agrees with the lack of significant differences in the measured distances. Finally, nemonapride and eticlopride, described as D2R/D3R selective antagonists, were located closest to D2-like DR residues compared to the D1-like DR, however it seemed as these two ligands demonstrated preference for D4R.
2.5. The Type of Pairwise Interactions Between Receptor Amino-Acids and Ligand is Relevant for Binding
In-house scripts using the BINANA algorithm (software used in other non-GPCR studies [39,75,76,77,78]) were constructed to identify the type of interactions established between the ligands and binding pocket amino-acids [39]. We measured close contacts between receptor and ligands below or equal 2.5 Å and below or equal 4.0 Å, hydrogen bonds (HB), hydrophobic contacts (hydrocontacts) and salt-bridges (SB) as well as π-interactions, further subdivided into cation-π-interactions (cat-π), aromatic superpositions (π-π-stack) and perpendicular interactions of aromatic rings also referred to as edge-face-interactions (T-stack) [39]. For a first overview, all interactions despite their type and ligand were summarized and compared between the DR-subtypes (Results section at SI and Figure S8). Moreover, detailed mapping of pairwise interactions for each receptor is displayed in Figure 6. Figures S9–S23 show the change of interaction pattern over time for each ligand. Furthermore, the pairwise analysis highlighted the role of key receptor residues. By assorting those for each ligand at all DRs (time points summarized), patterns but also unique receptor-ligand interactions were highlighted (Tables S10–S14).

Figure 6.
Interaction types counted for each ligand at DR-subtypes. Data are summarized for each ligand at all time points. Total numbers of the contacts for each interaction type are color-coded: few interactions were colored white, while many interactions were colored dark. Grey cells indicate that these values are outside the scale, which was only the case for bromocriptine at the D4R with 360 four Å-interactions.
2.6. 2.5 Å-Interactions
2.5 Å-interactions, very short (closer) contacts are especially relevant for ligand binding and are described in more detail herein. For dopamine the number of these interactions increased for D1-like DRs, while for 7-OH-DPAT the highest number of interactions observed in total only occurred for D3R. For bromocriptine, 2.5 Å-interactions were significantly higher for D4R. Also, haloperidole seemed to have a higher number of established interactions with D4R as well as eticlopride. Only risperidone had a higher number of interactions with D2R. Chlorpromazine had the lowest number of compared to all ligands with no preference for any DR-subtype. All in all, 2.5 Å-interactions seemed to be particularly relevant for the ligand binding to D4R.
2.7. Hydrogen Bonds and Hydrophobic Contacts
Charge-reinforced hydrogen bonds are reported to be much stronger than the neutral hydrophobic contacts [79]. Moreover, it was reported that HBs determine the specificity of receptor-ligand binding [79]. Nevertheless, hydrophobic contacts also contribute to ligand binding, and a balance between HB and hydro contacts is required for drug-like molecules [79]. It was not surprising that a large number of hydro contacts was observed for all ligands, while HB were less common. Hydro contacts were preferably formed for D1R and achieved their lowest value for D3R. These contacts were particularly relevant for one ligand, bromocriptine (Figure 6). Moreover, a large hydrophobic network involving conserved and non-conserved residues of all TMs were found for all DRs (less pronounced for D5R). The “dirty drugs” were the second in line with the highest number of hydro contacts.
Most interesting were the HB interactions. For dopamine a different set-up was presented at each DR. While the D2-like DRs and D5R HB were formed by the serine microdomain (5.42Ser, 5.43Ser and 5.46Ser), for D1R the serine microdomain was not involved at all. 3.32Asp appeared as interaction partner for all DRs. For D5R, an HB between 5.38Tyr and dopamine was stressed out as unique for all ligands. However, 5.38Tyr was found at the D4R to form HB with 7-OH-DPAT. Not more than 2 HB were found at any DR bounded to 7-OH-DPAT. Lastly, chlorpromazine does not seem to form any HB in any DR complex.
2.8. Salt-Bridges
Most stable SB interactions were unsurprisingly achieved by dopamine for all DR sub-types. 7-OH-DPAT, SB were found for D1R (three in total), while for the other subtypes, contacts ranged between one and three over time. The same trend was observed for nemonapride and SKF38393. SCH23390 formed the highest number of SB with D5R and with D2R between 70 and 85 ns. Haloperidole seemed to establish a higher number of SB with D1-like DR and D2R, while none were formed with D3R and D4R. Spiperone seemed to preferably form SB with D1-like DRs. The following ligands did not form any salt-bridges at any time point: apomorphine, bromocriptine, clozapine, risperidone, aripiprazole and chlorpromazine.
Undoubtedly, 3.32Asp was always involved in the establishment of SB in all DRs. However, at D1R, 74Pro located on ECL1 appeared also to establish relevant SB interactions. In addition, D3R SB-bonding for spiperone was found to occur involving 1.44Leu and 75Ser (ECL1) rather than 3.32Asp. All in all, salt-bridges were found to be highly conserved regarding the residues involved.
2.9. Cat-π- and π-π-Stacking Interactions
Cationic-π and π-π-stacking are considered as natural key non-covalent interactions [80]. They are important as solitary effects, but also their interplay omnipresent in many biological systems [81]. In the DR-ligand system frequent oscillations between different receptor conformations were noted for some ligands, depicted in Figures S9–S13.
Dopamine, for example, showed the highest cat-π-interactions for D2R, oscillating from 2–4 interactions/time point. Cation-π-interactions seemed to be more relevant for D4R and were less common and mainly formed by conserved residues on TM6 (6.42Gly, 6.31Thr, 6.30Glu, 6.39Val) for D1R. Bromocriptine (3.28Trp, 6.51Phe), nemonapride (6.48Trp, 6.51Phe, 6.52Phe), sulpiride (2.61Lys, 6.48Trp, 6.51Phe) and SCH23390 (6.48Trp) showed one cat-π-contacts to D5R each. For risperidone, cat-π-interactions were mainly formed with D4R, while π-π-stacking was mostly related to D3R complexes. Aripiprazole seemed to preferably form cat-π-interactions with D4R, while increasing π-π-stacking-interactions were observed with D1R between 65 and 80 ns. Haloperidole seemed to prefer π-π-stacking-interactions with D2R, maybe important for its selectivity towards this receptor. For chlorpromazine, no cat-π-interactions were observed at D1-like DRs (D1R and D5R), while many interactions were counted with D2R between 65 and 75 ns, with D3R at 95 ns and with D4R at 60 ns.
The π-π-interactions were rather rare compared to the other interaction types. Some ligands did not form π-π-stacking interactions with DR subtypes (e.g., D1R binding to dopamine, 7-OH-PAT and sulpiride; D2R binding to sulpiride either, D4R binding to eticlopride and haloperidole; D5R binding to nemonapride). It was also obvious that the residues of the aromatic microdomain (6.48Trp, 6.51Phe, 6.52Phe, 6.55His) were responsible for the majority of ligands interactions to all DRs. However, different residue partners were determined for π-π- compared to T-stacking such as residues from TM5 (5.38Tyr, 5.47Phe). For aripiprazole, residues 7.43Tyr (D2R-D4R) and 7.34Thr (D1R) seemed also to be important for this type of interaction. Most interesting was the interaction pattern for sulpiride: while for D1R and D2R no π-π-stacking was detected, for D3R and D5R only a few residues seemed to be relevant (2.43Val, 2.44Val, 2.48Val, 38Thr, 5.38Phe, 6.51Phe, 6.52Phe for D3R; 3.28Trp and 6.48Trp for D5R) while for D4R, 27 residues from all TMs were involved in contact network formation. This may be explained by the different possible binding poses of sulpiride on the different D4R conformations.
2.10. T-Stacking Interactions
T-stacking-interactions were similar to cat-π- and π-π-interactions, yet more frequent fluctuations in the number of interacts between ligands and receptor were observed in total. Especially for risperidone, which showed the highest number of T-stacking-contacts, preferably with D2R. Haloperidole and spiperone also seemed to have a D2R-preference, while chlorpromazine formed a large number of interactions with D5R. Despite the ligand, T-stack-contacts involved mainly conserved residues (6.39Val, 6.42Gly, 6.43Val) or residues from the aromatic microdomain (6.48Trp, 6.51Phe, 6.52Phe, 6.55His). An exception was bromocriptine and sulpiride for D2R, haloperidole for D4R and spiperone for D5R. Unique interactions were found for risperidone binding to D4R with 6.44Phe and for chlorpromazine binding to D1R with 6.30Glu. However, other residues from other TMs were also involved in forming T-stack-contacts: for example, 7-OH-DPAT unique interaction with 2.47Ala and SKF38393 with 35Ala (ICL1) were found at D3R. For risperidone, a unique interaction with 231Phe (ICL3) was determined for D1R. Whereas for spiperone 1.35Tyr and 159Ile (ECL2) seemed to be relevant for D4R, 2.14Tyr was relevant for chlorpromazine coupling.
However, TM7 residues were also involved in T-stack-formation: 7.34Thr (D1R) and 7.35Tyr (D2-like)/7.35Phe(D5R), 7.43Tyr(D2-like). Residues on TM2 were also relevant for T-stack-formation (2.41Tyr, 2.43Val, 2.45Ser, 2.46Leu, 2.47Ala, 2.50Asp) but only for D3R. For D4R and D5R, only residues from TM6 and TM7 were involved in T-stack-contacts, except for SKF38393 where 5.47Phe was relevant for binding to D4R. Lastly, for D1R and D2R TM3 (3.28Trp(D1R)/3.28Phe(D2R)) residues also established meaningful interactions with nemonapride, sulpiride, SCH23390, aripiprazole and spiperone. Although these residues (especially on TM2 and TM7) are more related to the SBP than to the OBP (herein TM6 is the most relevant TM), contact formation was also observed for smaller ligands (7OH-DPAT, SCH23390, SKF38393). It was not expected that these ligands would access the SBP. Noteworthy is also the fact, that dopamine exclusively formed T-stack-contacts with the conserved aromatic microdomain for all DR. Finally, it was also obvious that the variety of T-stack-contacts was also limited by the number of aromatic rings of the ligand (e.g., dopamine only contacted 3 different sequential residues). In brief, our results also pinpoint for the fact that T-stacking-interactions seem to be relevant for large ligands, primary in antagonists binding than in agonists case.
3. Discussion
One of the major research efforts in the research of dopamine receptors is the design of DR-subtype selective ligands [82]. However, most predictive studies have been performed on D2R ligand specificity, as this receptor is the most crucial in neurotransmission [17,57,83]. Herein, we present a comprehensive in silico approach, which reveals important interactions between DRs key residues and ligands in a more detailed way when compared with available literature [55,57,59,60,84,85].
3.1. Validation of the In Silico Pipeline
Homology modeling and TM definition of all DR subtypes showed that there were smaller structural differences among the “classical” TMs (TM3, TM5, TM6), which are known to be key for ligand binding. Yet, as expected, structural differences between the subtypes were observed in the intracellular and extracellular loops, where some are important for ligand binding (ECL2) or for intracellular signaling (ICL2 and ICL3) [86]. This was particularly true for D1-like receptors, due to their much larger ICL3. Although no crystal structure was available for the D1-like DRs, the high sequence similarity among all DR helped to find suitable models for molecular docking. Validating the docking performance by low binding energies and high NoC by cluster also showed that the homology modeling-docking approach was suitable and reproducible. In fact, the combination of the different software and in-house scripting resulted in a straightforward in silico approach which can certainly be applied for studying other GPCRs. Data is also in line with experimental information, which corroborates the conceptual framework of this analysis protocol [47].
3.2. Pairwise Interactions Analysis Was Able to Determine Key Amino-Acids and Types of Interaction
A clear D2-like selectivity or binding preference was only found for apomorphine, while for others either D2R and D5R seemed to form a lower number of 4 Å-interactions such as nemonapride (D2R/D3R-antagonist [87]), SCH23390 (D1-like antagonist [88]), SKF38393 (D1R-antagonist [30]) or D1R and D4R were highly preferred (higher number of meaningful interactions). In other cases, such as for eticlopride (D2R/D3R antagonist [37]) and spiperone (D2R-antagonist [64]), the D3R was the least attractive DR for interaction. It was shown that the “classical” conserved residues e.g., 3.32Asp, the serine microdomain 5.42Ser, 5.45Ser, 5.46Ser and the aromatic microdomain 6.48Trp, 6.51Phe, 6.52Phe, 6.55His were relevant for all ligands and formed specific interactions, electrostatic (cat-π, π-π, T-stack), salt-bridges and hydrogen bonds. These residues were omnipresent in all our analyses. Yet, the distances for the most conserved OBP residues (3.32Asp, serine residues and 6.48Trp), distinct differences were observed between agonists and antagonists. For example, dopamine was constantly close to OBP, indicating its receptor activating properties as described by Floresca and Schnetz [47], while risperidone was found distant from these residues according to its antagonistic properties. This was also the case for the other antagonists such as haloperidole, nemonapride and the biased ligand aripiprazole. In addition, we described other TM residues involved in binding of these ligands, as previously described by Kalani et al. for D2R [57].
It was not surprising that the “classical” TMs, e.g., TM3, TM5, TM6 and TM7 were involved in many different interaction types. TM3 residues such as 3.35Cys, 3.36Ser, 3.33Val or 3.33Ile and 3.39Ser were often found forming different interactions with different ligands. This was also in concordance with previous studies regarding the involvement of other conserved residues on TM2 and TM7 (and TM3) [57,82,83,85], which was also described as part of a SBP only assessable for ligands with piperazine-moieties [59]. Residues on TM4 were not contributing to receptor-ligand interaction, except for D4R complexes. By comparing large ligands such as spiperone or haloperidole with rather compact ligands such as dopamine, SCH23390 or clozapine, it was possible to point out a larger number of TM1 and TM2 residues involved in establishing meaningful interactions. Author’s had already hypothesized that these residues could belong to a SBP, only accessed by large ligands [57,85]. Furthermore, there was a clear higher network contact formation with D4R. Except for that fact that the D4R is physiologically distant compared to D2R and D3R [82], no further explanation could be found for this trend.
A systematic study by De Freitas and Schapira [89] showed that the most frequent type of non-covalent interactions for protein-ligand complexes were hydrophobic contacts, followed by hydrogen bonding, π-stacking, salt-bridges, amide-stacking (corresponds to T-stack) and lastly cation-π-stacking. The same ranking of frequency of interaction type was found in our study. As also described by Davis and Teague [79] hydrophobic contacts are the most common type of receptor-ligand-interactions as they not only enhance binding affinity but also are sometimes favored over tight, charged hydrogen bonds [79]. In addition, they can be formed with different ligand-atoms such as carbons, halogens or sulphurs [89]. As reviewed in Davis and Teague [79] most docking studies fail to count in the hydrophobicity for their ligands. However, the balance between polarity (causing hydrogen bonds) and lipophicity (causing hydrophobic contacts) is the main drive to make a ligand “drug-like” [79]. Our study was successful to determine not only the hydrogen bonds but also the large hydrophobic network of each “drug-like” ligand (as well as of the marketed drugs). Hydrophobic contacts appeared to form a huge network of conserved and non-conserved residues that stabilized ligand positions during binding. This network was spread across TM2-TM3-TM7. Residues from TM1 and TM2 were shown to be relevant for binding large ligands such as nemonapride. Lastly, T-stacking interactions revealed as especially relevant for some large ligands such as apomorphine, risperidone or aripiprazole.
Conserved residues in the OBP were found to be clustered in microdomains, stabilizing ligand-binding through the formation of a HB network. Indeed, HBs where mostly mediated by the serine microdomain (5.42Ser, 5.34Ser and 5.46Ser especially at D2R and D5R). Interestingly, these residues were not relevant for D1R, although a study by Hugo et al. mentioned 5.46Ser as key residue for activating D1R [90]. In this study, 3.37Trp was also proposed to be mediator of the D1R-activation [90]. We were not able to confirm these findings in our study, only bromocriptine and spiperone were interacting 3.37Trp at D1R, while at D5R we did not observe any interaction with this residue. 3.37Thr D2R was found to interact with 7-OH-DPAT, indicating that these residues may not be D1R-specific. Salt-bridges were exclusively formed by 3.32Asp but appeared to involve also residues from ECL1 for spiperdone for D1R and D3R. For “bulky” ligands such as clozapine or bromocriptine no salt-bridges were formed.
Frontera et al. reported that the strength of cation-π-interactions is also influenced by the presence of weaker interactions such as hydrogen or hydrophobic bonds [81]. For instance, it is well known that H-bonding is highly contributing to the bond strength of π-stacking [81]. But not only weaker interactions benefit π-interactions, cat-π and π-π-stacking were also found to be cooperative for each other [81]. Such combinations where cat-π and π-π-stacking were simultaneously present, were indisputably found for D2-like rather than for D1-like DRs. In addition, these residues and those of the TM6 aromatic microdomain (6.48Trp, 6.51Phe, 6.52Phe, 6.55His/Asn) were mostly involved in forming π-interactions (cat-π, π-π or T-stack). Phenylalanine, tyrosine and tryptophan interacting ligands could indeed be further extended in order to design a new selective SAR for D5R, as they were found to be exclusively involved in π-interactions and π-stacking formation at this DR subtype. Since for the D1R-like DR SCH23390 and SKF38393 are the only known selective ligands, a closer look at the interacting residues of these ligands revealed that cat-π-interactions (6.30Glu, 6.39Val, 6.42Gly) were only present at the D1R for SCH23390, the antagonist at the D1-like DR [88]. Moreover, these residues were not the “classical” TM6 residues usually involved in binding, while this was true for the other ligands. This encourages the search for D1R- or D5R-selective ligands, which should ideally form cat-π-interactions with certain amino-acids, as they were found in this ligand set. From a structural basis SCH23390 and SKF38393 are more related to the benzodiazepines, compared to the other ligands that are either small molecules or longer ligands with piperidine moieties [91]. Lastly, another difference found between SCH23390 and SKF38393 binding to D5R were that SKF38393 established more interactions with residues from different TMs and a variety of neighboring residues of the “classical” interacting residues; whereas SCH23390-receptor-interactions were more limited to a smaller number of residues. These observations were not found for both ligands at the D1R. Reported by Bourne, who discovered SCH23390, this compound is the 3-methyl, 7-chloro analogue of the D1R agonist SKF38393, which is furthermore enantioselective [88]. In addition, it was stated that the phenyl ring in the benzodiazepine-derivatives and the receptors was involved in electrostatic forces, important for binding [88,92]. Mapping the full electrostatic potential of the D5R using ligands with benzodiazepine properties may be useful to find D5R-selective SAR.
In order to find future SARs for DRs and improve subtype selectivity, we should not only considerer the known “classical” residues and binding motifs such as the “DRY” motif, but also conserved neighboring amino acids as shown herein. For sure, this would improve the treatment with antipsychotics of many patients.
4. Materials and Methods
4.1. Homology Modeling
4.1.1. General Approach
The apo-DR models were generated with MODELLER 9.19 (version MODELLER 9.19, released Jul 25th, 2017) (University of California San Francisco, San Francisco, CA, USA) [93]. For D2-like receptors we used their corresponding crystal structures as templates: the D2R complexed with risperidone (PDBid: 6CM4) [36], the D3R complexed with D2R-antagonist eticlopride, (PDBid: 3PBL) [37] or D4R complexed with D2R/D3R-antagonist nemonapride (PDBid: 5WIU) [38]. Depending on the sequence similarity obtained with Basic Local Alignment Search Tool (BLAST, NCBI, Rockville, MD, USA) [40] and ClustalOmega (EMBL-EBI, Cambridgeshire, UK) [41] and listed in Table 2, either D3R (for the D1R) or D4R (for the D5R) were chosen as template to model the D1-like DRs. Due to the length of the ICL3, this was cut and substituted with two or four alanine residues, for D2- and D1-like receptors. Water and co-crystalized compounds were removed from the template structures. In the modeling protocol the lengths of the TMs and the perimembrane intracellular helix (HX8) were specified. In addition, disulphide bonds were constricted in the known pairs of cysteines, in particular between 3.25Cys and a non-conserved cysteine in ECL2 and between two non-conserved cysteines in the ECL3. Furthermore, loop refinement was performed for extracellular and intracellular loops for all DR using the module “loop refinement” of MODELLER 9.19. The number of models calculated with MODELLER [93] was set to 100.
Table 2.
Identity between DRs in study and their corresponding templates calculated with BLAST [40] and ClustalOmega [41].
4.1.2. Model Evaluation/Methods of Quality
There are several approaches to validate homology models such as built-in metrics of open-source [52] and licensed softwares [94]. In a preliminary study we experienced [50] that the combination of different independent metrics provided adequate models suitable for molecular docking. For instance, the combination of MODELLER’s metrics [95], ProSA-web [43,44] and ProQ [45,46] revealed to be a promising and reliable protocol to create valid models for molecular docking.
Discrete Optimized Protein Energy (DOPE) [42] scores are MODELLER’s standard metrics and were utilized in combination with visual inspection to initially remove incorrect models. DOPE is specific for a given target sequence, e.g., it accounts for the finite and spherical shape of native protein states with the lowest free energy [42]. It should be noted, that although DOPE is not an absolute measure, it helps to rank the proposed models. Then, out of a small set of potential candidates (selection of 5–10), ProSA web service [44] and the online ProQ prediction server [46] were used to determine the final models with the best combination of scores. For the z-score provided by ProSA-web analysis values around −4 are suggested as acceptable. It was only used for error recognition, as it indicates overall model quality with respect to an energy distribution derived from random conformations for globular proteins [44] The ProQ analysis (LGscore [95] and MaxSub [96]) provides absolute measures based on a neural network, which were set as base for the more detailed evaluation of the models. Regarding the LGscore, values > 3, for MaxSub values > 0.5 are typically considered as “good”. Additionally, ProQ allows to include secondary structure information calculated with PSIPRED [97], improving the prediction accuracy and increasing the model quality up to 15%. The ProQ analysis was only carried out, if z-scores around 2–4 were achieved using the ProQ protocol.
We could not compare our models with other authors as metrics scores are mostly not shown [43,98]. D1-like models, without a known crystal structure and D2-like models for which there are 3D crystal structure, showed similar quality (Table 3).
Table 3.
Metrics and scores of the final DR homology models used herein.
4.2. Molecular Dynamics
4.2.1. System Setup
It is well known that GPCRs take an infinite number of conformations over time. As such we performed MD simulations of modelled apo-forms to verify the effect of punctual fluctuations into the overall binding arrangements of ligands. Before setting up the system, the selected DR models were subjected to the Orientations of Proteins in Membranes (OPM) web-server [99,100,101,102] to calculate spatial orientations respecting to the Membrane Normal defined by the z-axis. In addition, the state of titratable residues was calculated by Propka 3.1 [103,104] within the PDB2PQR web-server [105] at a pH of 7.0. The prepared receptor structures were inserted into a previously constructed lipid bilayer of POPC: Cholesterol (9:1). Insertion of the receptors in the membrane was performed with g_membed package of GROMACS [106]. System was then solvated with explicitly represented water. Sodium and chloride ions were added to neutralize the system until it reached a total concentration of 0.15 M. The final systems dimensions were 114 × 114 × 107 Å and included approximately 370 POPC, 40 cholesterols, 125 sodium ions, 139 chloride ions and 28500 water molecules, with small variations from receptor to receptor.
4.2.2. Molecular Dynamics Simulation Protocol
CHARMM36 force field, with an implemented CMAP correction, was used for ions, water (TIP3P model), lipids and protein parameters [107]. Prior to MD simulation, the systems were relaxed to remove any possible steric clashes by a set of 50,000 steps of Steepest Descent energy minimization. Equilibration was performed afterwards as following: the system was heated using Nosé-Hoover thermostat from 0 to 310.15 K in the NVT ensemble over 100 ps with harmonic restraints of 10.0 kcal/mol. Then systems were subjected through a first step of NPT ensemble of 1 ns with semi isotropic pressure coupling and a pressure of one bar. Further equilibration was performed with sequential release of membrane lipids and protein’s atoms with a final step of NPT ensemble with harmonic restraints on the protein of 1.0 kcal/mol, for a total of 5 ns of restrained equilibration.
MD simulations of all DR models were performed with the periodic boundary condition to produce isothermical-isobaric ensembles using GROMACS 5.1.1 [106]. The Particle Mesh Ewald (PME) method [108] was used to calculate the full electrostatic energy of a unit cell in a macroscopic lattice of repeating images. Temperature was regulated using the Nosé-Hoover thermostat at 310.15 K. Pressure was regulated using the Parrinello-Rahman algorithm. The equations of motion were integrated using leapfrog algorithm with a time step of 2 fs. All bonds, involving hydrogen atoms within protein and lipid molecules were constrained using the LINear Constraint Solver (LINCS) algorithm [109]. Additionally, a cut-off distance of 12 Å was attributed for Coulombic and van der Waals interactions. Then a single independent simulation of 100 ns was initialized from the final snapshot of the restrained equilibration from each DR, for a total of 5 simulations. Trajectory analysis was performed by in-house scripting using GROMACS [106] and Visual Molecular Dynamics (VMD) [110]. Trajectory snapshots were saved every 5 ns. The snapshots after the first 50 ns MD stabilization were used for molecular docking studies.
4.3. Molecular Docking
4.3.1. Ligand Dataset
The following ligands were docked into the receptor decoys: dopamine, 7-hydroxy-N,N-dipropyl-2-aminotetralin (7-OH-DPAT), apomorphine, bromocriptine, clozapine, nemonapride, sulpiride, SCH23390, SKF38393, eticlopride, risperidone, aripiprazole, haloperidole, spiperone and chlorpromazine (Table 1). All structures were obtained from the DrugBank database (https://www.drugbank.ca) or from ChemSpider (http://www.chemspider.com) [111].
4.3.2. Docking Procedure
DR binding pocket was defined in several experimental and computational studies [2,47,52,55,57,59,85]. Herein, we used the comprehensive review by Floresca and Schetz [47] as a base for exploration of the DR binding pocket, since it contains detailed experimental data. A summary of the procedure can be better reviewed in Bueschbell et al. [50]. AutoDock4.2 (version AutoDock 4.2.6, released in 2009) was used to perform ligand docking [112]. DR hydrogens were added and Kollman united atom charges were assigned. Hydrogens were also added to the ligand and Gasteiger-Marsili was used to calculate charges. Before docking an energy, grid was created using AutoGrid (version AutoGrid 4.2.6, released 2009) with a box-size varying with the times step and ligand. For each docking simulation 100 independent Lamarckian genetic algorithm (LGA) runs were performed with the number of energy evaluations set to 10.000.000, the population size set to 200 and the maximum number of generations set to 27.000. Default settings were maintained for the rest of the parameters. Docked conformations within a RMSD of 2 Å were clustered. The most populated and lowest energy cluster (Gibbs free energy of binding) was used for conformational analysis. To find the local energy minimum of the binding site with a limited search space to that region, a low-frequency local search method was used. The 100 conformations obtained from docking were clustered by low-energy and RMSD. The top-ranked conformations within the best 3 clusters were visually inspected. The docking parameters were not changed for any ligand, only the residues treated as flexible in the docking protocol differed between the ligands. The flexible residues for each DR model are summarized in Table 4.
Table 4.
Flexible residues used in the molecular docking different ligands.
4.3.3. Analysis of Molecular Docking
In this study, 15 DR ligands were docked to the homology model and to different conformational arrangements retrieved at every 5 ns for the 55–100 ns range for each DR simulation (825 dockings in total). All distances between the center of mass of the ligand and the alpha-C-atom (Cα) of the residues, treated as flexible in the docking protocol, were calculated using in-house PyMOL scripts [2,17,47,52,57,59] as well as previously published work [50]. We also develop in-house BINANA scripts to predict the main receptor-ligand interactions [39]. BINANA is an open-source python-implemented algorithm which uses output files from AutoDock [112] for the analysis of interactions and visualizes them in the free molecular-visualization program VMD [110]. Key binding characteristics such as hydrogen bonds, hydrophobic contacts, salt-bridges and π-interactions were calculated with BINANA.
Supplementary Materials
The following are available online. Figure S1—RMSD throughout the 100 ns of simulation for all DR models; Figure S2—Redocking of ligands with their respective DR and bound ligand; Figure S3—Molecular docking of Dopamine at the D1R–D5R during 0–60 ns; Figure S4—Molecular docking of Dopamine at the D1R–D5R during 65–75 ns; Figure S5—Molecular docking of Dopamine at the D1R–D5R during 80–90 ns; Figure S6—Molecular docking of Dopamine at the D1R–D5R during 95 and 100 ns; Figure S7—Results of the molecular docking of bromocriptine, clozapine, sulpiride, eticlopride, risperidone, aripiprazole and risperidone for all DR subtypes at time points [ns]; Figure S8—Total interactions counted for each DR over time points [ns]; Figure S9—Pairwise interaction results for dopamine; Figure S10—Pairwise interaction results for 7-OH-DPAT; Figure S11—Pairwise interaction results for apomorphine; Figure S12—Pairwise interaction results for bromocriptine; Figure S13—Pairwise interaction results for clozapine; Figure S14—Pairwise interaction results for nemonapride; Figure S15—Pairwise interaction results for sulpiride; Figure S16—Pairwise interaction results for SCH23390; Figure S17—Pairwise interaction results for SKF38393; Figure S18—Pairwise interaction results for eticlopride; Figure S19—Pairwise interaction results for risperidone; Figure S20—Pairwise interaction results for aripiprazole; Figure S21—Pairwise interaction results for haloperidole; Figure S22—Pairwise interaction results for spiperone; Figure S23—Pairwise interaction results for chlorpromazine. Table S1—Comparison between the total and transmembrane specific identity [%] of the DR model with their crystal structure templates calculated with Clustal Omega; Table S2—Averages RMSD values for TM throughout the simulations; Table S3—Summary of the structures used in literature for defining the binding pocket for the D2R and source (experimental and computational); Table S4—Docking results for the D1R; Table S5—Docking results for the D2R; Table S6—Docking results for the D3R; Table S7—Docking results for the D4R; Table S8—Docking results for the D5R; Table S9—Docking results for the crystal structure templates of D2R (PDBid: 6CM4), D3R (PDBid: 3PBL) and D4R (PDBid: 5WIU) docked with their co-crystalized ligands; Table S10—D1R residues with Ballesteros & Weinstein-numbering participating in different interaction types sorted by ligands; Table S11—D2R residues with Ballesteros & Weinstein-numbering participating in different interaction types sorted by ligands; Table S12—D3R residues with Ballesteros & Weinstein-numbering participating in different interaction types sorted by ligands; Table S13—D4R residues with Ballesteros & Weinstein-numbering participating in different interaction types sorted by ligands; Table S14—D5R residues with Ballesteros & Weinstein-numbering participating in different interaction types sorted by ligands.
Author Contributions
B.B., A.J.P. and C.A.V.B. performed the experiments; B.B, A.J.P. and C.A.V.B. analyzed the data; B.B., A.C.S and I.S.M conceived and designed the experiments; all authors wrote the paper.
Funding
B.B. and A.C.S. were supported by the German Federal Ministry of Education and Research (BMBF project) of the Bonn International Graduate School in Drug Sciences (BIGS DrugS). Irina S. Moreira acknowledges support by the Fundação para a Ciência e a Tecnologia (FCT) Investigator programme - IF/00578/2014 (co-financed by European Social Fund and Programa Operacional Potencial Humano). This work was also financed by the European Regional Development Fund (ERDF), through the Centro 2020 Regional Operational Programme under project CENTRO-01-0145-FEDER-000008: BrainHealth 2020. We also acknowledge the grant PTDC/QUI-OUT/32243/2017 financed by national funds through the FCT / MCTES and/or State Budget.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Beaulieu, J.-M.; Gainetdinov, R.R. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef]
- Platania, C.B.M.; Salomone, S.; Leggio, G.M.; Drago, F.; Bucolo, C. Homology Modeling of Dopamine D2 and D3 Receptors: Molecular Dynamics Refinement and Docking Evaluation. PLoS ONE 2012, 7, e44316. [Google Scholar] [CrossRef]
- Marsden, C.A. Dopamine: The rewarding years. Br. J. Pharmacol. 2006, 147 (Suppl. 1), 136–144. [Google Scholar] [CrossRef]
- Leggio, G.M.; Bucolo, C.; Platania, C.B.M.; Salomone, S.; Drago, F. Current drug treatments targeting dopamine D3 receptor. Pharmacol. Ther. 2016, 165, 164–177. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The structure and function of G protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef]
- Maurice, P.; Guillaume, J.L.; Benleulmi-Chaachoua, A.; Daulat, A.M.; Kamal, M.; Jockers, R. GPCR-Interacting Proteins, Major Players of GPCR Function. In GPCR—Interacting Proteins, Major Players of GPCR Function, 1st ed.; Elsevier Inc.: New York, NY, USA, 2011; Volume 62. [Google Scholar]
- Thimm, D.; Funke, M.; Meyer, A.; Müller, C.E. 6-Bromo-8-(4-methoxybenzamido)-4-oxo-4 H-chromene-2-carboxylic Acid: A powerful tool for studying orphan G protein-coupled receptor GPR35. J. Med. Chem. 2013, 56, 7084–7099. [Google Scholar] [CrossRef]
- Jaber, M.; Robinson, S.W.; Missale, C.; Caron, M.G. Dopamine receptors and brain function. Neuropharmacology 1997, 35, 1503–1519. [Google Scholar] [CrossRef]
- Seeman, P. Atypical Antipsychotics: Mechanism of Action. Focus 2002, 47, 27–38. [Google Scholar] [CrossRef]
- Rangel-Barajas, C.; Coronel, I.; Florán, B. Dopamine Receptors and Neurodegeneration. Aging Dis. 2015, 6, 349. [Google Scholar] [CrossRef]
- Amato, D.; Vernon, A.C.; Papaleo, F. Dopamine, the antipsychotic molecule: A perspective on mechanisms underlying antipsychotic response variability. Neurosci. Biobehav. Rev. 2017, 85, 146–159. [Google Scholar] [CrossRef]
- Noble, E.P. D2 dopamine receptor gene in psychiatric and neurologic disorders and its phenotypes. Am. J. Med. Genet. 2003, 116B, 103–125. [Google Scholar] [CrossRef]
- Zhang, A.; Neumeyer, J.L.; Baldessarini, R.J. Recent progress in development of dopamine receptor subtype-selective agents: Potential therapeutics for neurological and psychiatric disorders. Chem. Rev. 2007, 107, 274–302. [Google Scholar] [CrossRef]
- Miller, R. Mechanisms of action of antipsychotic drugs of different classes, refractoriness to therapeutic effects of classical neuroleptics, and individual variation in sensitivity to their actions: Part II. Curr. Neuropharmacol. 2009, 7, 315–330. [Google Scholar] [CrossRef]
- Mauri, M.C.; Paletta, S.; Maffini, M.; Colasanti, A.; Dragogna, F.; Di Pace, C.; Altamura, A.C. Clinical pharmacology of atypical antipsychotics: An update. EXCLI J. 2014, 13, 1163–1191. [Google Scholar]
- Sykes, D.A.; Moore, H.; Stott, L.; Holliday, N.; Javitch, J.A.; Lane, J.R.; Charlton, S.J. Extrapyramidal side effects of antipsychotics are linked to their association kinetics at dopamine D2 receptors. Nat. Commun. 2017, 8, 763. [Google Scholar] [CrossRef]
- Salmas, R.; Serhat Is, Y.; Durdagi, S.; Stein, M.; Yurtsever, M. A QM protein–ligand investigation of antipsychotic drugs with the dopamine D2 Receptor (D2R). J. Biomol. Struct. Dyn. 2018, 36, 2668–2677. [Google Scholar] [CrossRef]
- Loebel, A.; Citrome, L.; Correll, C.U.; Xu, J.; Cucchiaro, J.; Kane, J.M. Treatment of early non-response in patients with schizophrenia: Assessing the efficacy of antipsychotic dose escalation. BMC Psychiatry 2015, 15, 1–7. [Google Scholar] [CrossRef]
- Behere, B.P.; Das, A.; Behere, A.P. Antipsychotics. In Clinical Psychopharmacology; Springer: Singapore, 2019; pp. 39–87. [Google Scholar]
- Moritz, A.E.; Benjamin Free, R.; Sibley, D.R. Advances and challenges in the search for D2 and D3 dopamine receptor-selective compounds. Cell. Signal. 2018, 41, 75–81. [Google Scholar] [CrossRef]
- Banala, A.K.; Levy, B.A.; Khatri, S.S.; Furman, C.A.; Roof, R.A.; Mishra, Y.; Gri, S.A.; Sibley, D.R.; Luedtke, R.R.; Newman, A.H. N-(3-Fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)arylcarboxamides as selective dopamine D3 receptor ligands: Critical role of the carboxamide linker for D3 recetpor selectivity. J. Med. Chem. 2011, 54, 3581–3594. [Google Scholar] [CrossRef]
- Newman, A.H.; Beuming, T.; Banala, A.K.; Donthamsetti, P.; Pongetti, K.; LaBounty, A.; Levy, B.A.; Cao, J.; Michino, M.; Luedtke, R.R.; et al. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J. Med. Chem. 2012, 55, 6689–6699. [Google Scholar] [CrossRef]
- Damsma, G.; Bottema, T.; Westerink, B.H.C.; Tepper, P.G.; Dijkstra, D.; Pugsley, T.A.; MacKenzie, R.G.; Heffner, T.G.; Wikström, H. Pharmacological aspects of R-(+)-7-OH-DPAT, a putative dopamine D3 receptor ligand. Eur. J. Pharmacol. 1993, 249, 9–10. [Google Scholar] [CrossRef]
- Lévesque, D.; Diaz, J.; Pilon, C.; Martres, M.P.; Giros, B.; Souil, E.; Schott, D.; Morgat, J.L.; Schwartz, J.C.; Sokoloff, P. Identification, characterization, and localization of the dopamine D3 receptor in rat brain using 7-[3H]hydroxy-N,N-di-n-propyl-2-aminotetralin. Proc. Natl. Acad. Sci. USA 1992, 89, 8155–8159. [Google Scholar] [CrossRef]
- Sampson, D.; Zhu, X.Y.; Eyunni, S.V.K.; Etukala, J.R.; Ofori, E.; Bricker, B.; Lamango, N.S.; Setola, V.; Roth, B.L.; Ablordeppey, S.Y. Identification of a new selective dopamine D4receptor ligand. Bioorg. Med. Chem. 2014, 22, 3105–3114. [Google Scholar] [CrossRef]
- Zhang, J.; Xiong, B.; Zhen, X.; Zhang, A. Dopamine D1 receptor ligands: Where are we now and where are we going. Med. Res. Rev. 2009, 29, 272–294. [Google Scholar] [CrossRef]
- Conroy, J.L.; Free, R.B.; Sibley, D.R. Identification of G Protein-Biased Agonists That Fail to Recruit β-Arrestin or Promote Internalization of the D1 Dopamine Receptor. ACS Chem. Neurosci. 2015, 6, 681–692. [Google Scholar] [CrossRef]
- O’Sullivan, G.J.; Roth, B.L.; Kinsella, A.; Waddington, J.L. SK&F 83822 distinguishes adenylyl cyclase from phospholipase C-coupled dopamine D1-like receptors: Behavioural topography. Eur. J. Pharmacol. 2004, 486, 273–280. [Google Scholar]
- Butini, S.; Nikolic, K.; Kassel, S.; Brückmann, H.; Filipic, S.; Agbaba, D.; Gemma, S.; Brogi, S.; Brindisi, M.; Campiani, G.; et al. Polypharmacology of dopamine receptor ligands. Prog. Neurobiol. 2016, 142, 68–103. [Google Scholar] [CrossRef]
- Lee, S.M.; Kant, A.; Blake, D.; Murthy, V.; Boyd, K.; Wyrick, S.J.; Mailman, R.B. SKF-83959 is not a highly-biased functionally selective D1dopamine receptor ligand with activity at phospholipase C. Neuropharmacology 2014, 86, 145–154. [Google Scholar] [CrossRef]
- Arimitsu, E.; Ogasawara, T.; Takeda, H.; Sawasaki, T.; Ikeda, Y.; Hiasa, Y.; Maeyama, K. The ligand binding ability of dopamine D1 receptors synthesized using a wheat germ cell-free protein synthesis system with liposomes. Eur. J. Pharmacol. 2014, 745, 117–122. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Comput. Methods Drug Discov. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Jain, A. Computer Aided Drug Design & QSAR. J. Phys. Conf. Ser. 2017, 884, 012072. [Google Scholar]
- Lemos, A.; Melo, R.; Preto, A.J.; Almeida, J.G.; Moreira, I.S.; Natália, M.D.S. In silico studies targeting G-protein coupled receptors for drug research against Parkinson’s disease. Curr. Neuropharmacol. 2018, 16, 786–848. [Google Scholar] [CrossRef]
- Shin, W.H.; Christoffer, C.W.; Kihara, D. In silico structure-based approaches to discover protein-protein interaction-targeting drugs. Methods 2017, 131, 22–32. [Google Scholar] [CrossRef]
- Wang, S.; Che, T.; Levit, A.; Shoichet, B.K.; Wacker, D.; Roth, B.L. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269. [Google Scholar] [CrossRef]
- Chien, E.Y.T.; Liu, W.; Zhao, Q.; Katritch, V.; Won Han, G.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the Human Dopamine D3 Receptor in Complex with a D2/D3 Selective Antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef]
- Wang, S.; Wacker, D.; Levit, A.; Che, T.; Betz, R.M.; Mccorvy, J.D.; Venkatakrishnan, A.J.; Huang, X.-P.; Dror, R.O.; Shoichet, B.K.; et al. D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 2017, 358, 381–386. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. BINANA: A novel algorithm for ligand-binding characterization. J. Mol. Graph. Model. 2011, 29, 888–893. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Shen, M.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef]
- Hou, J.; Charron, C.L.; Fowkes, M.M.; Luyt, L.G. Bridging computational modeling with amino acid replacements to investigate GHS-R1a-peptidomimetic recognition. Eur. J. Med. Chem. 2016, 123, 822–833. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, 407–410. [Google Scholar] [CrossRef]
- Wallner, B.; Elofsson, A. Can correct protein models be identified? Protein Sci. 2003, 12, 1073–1086. [Google Scholar] [CrossRef]
- Wallner, B.; Elofsson, A. Identification of correct regions in protein models using structural, alignment, and consensus information. Protein Sci. 2006, 15, 900–913. [Google Scholar] [CrossRef]
- Floresca, C.Z.; Schetz, J.A. Dopamine Receptor Microdomains Involved in Molecular Recognition and the Regulation of Drug Affinity and Function. J. Recept. Signal Transduct. 2004, 24, 207–239. [Google Scholar] [CrossRef]
- Cummings, D.F.; Ericksen, S.S.; Goetz, A.; Schetz, J.A. Transmembrane Segment Five Serines of the D4 Dopamine Receptor Uniquely Influence the Interactions of Dopamine, Norepinephrine, and Ro10-4548. J. Pharmacol. Exp. Ther. 2010, 333, 682–695. [Google Scholar] [CrossRef]
- Ericksen, S.S.; Cummings, D.F.; Teer, M.E.; Amdani, S.; Schetz, J.A. Ring Substituents on Substituted Benzamide Ligands Indirectly Mediate Interactions with Position 7.39 of Transmembrane Helix 7 of the D4 Dopamine Receptor. J. Pharmacol. Exp. Ther. 2012, 342, 472–485. [Google Scholar] [CrossRef]
- Bueschbell, B.; Preto, A.J.; Barreto, C.A.V.; Schiedel, A.C.; Moreira, I.S. Creating a valid in silico Dopamine D2-receptor model for small molecular docking studies. In MOL2NET, International Conference Series on Multidisciplinary Sciences; SCIFORUM: Basel, Switzerland, 2017; Volume 3, pp. 1–6. [Google Scholar]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Salmas, R.E.; Yurtsever, M.; Stein, M.; Durdagi, S. Modeling and protein engineering studies of active and inactive states of human dopamine D2 receptor (D2R) and investigation of drug/receptor interactions. Mol. Divers. 2015, 19, 321–332. [Google Scholar] [CrossRef]
- Moreira, I.S.; Shi, L.; Freyberg, Z.; Ericksen, S.S.; Weinstein, H.; Javitch, J.A. Structural Basis of Dopamine Receptor Activation. In The Dopamine Receptors; Neve, K.A., Ed.; Humana/Springer: Berlin, Germany, 2010; pp. 47–73. [Google Scholar]
- Huang, E.S. Construction of a sequence motif characteristic of aminergic G protein-coupled receptors. Protein Sci. 2003, 12, 1360–1367. [Google Scholar] [CrossRef]
- Tschammer, N.; Dörfler, M.; Hübner, H.; Gmeiner, P. Engineering a GPCR-ligand pair that simulates the activation of D 2L by dopamine. ACS Chem. Neurosci. 2010, 1, 25–35. [Google Scholar] [CrossRef]
- Kling, R.C.; Tschammer, N.; Lanig, H.; Clark, T.; Gmeiner, P. Active-state model of a dopamine D2 receptor—Galpha-i complex stabilized by aripiprazole-type partial agonists. PLoS ONE 2014, 9, e100069. [Google Scholar] [CrossRef]
- Kalani, M.Y.S.; Vaidehi, N.; Hall, S.E.; Trabanino, R.J.; Freddolino, P.L.; Kalani, M.A.; Floriano, W.B.; Kam, V.W.T.; Goddard, W.A. The predicted 3D structure of the human D2 dopamine receptor and the binding site and binding affinities for agonists and antagonists. Proc. Natl. Acad. Sci. USA 2004, 101, 3815–3820. [Google Scholar] [CrossRef]
- Holst, B.; Nygaard, R.; Valentin-Hansen, L.; Bach, A.; Engelstoft, M.S.; Petersen, P.S.; Frimurer, T.M.; Schwartz, T.W. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J. Biol. Chem. 2010, 285, 3973–3985. [Google Scholar] [CrossRef]
- Männel, B.; Jaiteh, M.; Zeifman, A.; Randakova, A.; Möller, D.; Hübner, H.; Gmeiner, P.; Carlsson, J. Structure-guided screening for functionally selective D2 dopamine receptor ligands from a virtual chemical library. ACS Chem. Biol. 2017, 12, 2652–2661. [Google Scholar] [CrossRef] [PubMed]
- Durdagi, S.; Salmas, R.E.; Stein, M.; Yurtsever, M.; Seeman, P. Binding Interactions of Dopamine and Apomorphine in D2High and D2Low States of Human Dopamine D2 Receptor Using Computational and Experimental Techniques. ACS Chem. Neurosci. 2016, 7, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Boyd, K.N.; Mailman, R.B. Dopamine receptor signaling and cirrent and future antipyschotic drugs. Handb. Exp. Pharmacol. 2012, 212, 53–86. [Google Scholar]
- Bergman, J.; Madras, B.K.; Spealman, R.D. Behavioral effects of D1 and D2 dopamine receptor antagonists in squirrel monkeys. J. Pharmacol. Exp. Ther. 1991, 258, 910–917. [Google Scholar] [PubMed]
- Chen, T.; Hu, Y.; Lin, X.; Huang, X.; Liu, B.; Leung, P.; Chan, S.O.; Guo, D.; Jin, G. Dopamine signaling regulates the projection patterns in the mouse chiasm. Brain Res. 2015, 1625, 324–336. [Google Scholar] [CrossRef]
- Hidaka, K.; Matsumoto, M.; Tada, S.; Tasaki, Y.; Yamaguchi, T. Differential effects of [3H]nemonapride and [3H]spiperone binding on human dopamine D4 receptors. Neurosci. Lett. 1995, 186, 145–148. [Google Scholar] [CrossRef]
- Seeman, P.; Van Tol, H.H. Dopamine receptor pharmacology. Trends Pharmacol. Sci. 1994, 15, 264–270. [Google Scholar] [CrossRef]
- Lawler, C.P.; Prioleau, C.; Lewis, M.M.; Mak, C.; Jiang, D.; Schetz, J.A.; Gonzalez, A.M.; Sibley, D.R.; Mailman, R.B. Interactions of the novel antipsychotic aripiprazole (OPC-14597) with dopamine and serotonin receptor subtypes. Neuropsychopharmacology 1999, 20, 612–627. [Google Scholar] [CrossRef]
- Lindsley, C.W.; Hopkins, C.R. Return of D4 Dopamine Receptor Antagonists in Drug Discovery. J. Med. Chem. 2017, 60, 7233–7243. [Google Scholar] [CrossRef]
- Newton, C.L.; Wood, M.D.; Strange, P.G. Examining the effects of sodium ions on the binding of antagonists to dopamine D2 and D3 receptors. PLoS ONE 2016, 11, e0158808. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, X.; Tiberi, M. Functional importance of two conserved residues in intracellular loop 1 and transmembrane region 2 of Family A GPCRs: Insights from ligand binding and signal transduction responses of D1 and D5 dopaminergic receptor mutants. Cell. Signal. 2015, 27, 2014–2025. [Google Scholar] [CrossRef]
- Andringa, G.; Drukarch, B.; Leysen, J.E.; Cools, A.R.; Stoof, J.C. The alleged dopamine D1 receptor agonist SKF 83959 is a dopamine D1 receptor antagonist in primate cells and interacts with other receptors. Eur. J. Pharmacol. 1999, 364, 33–41. [Google Scholar] [CrossRef]
- Burris, K.D.; Molski, T.F.; Xu, C.; Ryan, E.; Tottori, K.; Kikuchi, T.; Yocca, F.D.; Molinoff, P.B. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J. Pharmacol. Exp. Ther. 2002, 302, 381–389. [Google Scholar] [CrossRef]
- López-Muñoz, F.; Alamo, C. The consolidation of neuroleptic therapy: Janssen, the discovery of haloperidol and its introduction into clinical practice. Brain Res. Bull. 2009, 79, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Madras, B.K. History of the discovery of the antipsychotic dopamine D2 receptor: A basis for the dopamine hypothesis of schizophrenia. J. Hist. Neurosci. 2013, 22, 62–78. [Google Scholar] [CrossRef]
- Abhijnhan, A.; Adams, C.E.; David, A.; Ozbilen, M. Depot fluspirilene for schizophrenia. Cochrane Database Syst. Rev. 2007, 1, 1–45. [Google Scholar] [CrossRef] [PubMed]
- Valizade Hasanloei, M.A.; Sheikhpour, R.; Sarram, M.A.; Sheikhpour, E.; Sharifi, H. A combined Fisher and Laplacian score for feature selection in QSAR based drug design using compounds with known and unknown activities. J. Comput. Aided Mol. Des. 2018, 32, 375–384. [Google Scholar] [CrossRef]
- Kumar, S.P. PLHINT: A knowledge-driven computational approach based on the intermolecular H bond interactions at the protein-ligand interface from docking solutions. J. Mol. Graph. Model. 2018, 79, 194–212. [Google Scholar] [CrossRef]
- Trisciuzzi, D.; Nicolotti, O.; Miteva, M.A.; Villoutreix, B.O. Analysis of solvent-exposed and buried co-crystallized ligands: A case study to support the design of novel protein–protein interaction inhibitors. Drug Discov. Today 2018, 24, 551–559. [Google Scholar] [CrossRef]
- Tanina, A.; Wohlkönig, A.; Soror, S.H.; Flipo, M.; Villemagne, B.; Prevet, H.; Déprez, B.; Moune, M.; Perée, H.; Meyer, F.; et al. A comprehensive analysis of the protein-ligand interactions in crystal structures of Mycobacterium tuberculosis EthR. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 248–258. [Google Scholar] [CrossRef]
- Davis, A.M.; Teague, S.J. Hydrogen bonding, hydrophobic interactions, and failure of the rigid receptor hypothesis. Angew. Chem. Int. Ed. 1999, 38, 736–749. [Google Scholar] [CrossRef]
- Bosch, E.; Barnes, C.L.; Brennan, N.L.; Eakins, G.L.; Breyfogle, B.E. Cation-Induced π-stacking. J. Org. Chem. 2008, 73, 3931–3934. [Google Scholar] [CrossRef]
- Frontera, A.; Quiñonero, D.; Deyà, P.M. Cation-π and anion-π interactions. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 440–459. [Google Scholar] [CrossRef]
- Wang, Q.; MacH, R.H.; Luedtke, R.R.; Reichert, D.E. Subtype selectivity of dopamine receptor ligands: Insights from structure and ligand-based methods. J. Chem. Inf. Model. 2010, 50, 1970–1985. [Google Scholar] [CrossRef]
- Simpson, M.M.; Ballesteros, J.A.; Chiappa, V.; Chen, J.; Suehiro, M.; Hartman, D.S.; Godel, T.; Snyder, L.A.; Sakmar, T.P.; Javitch, J.A. Dopamine D4/D2 receptor selectivity is determined by A divergent aromatic microdomain contained within the second, third, and seventh membrane-spanning segments. Mol. Pharmacol. 1999, 56, 1116–1126. [Google Scholar] [CrossRef]
- Salmas, R.E.; Yurtsever, M.; Durdagi, S. Atomistic molecular dynamics simulations of typical and atypical antipsychotic drugs at the dopamine D2 receptor (D2R) elucidates their inhibition mechanism. J. Biomol. Struct. Dyn. 2016, 35, 1–17. [Google Scholar] [CrossRef]
- Sukalovic, V.; Soskic, V.; Sencanski, M.; Andric, D.; Kostic-Rajacic, S. Determination of key receptor-ligand interactions of dopaminergic arylpiperazines and the dopamine D2 receptor homology model. J. Mol. Model. 2013, 19, 1751–1762. [Google Scholar] [CrossRef]
- Moreira, I.S. Structural features of the G-protein/GPCR interactions. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 16–33. [Google Scholar] [CrossRef]
- Scarselli, M.; Novi, F.; Schallmach, E.; Lin, R.; Baragli, A.; Colzi, A.; Griffon, N.; Corsini, G.U.; Sokoloff, P.; Levenson, R.; et al. D2/D3 Dopamine Receptor Heterodimers Exhibit Unique Functional Properties. J. Biol. Chem. 2001, 276, 30308–30314. [Google Scholar] [CrossRef]
- Bourne, J.A. SCH23390: The First Selective Dopamine D1-Like Receptor Antagonist. CNS Drug Rev. 2006, 7, 399–414. [Google Scholar] [CrossRef]
- Ferreira De Freitas, R.; Schapira, M. A systematic analysis of atomic protein-ligand interactions in the PDB. Medchemcomm 2017, 8, 1970–1981. [Google Scholar] [CrossRef]
- Hugo, E.A.; Cassels, B.K.; Fierro, A. Functional roles of T3.37 and S5.46 in the activation mechanism of the dopamine D1 receptor. J. Mol. Model. 2017, 23, 142. [Google Scholar] [CrossRef]
- Zarrindast, M.R.; Honardar, Z.; Sanea, F.; Owji, A.A. SKF 38393 and SCH 23390 inhibit reuptake of serotonin by rat hypothalamic synaptosomes. Pharmacology 2011, 87, 85–89. [Google Scholar] [CrossRef]
- Pettersson, I.; Gundertofte, K.; Palm, J.; Liljefors, T. A Study on the Contribution of the 1-Phenyl Substituent to the Molecular Electrostatic Potentials of Some Benzazepines in Relation to Selective Dopamine D-1 Receptor Activity. J. Med. Chem. 1992, 35, 502–507. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A.; Francisco, S. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinforma 2017, 54, 1–55. [Google Scholar]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Hege, S.B.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Cristobal, S.; Zemla, A.; Fischer, D.; Rychlewski, L.; Elofsson, A. A study of quality measures for protein threading models. BMC Bioinform. 2001, 2, 5. [Google Scholar] [CrossRef]
- Siew, N.; Elofsson, A.; Rychlewski, L.; Fischer, D. MaxSub: An automated measure for the assessment of protein structure prediction quality. Bioinformatics 2000, 16, 776–785. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Gaete-Eastman, C.; Morales-Quintana, L.; Herrera, R.; Moya-León, M.A. In-silico analysis of the structure and binding site features of an α-expansin protein from mountain papaya fruit (VpEXPA2), through molecular modeling, docking, and dynamics simulation studies. J. Mol. Model. 2015, 21, 1–12. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, 370–376. [Google Scholar] [CrossRef]
- Lomize, A.L.; Pogozheva, I.D.; Lomize, M.A.; Mosberg, H.I. Positioning of proteins in membranes: A computational approach. Protein Sci. 2006, 15, 1318–1333. [Google Scholar] [CrossRef]
- Lomize, A.L.; Pogozheva, I.D.; Lomize, M.A.; Mosberg, H.I. The role of hydrophobic interactions in positioning of peripheral proteins in membranes. BMC Struct. Biol. 2007, 7, 1–30. [Google Scholar] [CrossRef]
- Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. Anisotropic solvent model of the lipid bilayer. 2. Energetics of insertion of small molecules, peptides and proteins in membranes Andrei. J. Chem. Inf. Model. 2011, 51, 930–946. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, 665–667. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N*log(N) method for Ewald sums in large systems Tom. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schultern, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open chemical toolbox. J. Cheminform. 2011, 3, 1–14. [Google Scholar] [CrossRef]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).