CtACO1 Overexpression Resulted in the Alteration of the Flavonoids Profile of Safflower

Abstract
:1. Introduction
2. Results
2.1. Discovery and Characterization of CtACOs from the Transcription Profiling Database of Safflower
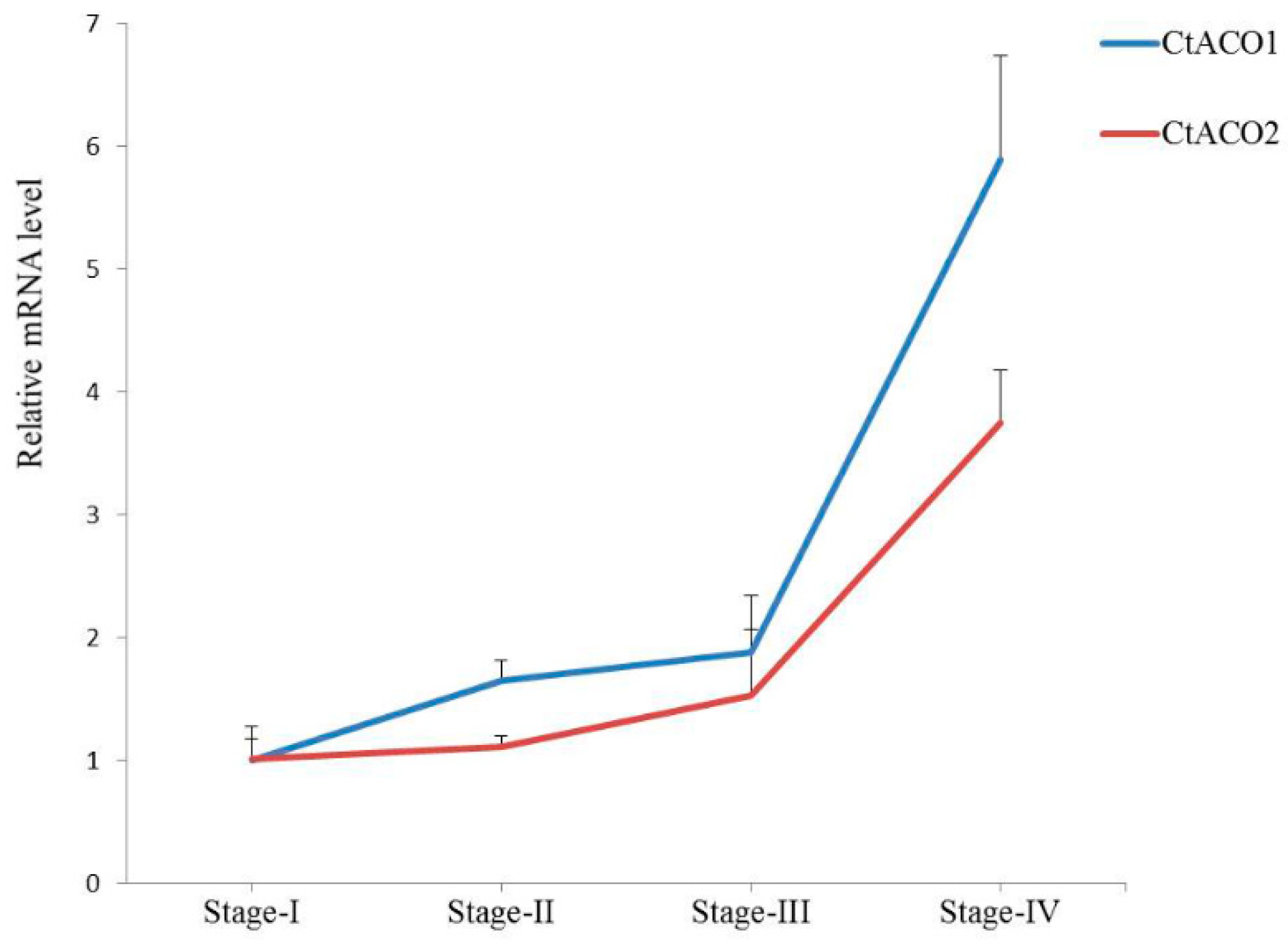
2.2. Expression of CtACO1 and CtACO2 in Flowers at Different Flowering Times
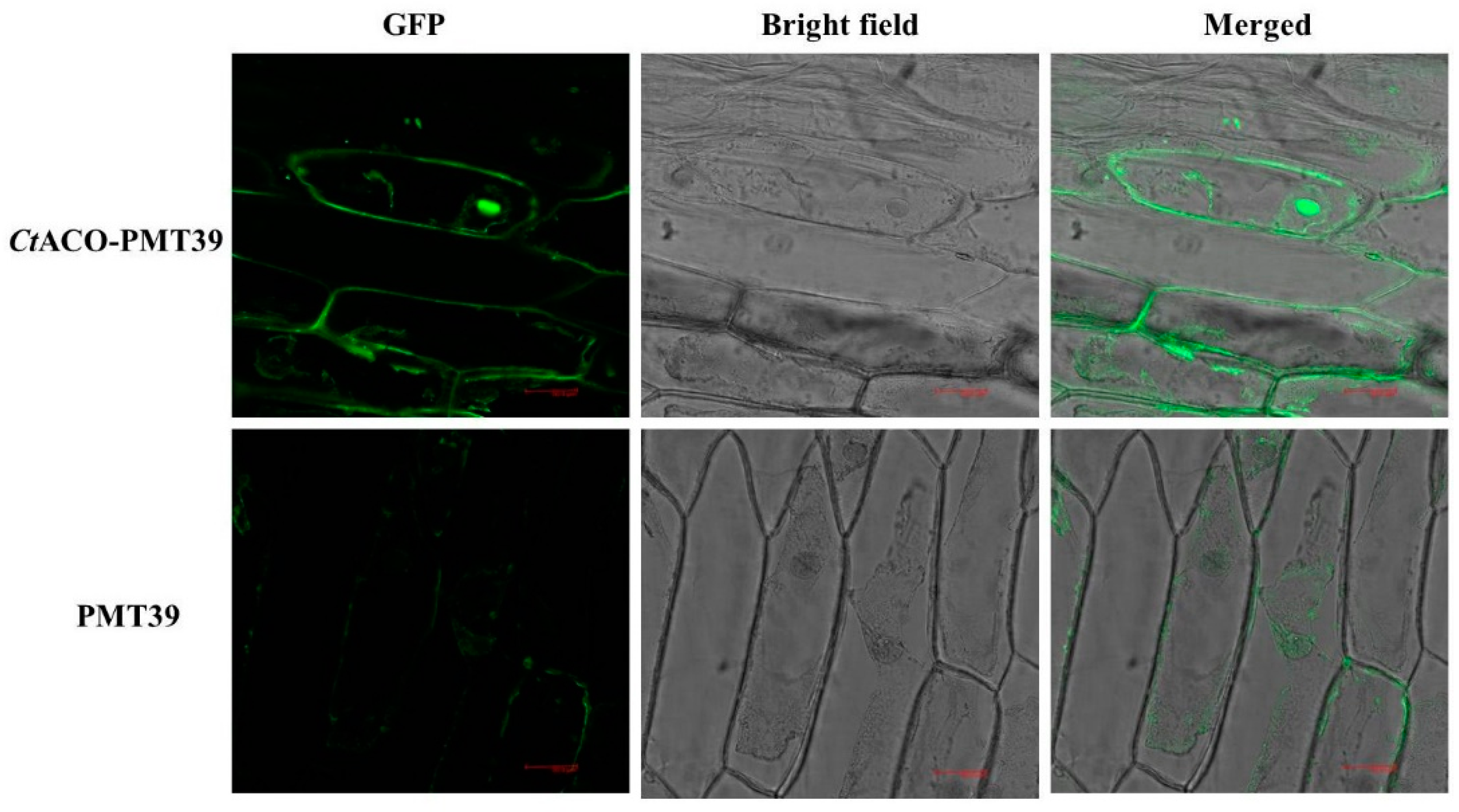
2.3. Subcellular Localization of CtACO1 Protein
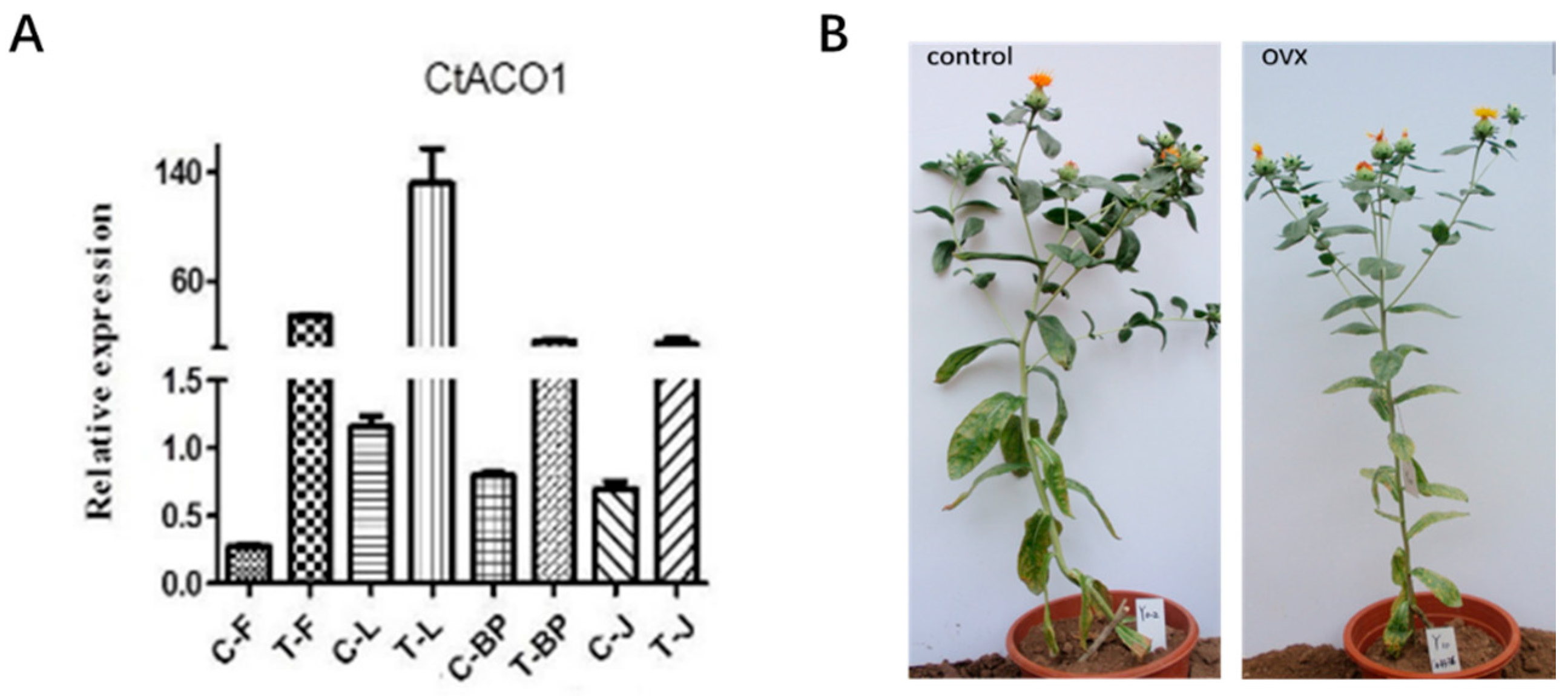
2.4. Transcript Expression Patterns of CtACO1 in Safflower
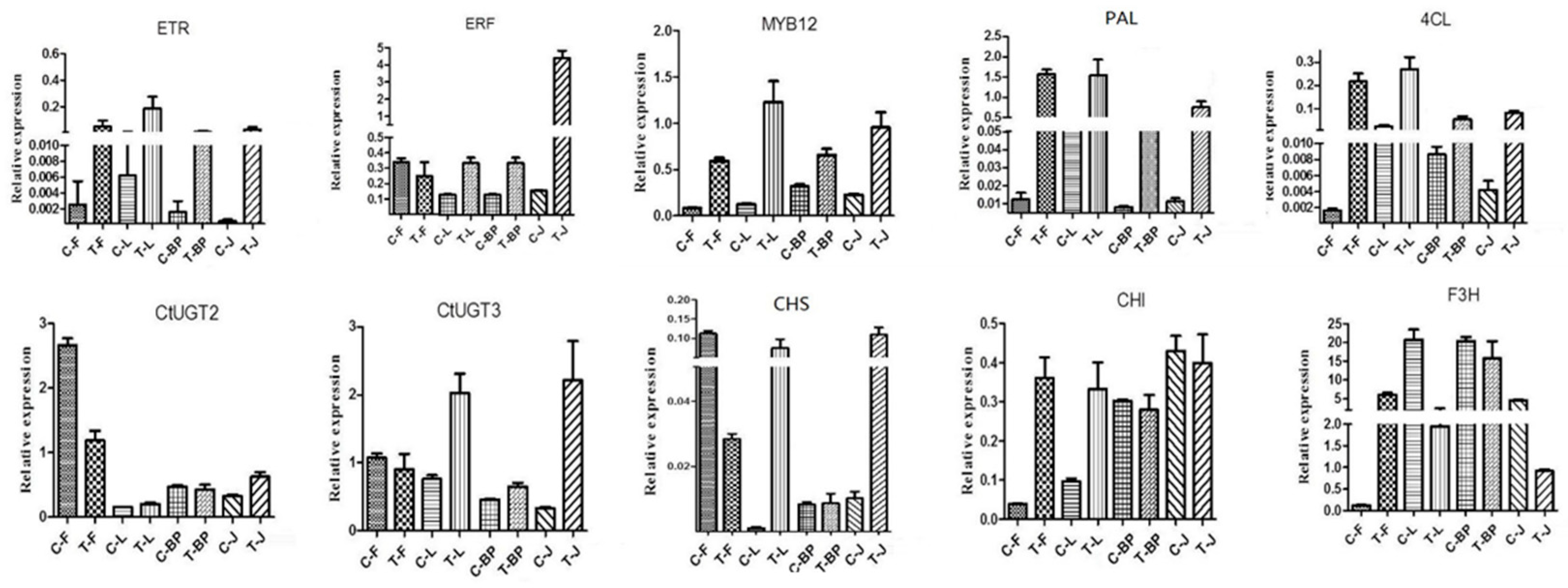
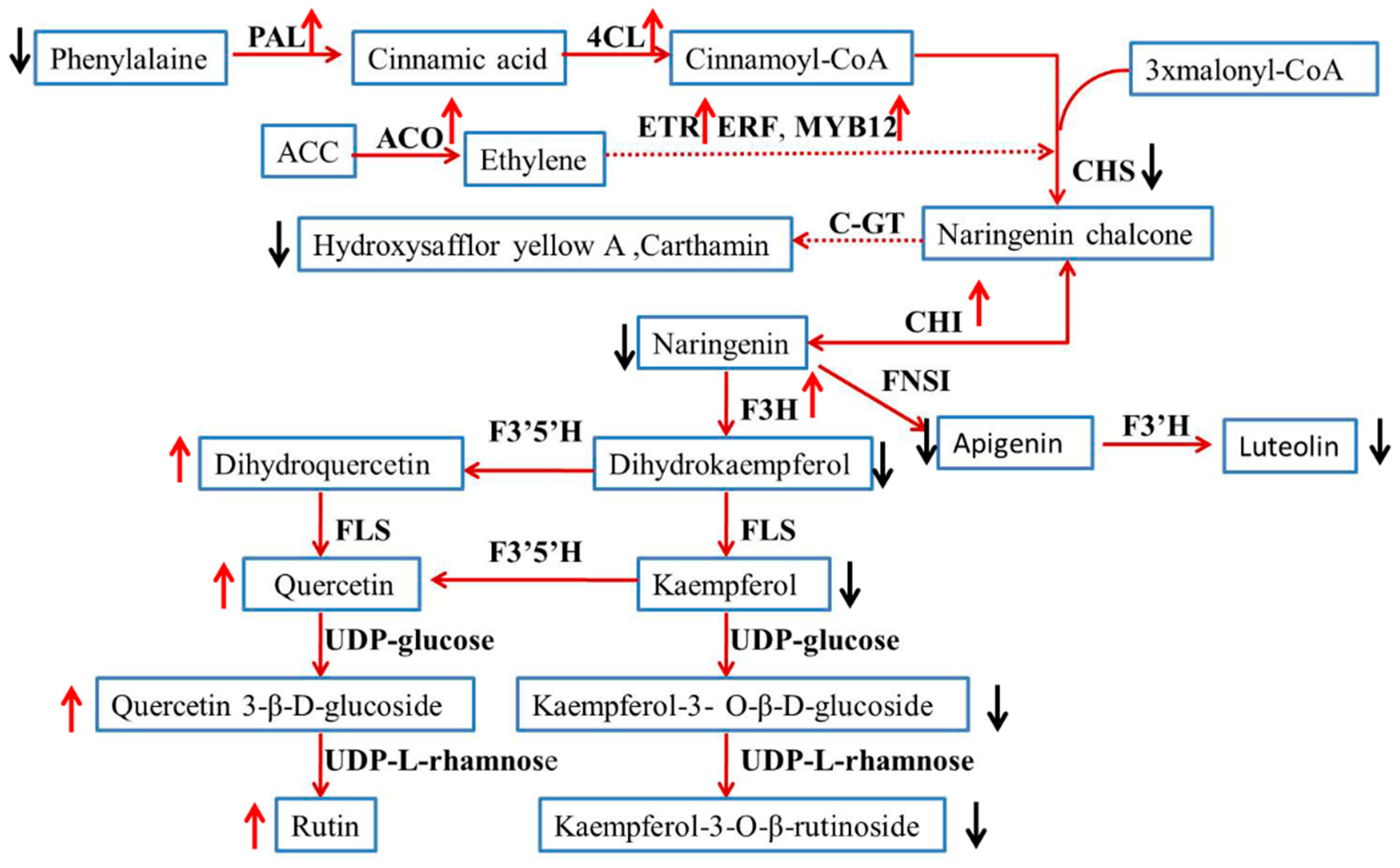
2.5. Expression of Genes Involved in Ethylene and Flavonoid Biosynthesis Pathway
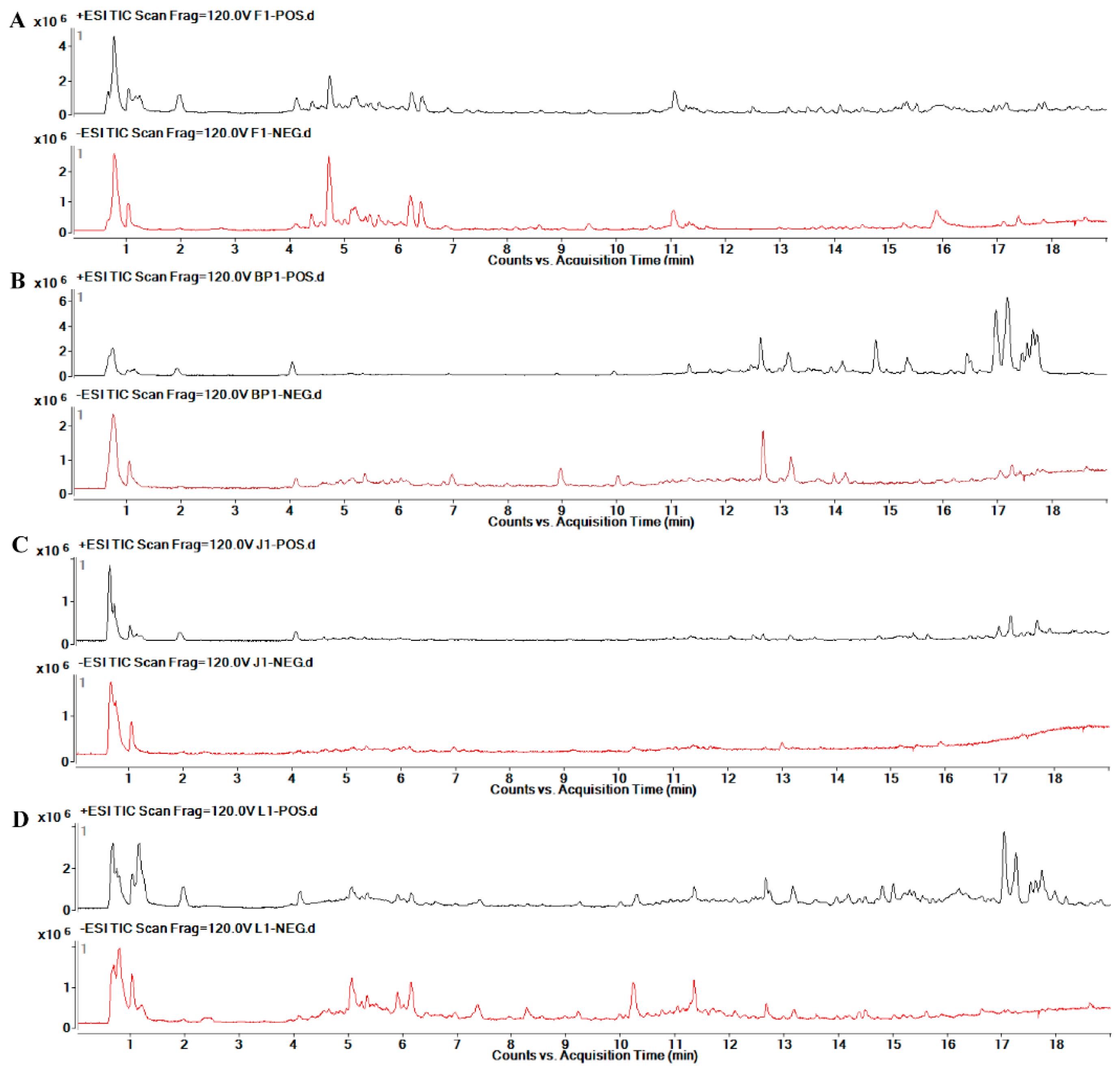
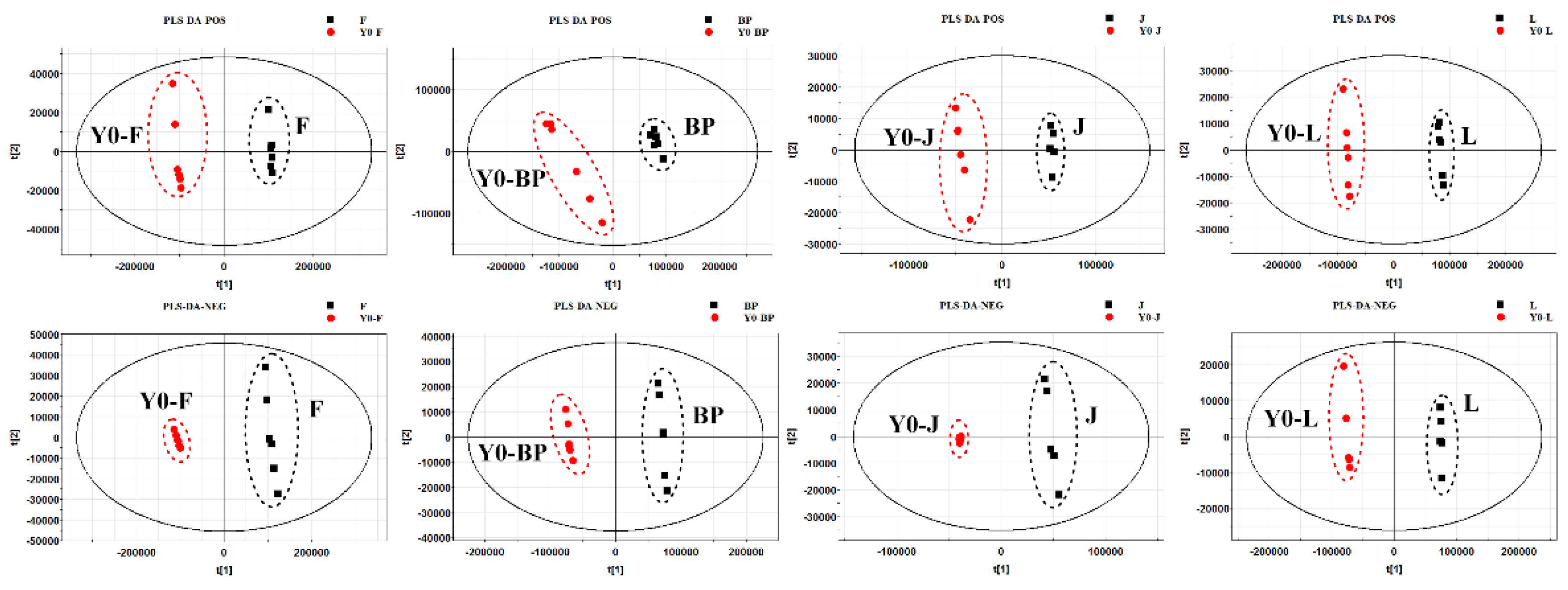
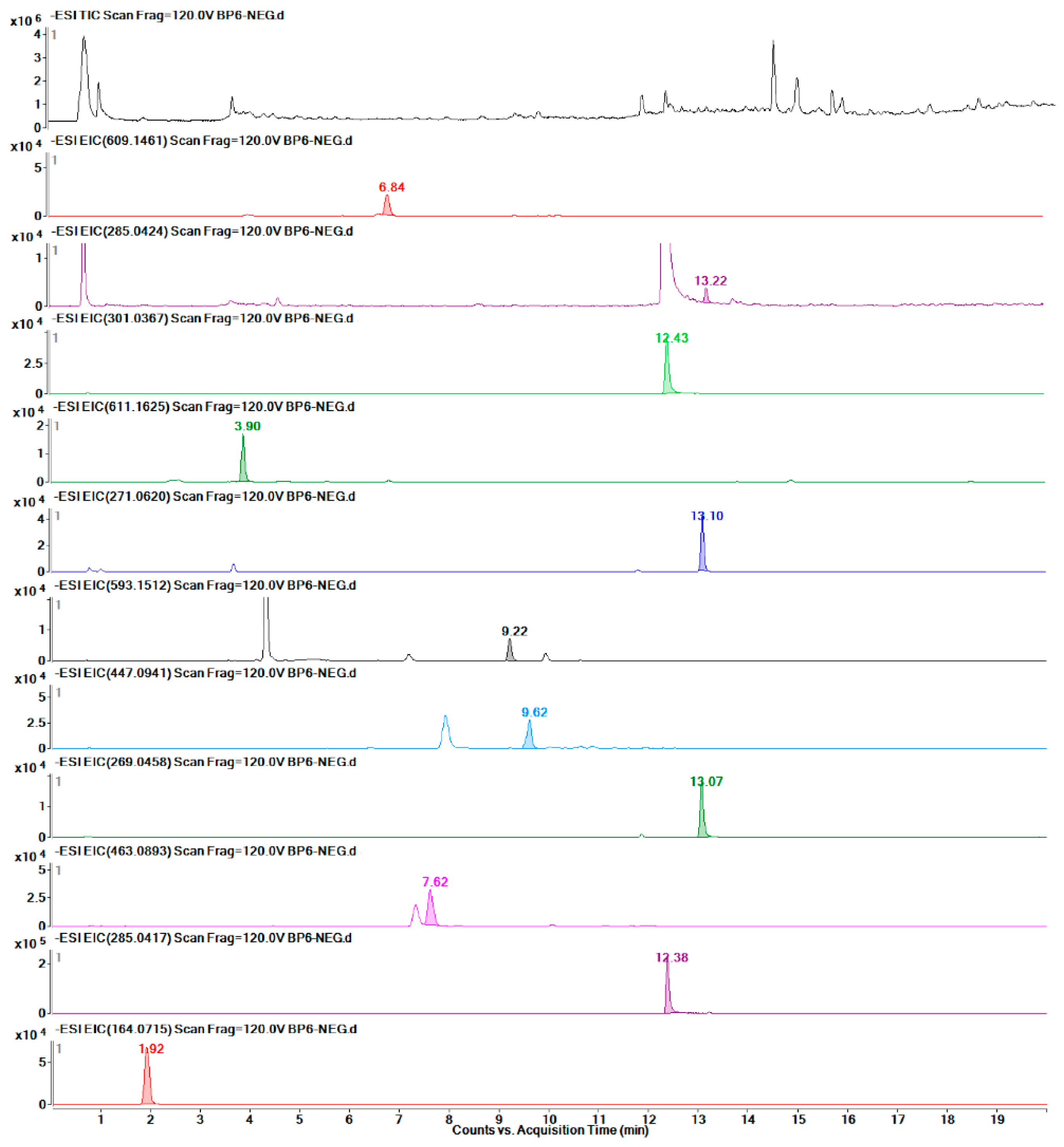
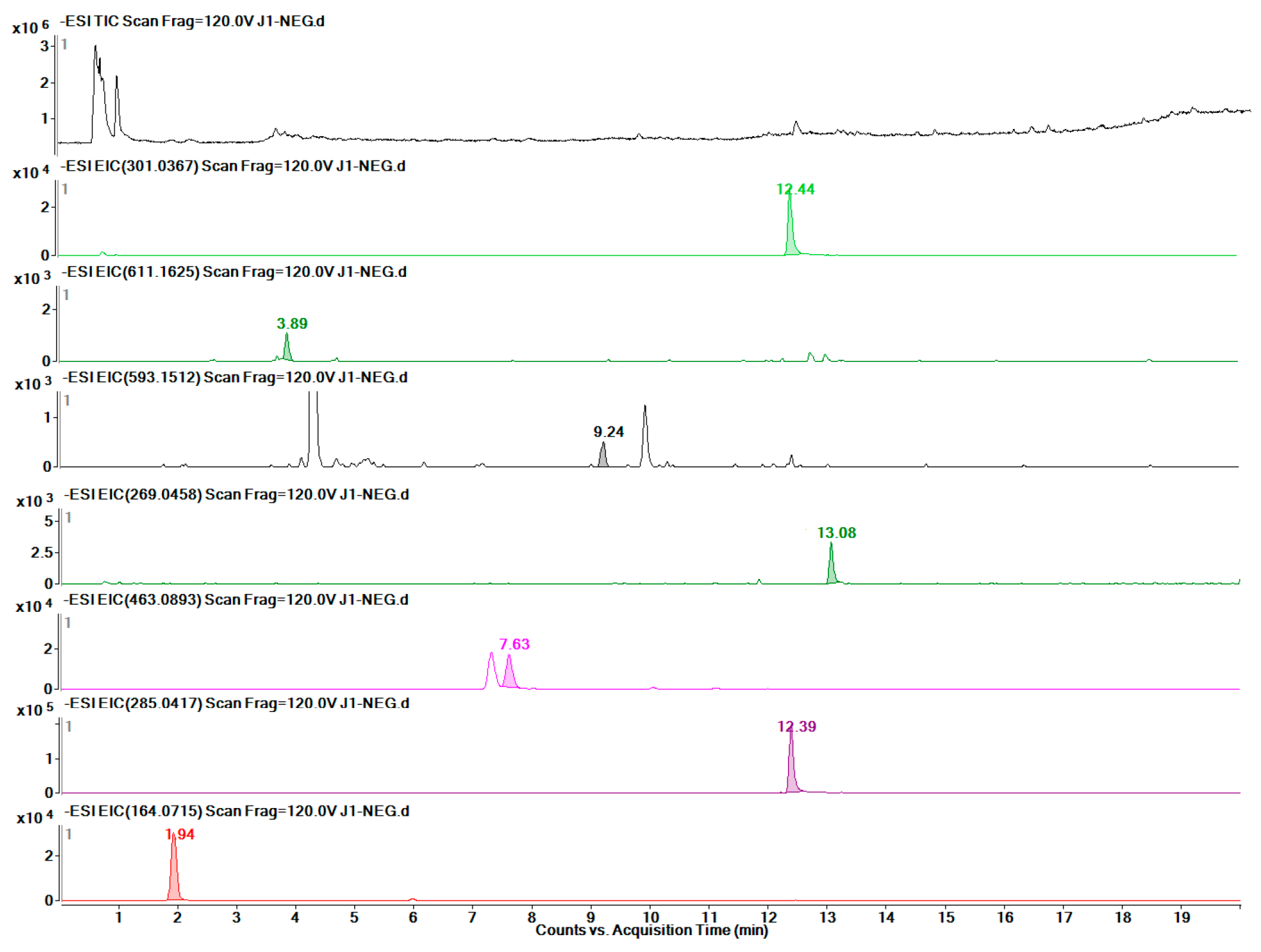
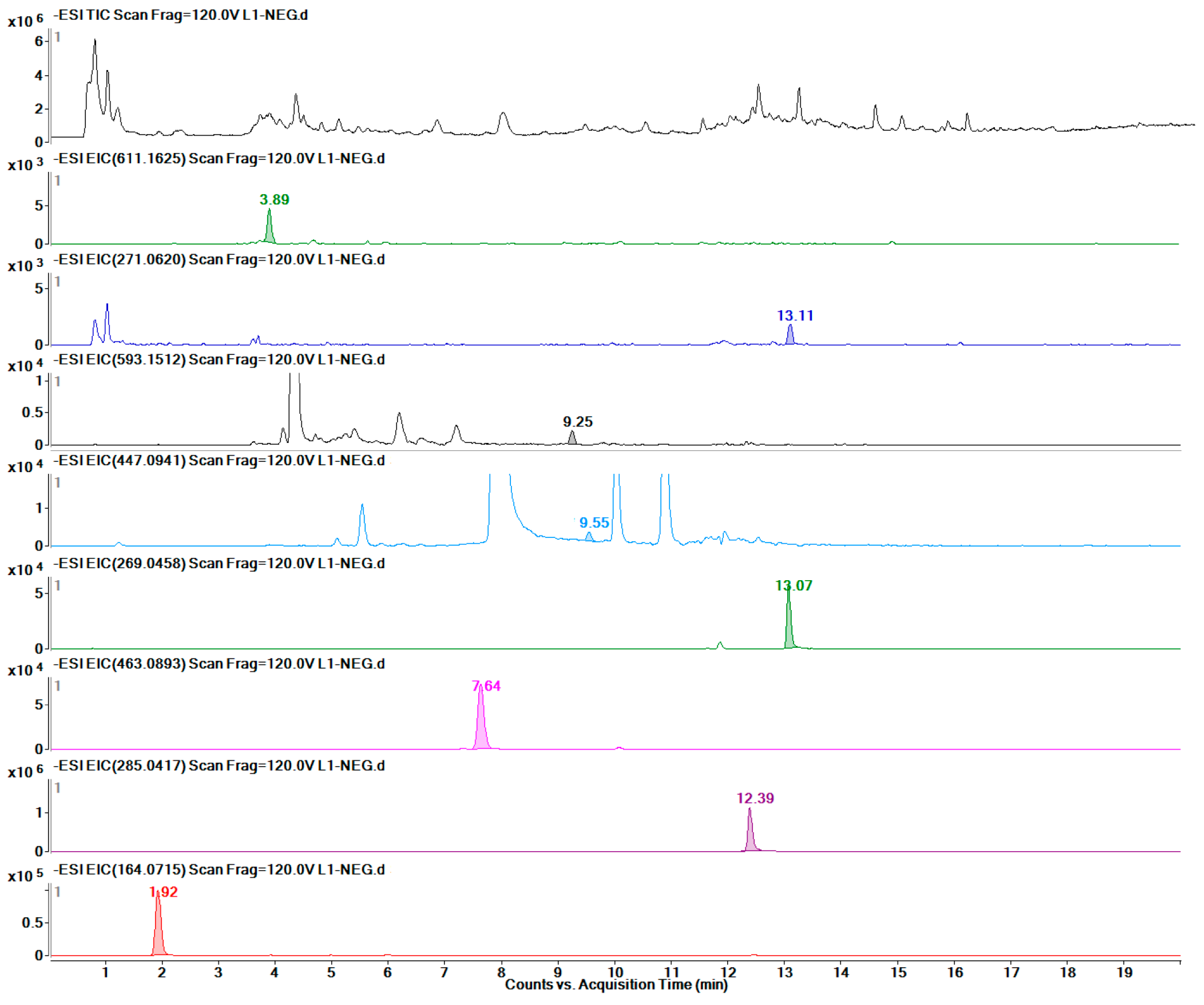
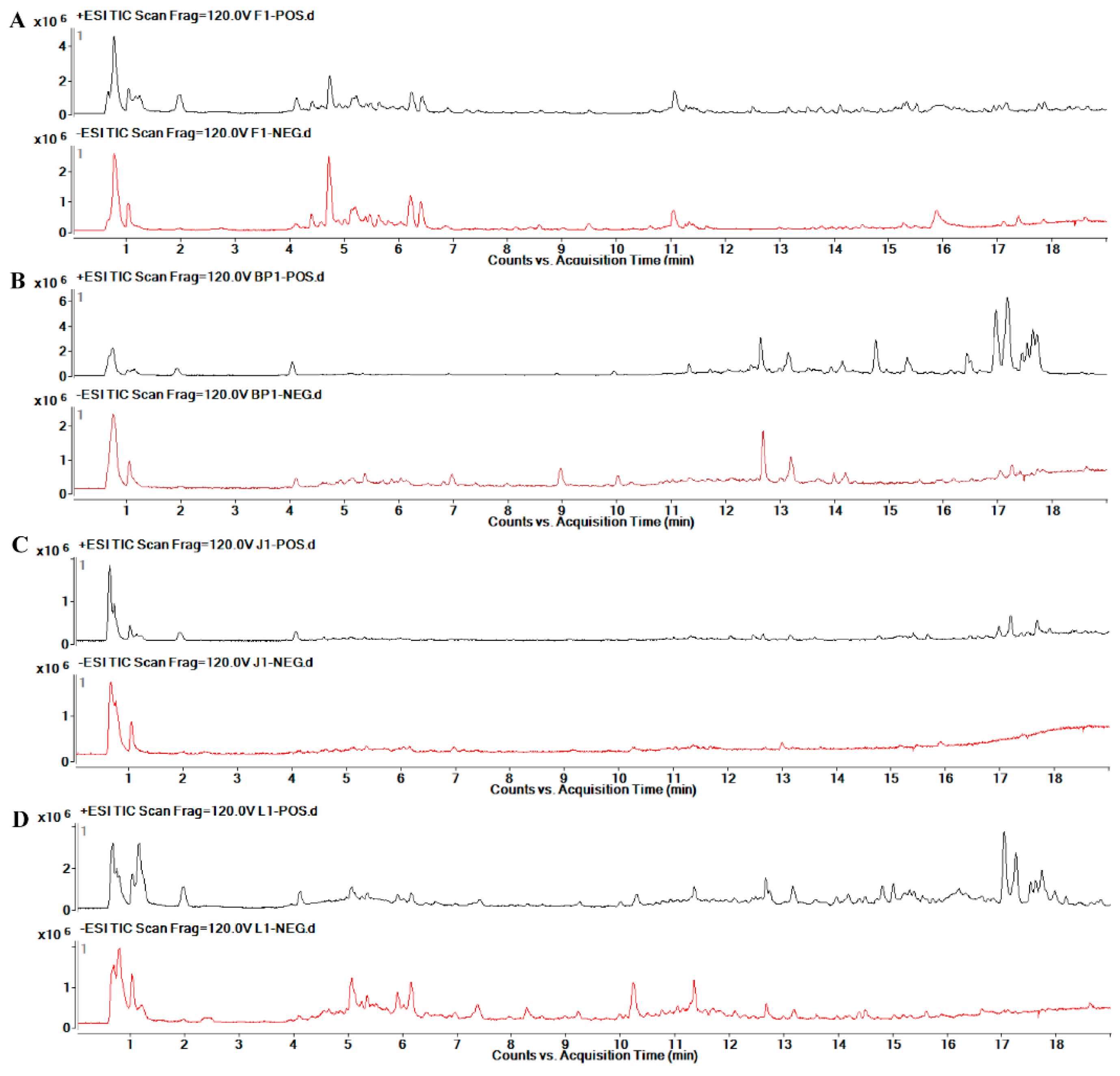
2.6. Metabolic Profile Analysis of Different Organs of Safflower
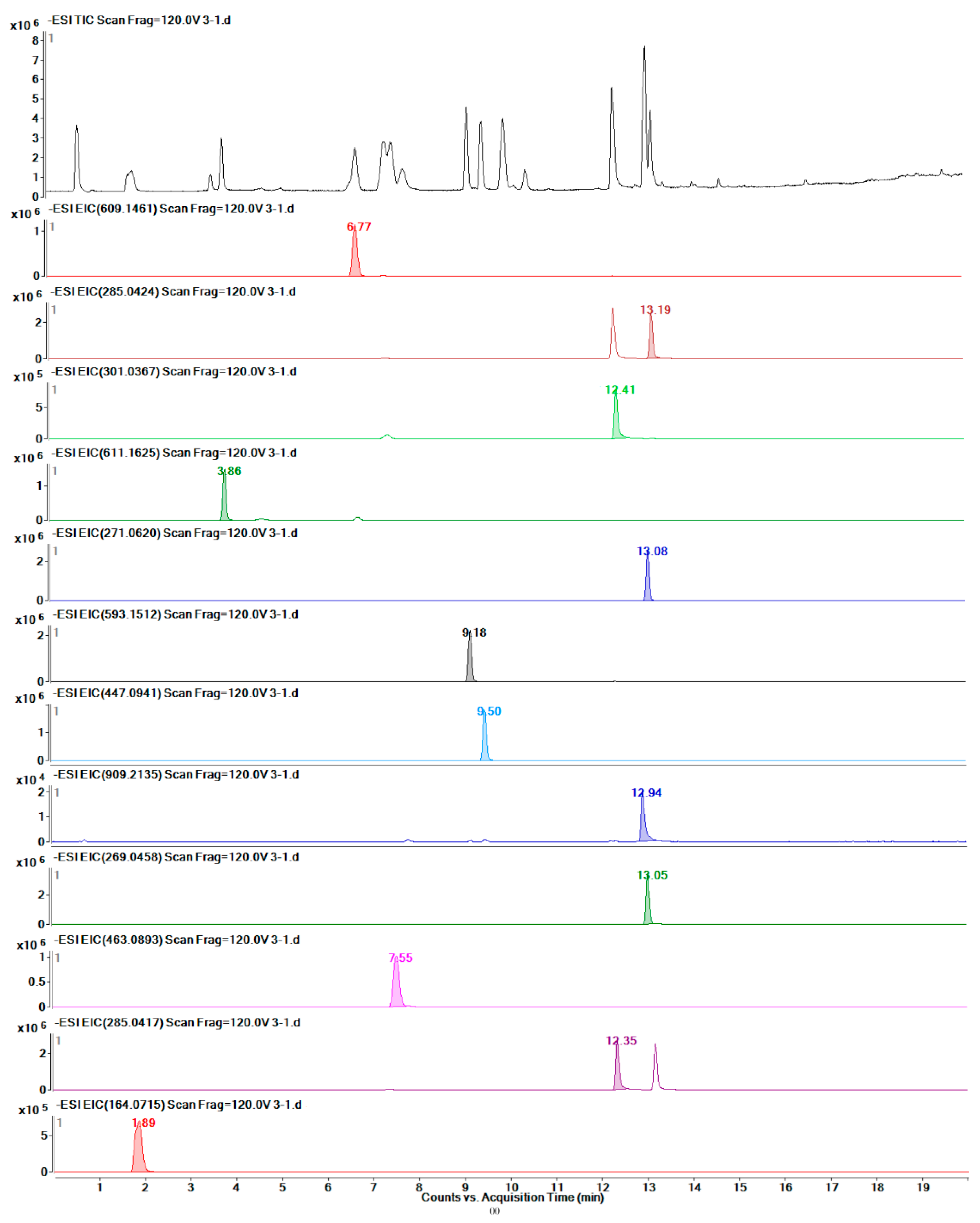
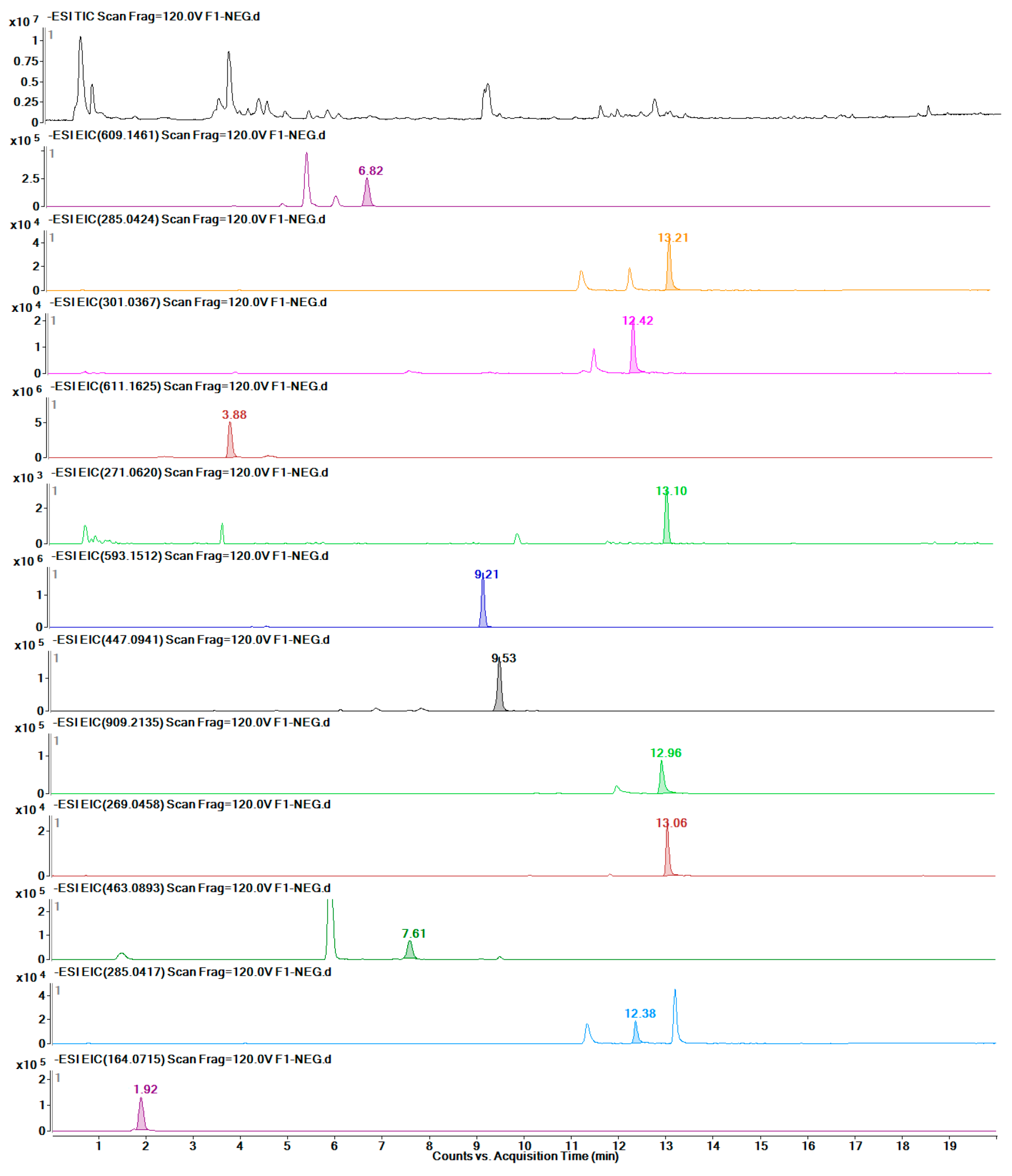
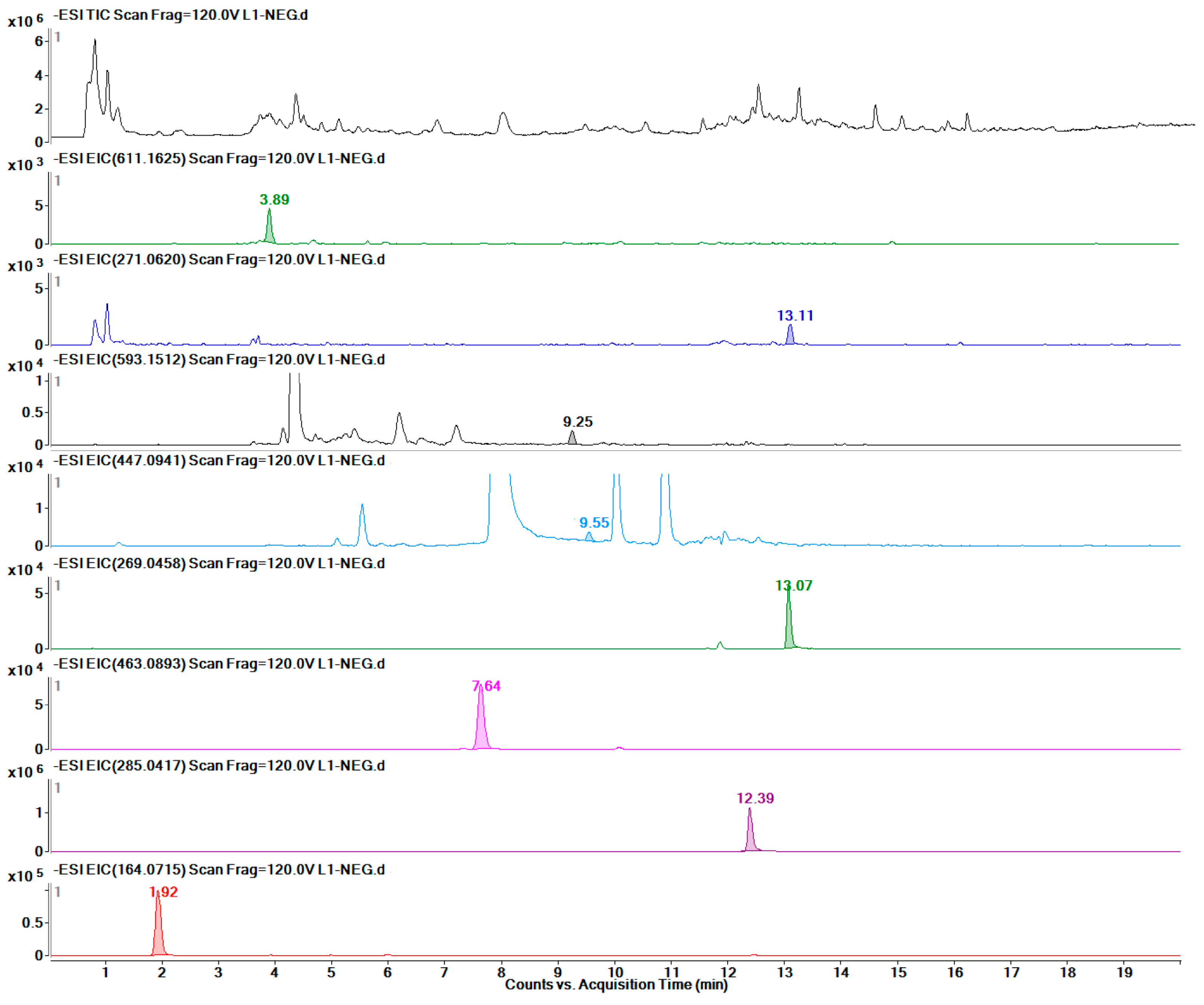
2.7. Quantitative Detection of 12 Target Metabolites
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Analysis of the Transcription Profiling Database of Safflower
4.3. Sequence Alignment and Phylogeny Analysis
4.4. Subcellular Localization of the CtACO1 Protein
4.5. Transformation of Safflower and Screening
4.6. RNA Isolation and qPCR
4.7. Metabolite Extraction and UHPLC-MS/TOF Analysis
4.8. Preparation of Sample
4.9. UHPLC-Q-TOF/MS Profiling Analysis of Different Safflower Parts between the Transgenic and Control Groups
4.10. Quantitative Determination of Target Differential Metabolites
4.11. Chromatography and Mass Spectrometry
4.12. Preparation of Standards
4.13. Validation of the Quantitative Measurement Method
4.14. Quantitative Measurement of Objective Metabolites
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACO | 1-aminocyclopropane carboxylic acid oxidase |
| GFP | green fluorescent protein |
| PMT-39 | pCAMBIA-1380-CaMV35S-MCS-EGFP-NOS |
| LB | Luria-Bertani |
| MS | Murashige and Skoog |
| UHPLC-Q-TOF/MS | Ultra performance liquid chromatography-quadrupole-time-of-flight massspectrometry |
| F | flower |
| L | leaf |
| BP | bracteole |
| J | stem |
| C | control |
References
- Jian, Z. Flavonoid transport mechanisms: How to go, and with whom. Trends Plant Sci. 2015, 20, 576–585. [Google Scholar]
- Shi, Y.H.; Zhu, S.W.; Mao, X.Z.; Feng, J.X.; Qin, Y.M.; Zhang, L.; Cheng, J.; Wei, L.P.; Wang, Z.Y.; Zhu, Y.X.; et al. Transcriptome Profiling, Molecular Biological, and Physiological Studies Reveal a Major Role for Ethylene in Cotton Fiber Cell Elongation. Plant Cell. 2006, 18, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Alonso, J.M.; Stepanova, A.N. The ethylene signaling pathway. Science 2004, 306, 1513–1515. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.P.; Nourizadeh, S.D.; Peer, W.A.; Xu, J.; Bandyopadhyay, A.; Murphy, A.S.; Goldsbrough, P.B. Arabidopsis AtGSTF2 is regulated by ethylene and auxin, and encodes a glutathione S-transferase that interacts with flavonoids. Plant J. 2003, 36, 433–442. [Google Scholar] [CrossRef]
- Woltering, E.J.; Somhorst, D. Regulation of Anthocyanin Synthesis in Cymbidium Flowers: Effects of Emasculation and Ethylene. J. Plant Physiol. 1990, 136, 295–299. [Google Scholar] [CrossRef]
- Adams, D.O.; Yang, S.F. Ethylene biosynthesis: Identification of 1-aminocyclopropane-l-carboxylic acid as an intermediate in the conversion of methionine to ethylene. Proc. Nat. 1979, 76, 170–174. [Google Scholar] [CrossRef]
- Wang, K.L.; Li, H.; Ecker, J.R. Ethylene biosynthesis and signaling networks. Plant Cell. 2002, 14, S131–S151. [Google Scholar] [CrossRef]
- Lycett, G.W.; Roberts, J.A.; Jackson, M.B. Increased 1-Aminocyclopropane-1-Carboxylic Acid Oxidase Activity in Shoots of Flooded Tomato Plants Raises Ethylene Production to Physiologically Active Levels. Plant Physiol. 1996, 109, 1435–1440. [Google Scholar]
- Binnie, J.E.; Mcmanus, M.T. Characterization of the 1-aminocyclopropane-1-carboxylic acid (ACC) oxidase multigene family of Malus domestica Borkh. Phytochemistry 2009, 70, 348–360. [Google Scholar] [CrossRef]
- Dupille, E.; Rombaldi, C.; Lelièvre, J.M.; Cleyetmarel, J.C.; Pech, J.C.; Latché, A. Purification, properties and partial amino-acid sequence of 1-aminocyclopropane-1-carboxylic acid oxidase from apple fruits. Plant 1993, 190, 65–70. [Google Scholar] [CrossRef]
- Fan, X.T.; Mattheis, J.P.; Fellman, J.K. Inhibition of apple fruit 1-aminocyclopropane-1-carboxylic acid oxidase activity and respiration by acetylsalicylic acid. J. Plant Physiol. 1996, 149, 469–471. [Google Scholar] [CrossRef]
- Sekeli, R.; Abdullah, J.; Namasivayam, P.; Muda, P.; Bakar, U.; Yeong, W.; Pillai, V. RNA Interference of 1-Aminocyclopropane-1-carboxylic Acid Oxidase (ACO1 and ACO2) Genes Expression Prolongs the Shelf Life of Eksotika (Carica papaya L.) Papaya Fruit. Molecules 2014, 19, 8350–8362. [Google Scholar] [CrossRef]
- Piquemal, J.; Chamayou, S.; Nadaud, I.; Beckert, M.; Barriere, Y.; Mila, I.; Lapierre, C.; Rigau, J.; Puigdomenech, P.; Jauneau, A.; et al. Down-regulation of caffeic acid o-methyltransferase in maize revisited using a transgenic approach. Plant Physiol. 2003, 130, 1675–1685. [Google Scholar] [CrossRef]
- Wang, Y.S.; Xu, Y.J.; Gao, L.P.; Yu, O.; Wang, X.Z.; He, X.J.; Jiang, X.L.; Liu, Y.J.; Xia, T. Functional analysis of Flavonoid 3′,5′-hydroxylase from Tea plant (Camellia sinensis): Critical role in the accumulation of catechins. BMC Plant Biol. 2014, 14, 347. [Google Scholar] [CrossRef]
- Mahjoub, A.; Hernould, M.; Joubès, J.; Decendit, A.; Mars, M.; Barrieu, F.; Hamdi, S.; Delrot, S. Overexpression of a grapevine R2R3-MYB factor in tomato affects vegetative development, flower morphology and flavonoid and terpenoid metabolism. Plant Physiol Biochem. 2009, 47, 551–561. [Google Scholar] [CrossRef]
- Zhou, X.; Tang, L.; Xu, Y.; Zhou, G.; Wang, Z. Towards a better understanding of medicinal uses of Carthamus tinctorius L. In traditional Chinese medicine: A phytochemical and pharmacological review. J. Ethnopharmacol. 2013, 151, 27–43. [Google Scholar] [CrossRef]
- Tu, Y.; Xue, Y.; Guo, D.; Sun, L.; Guo, M. Carthami flos: A review of its ethnopharmacology, pharmacology and clinical applications. Reva Bras Farmacogn. 2015, 2, 553–566. [Google Scholar] [CrossRef]
- Shilpa, K.S.; Kumar, V.D.; Sujatha, M. Agrobacterium-mediated genetic transformation of safflower (Carthamus tinctorius L.). Plant Cell Tissue Org. 2010, 103, 387–401. [Google Scholar] [CrossRef]
- Belide, S.; Hac, L.; Singh, S.P.; Green, A.G.; Wood, C.C. Agrobacterium -mediated transformation of safflower and the efficient recovery of transgenic plants via grafting. Plant Methods. 2011, 7, 12. [Google Scholar] [CrossRef]
- Guo, D.; Xue, Y.; Li, D.; He, B.; Jia, X.; Dong, X.; Guo, M. Overexpression of CtCHS1 Increases Accumulation of Quinochalcone in Safflower. Front. Plant Sci. 2017, 8, 1409. [Google Scholar] [CrossRef]
- Mierziak, J.; Kostyn, K.; Kulma, A. Flavonoids as important molecules of plant interactions with the environment. Molecules 2014, 19, 16240–16265. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.H.; Kuleck, G.; Hirsch, A.M.; Hawes, M.C. Flavonoids: Signal Molecules in Plant Development. Ad. Exp. Med. Biol. 2002, 505, 51–60. [Google Scholar]
- Bartwal, A.; Mall, R.; Lohani, P.; Guru, S.K.; Arora, S. Role of Secondary Metabolites and Brassinosteroids in Plant Defense Against Environmental Stresses. J. Plant Growth Regul. 2013, 32, 216–232. [Google Scholar] [CrossRef]
- Guo, H.; Ecker, J.R. The ethylene signaling pathway: New insights. Curr. Opin. Plant Biol. 2004, 7, 40–49. [Google Scholar] [CrossRef]
- Buer, C.S.; Sukumar, P.; Muday, G.K. Ethylene Modulates Flavonoid Accumulation and Gravitropic Responses in Roots of Arabidopsis. Plant Physiol. 2006, 140, 1384–1396. [Google Scholar] [CrossRef]
- Madleen, S.; Fabio, O.; Beatrice, B.; Marius, R.; AJalila, S.; Christian, L. A Structural and Functional Model for the 1-Aminocyclopropane-1-carboxylic Acid Oxidase. Angew. Chem. 2015, 54, 12325–12328. [Google Scholar]
- Reinhardt, D.; Kende, H.; Boller, T. Subcellular localization of 1-aminocyclopropane-1-carboxylate oxidase in tomato cells. Plant 1994, 195, 142–146. [Google Scholar] [CrossRef]
- Rombaldi, C.; Pech, J.C. Immunocytolocalization of 1-aminocyclopropane-1-carboxylic acid oxidase in tomato and apple fruit. Plant 1994, 192, 453–460. [Google Scholar] [CrossRef]
- Chung, M.C.; Chou, S.J.; Kuang, L.Y.; Charng, Y.Y.; Yang, S.F. Subcellular localization of 1-aminocyclopropane-1-carboxylic acid oxidase in apple fruit. Plant Cell Physiol. 2002, 43, 549. [Google Scholar] [CrossRef]
- Wang, Y.; Noguchi, K.; Ono, N.; Inoue, S.; Terashima, I.; Kinoshita, T. Overexpression of plasma membrane H+-ATPase in guard cells promotes light-induced stomatal opening and enhances plant growth. Proc. Natl. Acad. Sci. USA 2014, 111, 533. [Google Scholar] [CrossRef]
- Atkinson, R.G.; Gunaseelan, K.; Wang, M.Y.; Luo, L.; Wang, T.; Norling, C.L.; Johnston, S.L.; Maddumage, R.; Schröder, R.; Schaffer, R.J. Dissecting the role of climacteric ethylene in kiwifruit (Actinidia chinensis) ripening using an ACC-oxidase knockdown line. J. Exp. Bot. 2011, 62, 3821–3835. [Google Scholar] [CrossRef]
- Chen, Y.T.; Lee, Y.R.; Yang, C.Y.; Wang, Y.T.; Yang, S.F.; Shaw, J.F. A novel papaya acc oxidase gene (cp-aco2) associated with late stage fruit ripening and leaf senescence. Plant Sci. 2003, 164, 531–540. [Google Scholar] [CrossRef]
- He, X.J.; Mu, R.L.; Cao, W.H.; Zhang, Z.G.; Zhang, J.S.; Chen, S.Y. Atnac2, a transcription factor downstream of ethylene and auxin signaling pathways, is involved in salt stress response and lateral root development. Plant J. 2005, 44, 903. [Google Scholar] [CrossRef] [PubMed]
- Saslowsky, D.E.; Warek, U.; Winkel, B.S.J. Nuclear Localization of Flavonoid Enzymes in Arabidopsis. J. Biol. Chem. 2005, 280, 23735–23740. [Google Scholar] [CrossRef]
- Lewis, D.R.; Ramirez, M.V.; Miller, N.D.; Vallabhaneni, P.; Ray, W.K.; Helm, R.F.; Winkel, B.S.; Muday, G.K. Auxin and Ethylene Induce Flavonol Accumulation through Distinct Transcriptional Networks. Plant Physiol. 2011, 156, 144. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.M.; Muday, G.K. Ethylene-induced flavonol accumulation in guard cells suppresses reactive oxygen species and moderates stomatal aperture. Plant Physiol. 2014, 164, 1707. [Google Scholar] [CrossRef]
- Jie, T.; Lou, Z.Y.; Yan, W.; Guo, M.L. Expression of a small heat shock protein (CTL-hsyapr) screened by cDNA-AFLP approach is correlated with hydroxysafflor yellow A in safflower (Carthamus tinctorius L.). Biochem. Syst. Ecol. 2010, 38, 722–730. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Liu, F.; Guo, D.D.; Tu, Y.H.; Xue, Y.R.; Yue, G.; Li, G.M. Identification of reference genes for gene expression normalization in safflower (carthamus tinctorius). Rev. Bras. Farmacogn. 2016, 26, 564–570. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | F | BP | J | L | ||||
|---|---|---|---|---|---|---|---|---|
| POS | NEG | POS | NEG | POS | NEG | POS | NEG | |
| R2X | 0.97 | 0.979 | 0.86 | 0.899 | 0.857 | 0.979 | 0.891 | 0.927 |
| R2Y | 1 | 0.999 | 0.998 | 1 | 0.998 | 0.999 | 1 | 1 |
| Q2 | 1 | 0.999 | 0.995 | 0.999 | 0.993 | 0.999 | 0.998 | 0.999 |
| No. | Analytes | Linear Regression | LOQ (ng/mL) | Precision (RSD%, n = 6) | ||
|---|---|---|---|---|---|---|
| Regression Equations | Linear Ranges (ng/mL) | R2 | ||||
| 1 | Rutin | y = 0.3535x − 0.0059 | 20.3–5202.7 | 0.9997 | 20.3 | 1.56 |
| 2 | Kaempferol | y = 0.9063x + 0.0043 | 4.3–273.4 | 0.9985 | 4.3 | 1.42 |
| 3 | Quercetin | y = 0.3528x + 0.0142 | 16–1024.8 | 0.9951 | 16 | 1.86 |
| 4 | Hydroxysafflor yellow A | y = 0.2511x − 0.0167 | 20–20495.5 | 0.9999 | 20 | 2.49 |
| 5 | Naringenin | y = 0.9582x + 0.0057 | 2.6–335 | 0.9967 | 2.6 | 0.66 |
| 6 | Kaempferol-3-O-β-rutinoside | y = 0.4554x + 0.001 | 6.4–6582.2 | 1 | 6.4 | 1.34 |
| 7 | Kaempferol-3-O-β-d-glucoside | y = 0.5133x − 0.0006 | 4–4099.1 | 0.9999 | 4 | 1.26 |
| 8 | Carthamin | y = 0.0245x + 0.0013 | 262.1–16774.8 | 0.9999 | 262.1 | 3.37 |
| 9 | Apigenin | y = 1.45x + 0.0065 | 2–261.1 | 0.9971 | 2 | 1.43 |
| 10 | Quercetin 3-β-d-glucoside | y = 0.4194x + 0.0055 | 18.9–1212 | 0.9994 | 18.9 | 1.43 |
| 11 | Luteolin | y = 1.2568x + 0.0248 | 2–1034.6 | 0.9958 | 2 | 0.92 |
| 12 | Phenylalanine | y = 0.0925x + 0.0176 | 57.3–14662.2 | 0.9984 | 57.3 | 0.83 |
| Part | The Contents of Flavone in Different Organs (%) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rutin | Kaempferol | Quercetin | Hydroxysafflor Yellow A | Naringenin | Kaempferol-3-O-β-rutinoside | Kaempferol-3-O-β-d-glucoside | Carthamin | Apigenin | Quercetin 3-β-d-glucoside | Luteolin | Phenylalanine | |
| F | 0.0707 | 0.0032 | 0.0047 | 0.8289 | 0.0002 | 0.3898 | 0.0217 | 0.4125 | 0.0012 | 0.0151 | 0.0011 | 0.1654 |
| Y0-F | 0.0401 | 0.0154 | 0.0024 | 1.7159 | 0.0005 | 0.6276 | 0.0598 | 1.301 | 0.0036 | 0.0087 | 0.0013 | 0.2593 |
| FC | 1.76 | 0.21 | 1.93 | 0.48 | 0.30 | 0.62 | 0.36 | 0.32 | 0.34 | 1.72 | 0.84 | 0.64 |
| P | 9.30 × 10−5 | 2.38 × 10−8 | 3.73 × 10−3 | 5.39 × 10−7 | 7.68 × 10−5 | 5.50 × 10−6 | 3.85 × 10−9 | 1.29 × 10−4 | 3.69 × 10−6 | 8.19 × 10−4 | 5.02 × 10−1 | 2.08 × 10−3 |
| BP | 0.0083 | 0.0001 | 0.0094 | 0.0084 | 0.003 | 0.0015 | 0.0057 | —— | 0.0009 | 0.0098 | 0.0137 | 0.0796 |
| Y0-BP | 0.0019 | 0.0007 | 0.0001 | 0.0216 | 0.0032 | 0.0039 | 0.01 | 0.0002 | 0.0003 | 0.0033 | 0.1929 | |
| FC | 4.44 | 0.18 | 68.3 | 0.39 | 0.93 | 0.39 | 0.57 | 4.74 | 29.38 | 4.13 | 0.41 | |
| P | 5.64 × 10−5 | 3.36 × 10−3 | 1.27 × 10−3 | 9.23 × 10−5 | 5.76 × 10−1 | 2.56 × 10−5 | 4.78 × 10−4 | 1.66 × 10−3 | 4.03 × 10−7 | 1.26 × 10−4 | 2.26 × 10−6 | |
| J | —— | —— | 0.0038 | 0.0019 | —— | 0.0001 | —— | —— | 0.0002 | 0.0064 | 0.0149 | 0.0435 |
| Y0-J | 0.0083 | 0.0063 | 0.0007 | 0.0001 | 0.0049 | 0.0142 | 0.0565 | |||||
| FC | 0.46 | 0.31 | 0.1 | 2.26 | 1.31 | 1.05 | 0.77 | |||||
| P | 4.29 × 10−3 | 1.82 × 10−6 | 1.57 × 10−5 | 1.21 × 10−1 | 3.40 × 10−2 | 7.44 × 10−1 | 8.47 × 10−2 | |||||
| L | —— | —— | —— | 0.0019 | 0.0001 | 0.0003 | 0.0002 | —— | 0.002 | 0.0129 | 0.0711 | 0.066 |
| Y0-L | 0.006 | 0.0001 | 0.0007 | 0.0002 | 0.0033 | 0.0011 | 0.0051 | 0.0717 | ||||
| FC | 0.31 | 0.46 | 0.42 | 0.9 | 0.59 | 12.06 | 13.95 | 0.92 | ||||
| P | 3.52 × 10−9 | 1.66 × 10−2 | 3.69 × 10−5 | 4.60 × 10−1 | 1.92 × 10−2 | 7.77 × 10−4 | 2.39 × 10−5 | 2.76 × 10−1 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, Y.; He, B.; Gao, S.; Guo, D.; Jia, X.; Dong, X.; Guo, M. CtACO1 Overexpression Resulted in the Alteration of the Flavonoids Profile of Safflower. Molecules 2019, 24, 1128. https://doi.org/10.3390/molecules24061128
Tu Y, He B, Gao S, Guo D, Jia X, Dong X, Guo M. CtACO1 Overexpression Resulted in the Alteration of the Flavonoids Profile of Safflower. Molecules. 2019; 24(6):1128. https://doi.org/10.3390/molecules24061128
Chicago/Turabian StyleTu, Yanhua, Beixuan He, Songyan Gao, Dandan Guo, Xinlei Jia, Xin Dong, and Meili Guo. 2019. "CtACO1 Overexpression Resulted in the Alteration of the Flavonoids Profile of Safflower" Molecules 24, no. 6: 1128. https://doi.org/10.3390/molecules24061128
APA StyleTu, Y., He, B., Gao, S., Guo, D., Jia, X., Dong, X., & Guo, M. (2019). CtACO1 Overexpression Resulted in the Alteration of the Flavonoids Profile of Safflower. Molecules, 24(6), 1128. https://doi.org/10.3390/molecules24061128