
Computational Drug Design Applied to the Study of Metabotropic Glutamate Receptors


Abstract
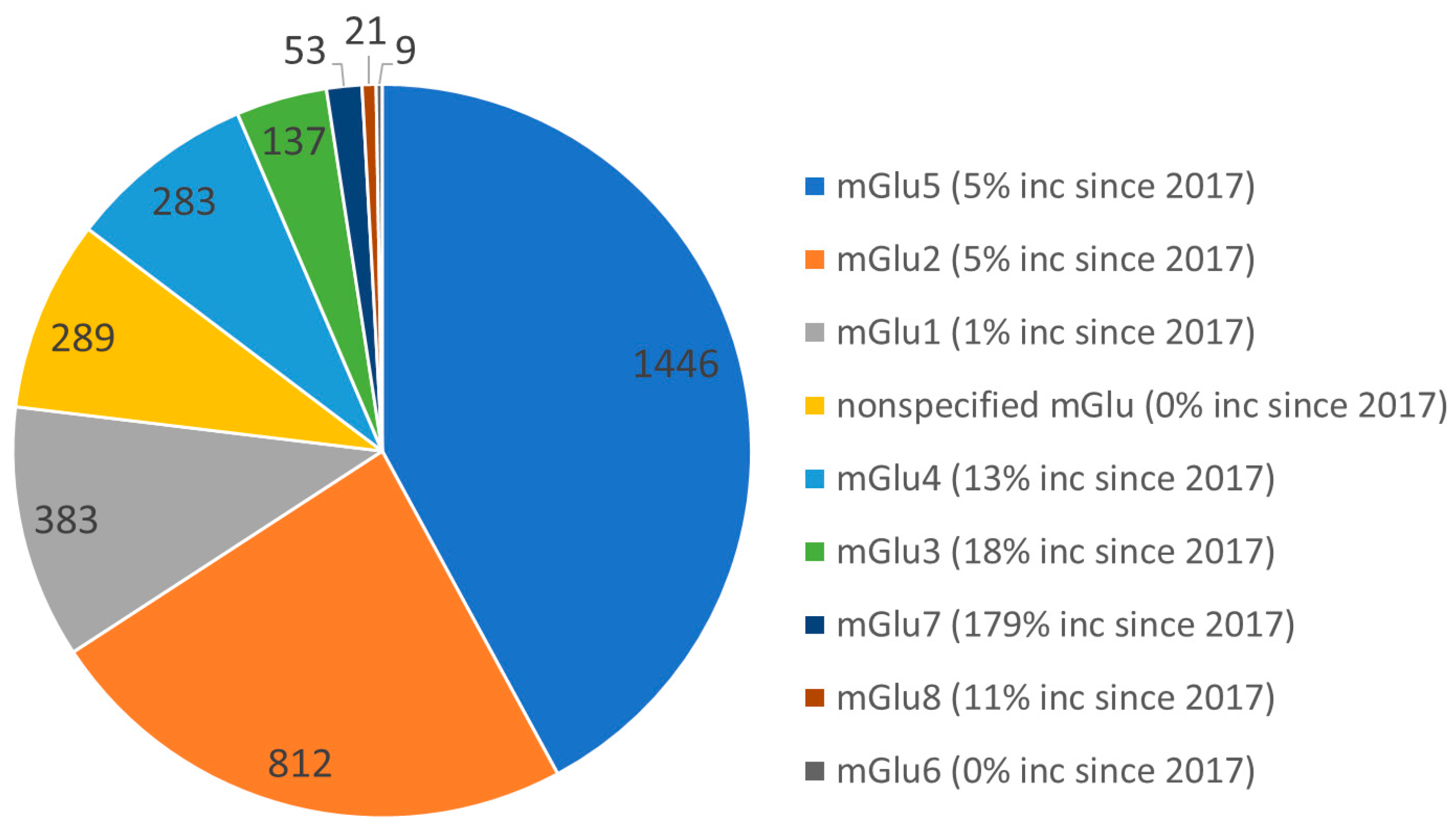
1. Introduction
2. Group I mGlu Receptors
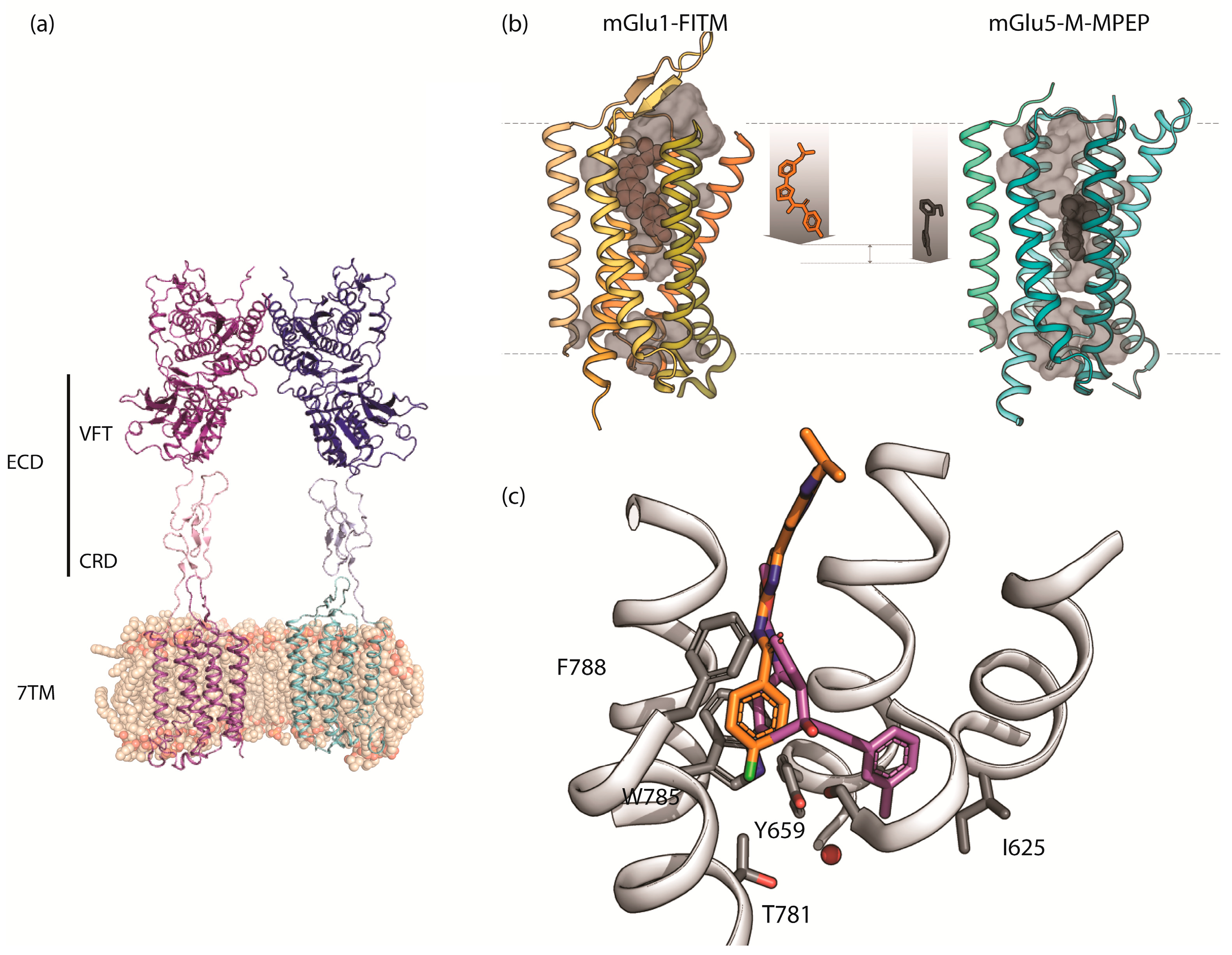
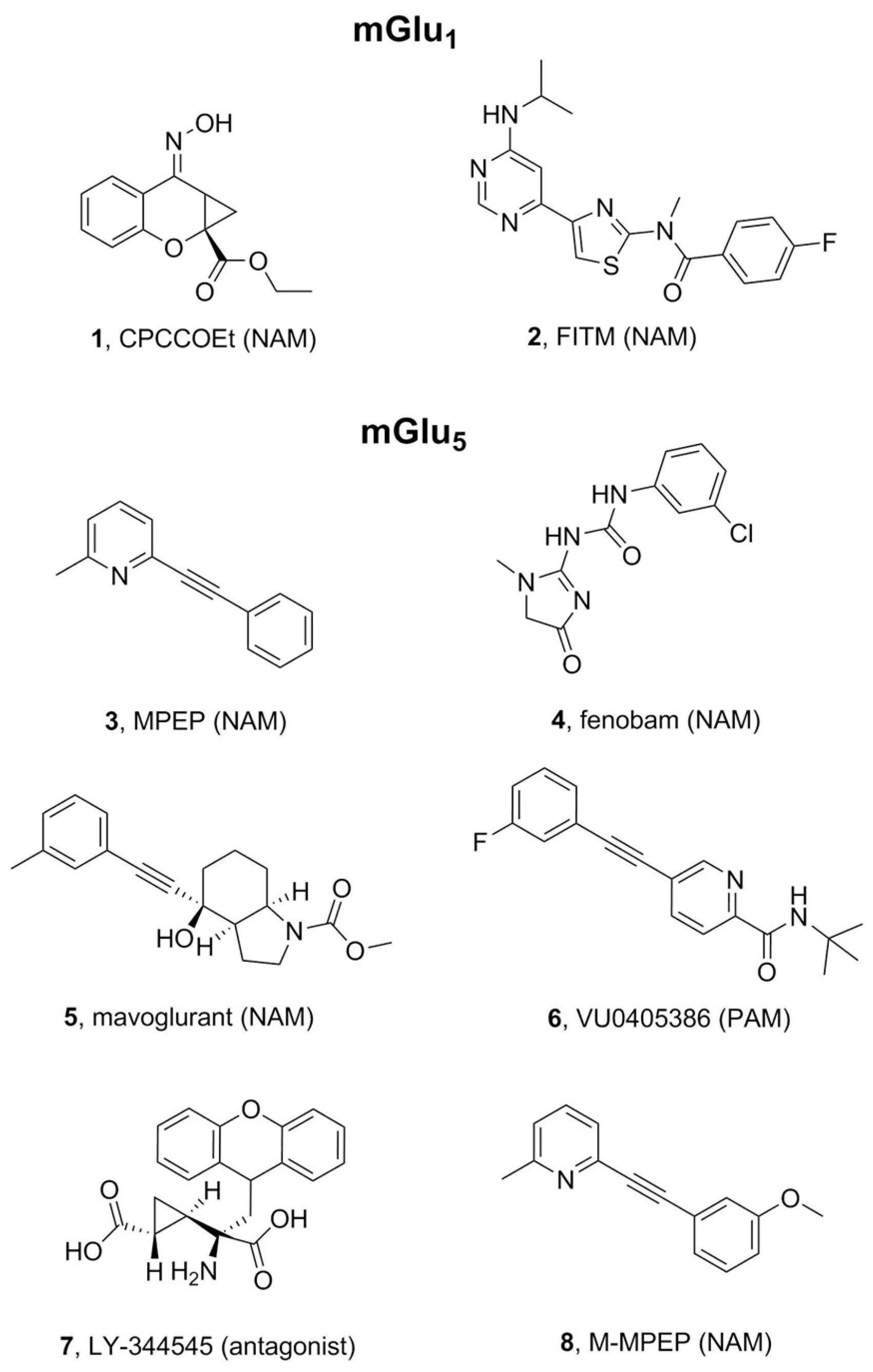
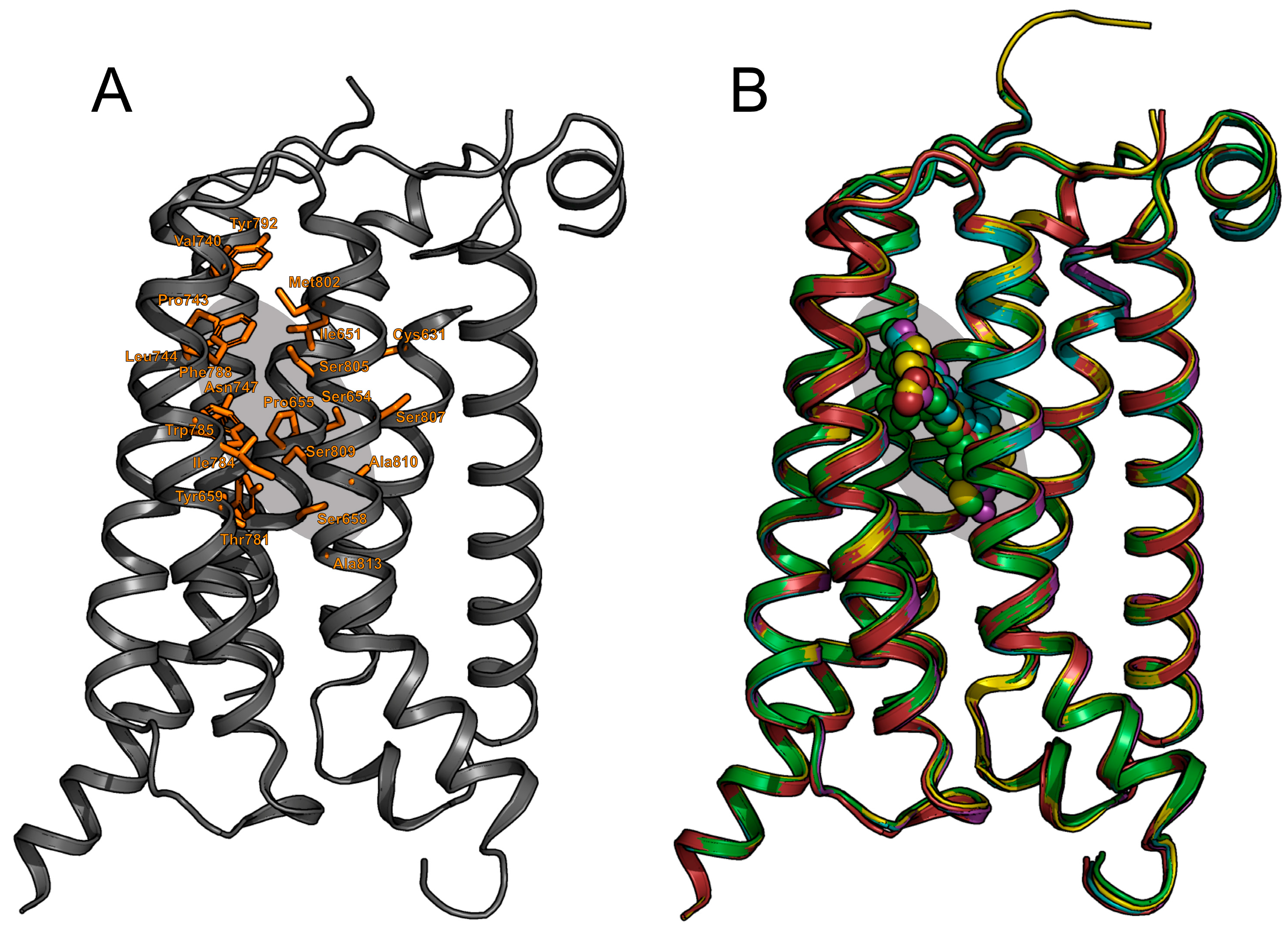
2.1. mGlu1 Receptor
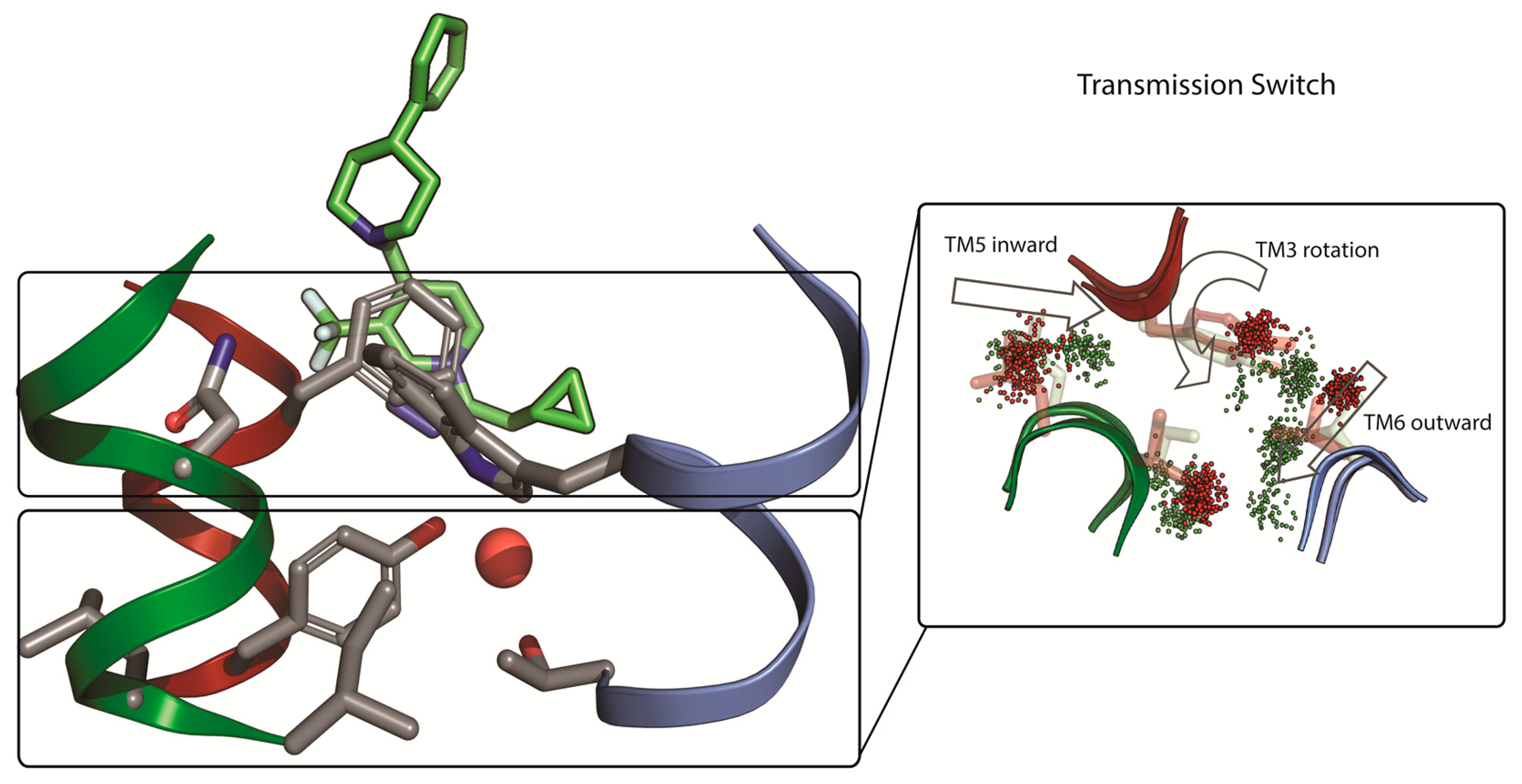
2.2. mGlu5 Receptor
3. Group II mGlu Receptors
3.1. mGlu2 Receptor
3.2. mGlu3 Receptor
4. Group III mGlu Receptors
4.1. mGlu4 Receptor
4.2. mGlu7 Receptor
4.3. mGlu8 Receptor
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fagerberg, L.; Jonasson, K.; von Heijne, G.; Uhlén, M.; Berglund, L. Prediction of the human membrane proteome. Proteomics 2010, 10, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Pin, J.-P.; Galvez, T.; Prézeau, L. Evolution, structure, and activation mechanism of family 3/C G-protein-coupled receptors. Pharmacol. Ther. 2003, 98, 325–354. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic Glutamate Receptors: Physiology, Pharmacology, and Disease. Annu. Rev. Pharmacool. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Lavreysen, H.; Dautzenberg, F.M. Therapeutic Potential of Group III Metabotropic Glutamate Receptors. Curr. Med. Chem. 2008, 15, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.J.; Lindsley, C.W.; Jones, C.K. Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharmacol. Sci. 2009, 30, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Shigemoto, R.; Mizuno, N. Chapter III Metabotropic glutamate receptors—Immunocytochemical and in situ hybridization analyses. In Handbook of Chemical Neuroanatomy; Ottersen, O.P., Storm-Mathisen, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2000; Volume 18, pp. 63–98. [Google Scholar]
- Cauli, B.; Porter, J.T.; Tsuzuki, K.; Lambolez, B.; Rossier, J.; Quenet, B.; Audinat, E. Classification of fusiform neocortical interneurons based on unsupervised clustering. Proc. Natl. Acad. Sci. USA 2000, 97, 6144. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-M.; Jia, Z.; Janus, C.; Henderson, J.T.; Gerlai, R.; Wojtowicz, J.M.; Roder, J.C. Mice Lacking Metabotropic Glutamate Receptor 5 Show Impaired Learning and Reduced CA1 Long-Term Potentiation (LTP) But Normal CA3 LTP. J. Neurosci. 1997, 17, 5196. [Google Scholar] [CrossRef]
- Wright, R.A.; Johnson, B.G.; Zhang, C.; Salhoff, C.; Kingston, A.E.; Calligaro, D.O.; Monn, J.A.; Schoepp, D.D.; Marek, G.J. CNS distribution of metabotropic glutamate 2 and 3 receptors: Transgenic mice and [3H]LY459477 autoradiography. Neuropharmacology 2013, 66, 89–98. [Google Scholar] [CrossRef]
- Mercier, M.S.; Lodge, D. Group III Metabotropic Glutamate Receptors: Pharmacology, Physiology and Therapeutic Potential. Neurochem. Res. 2014, 39, 1876–1894. [Google Scholar] [CrossRef]
- Fisher, N.M.; Seto, M.; Lindsley, C.W.; Niswender, C.M. Metabotropic Glutamate Receptor 7: A New Therapeutic Target in Neurodevelopmental Disorders. Front. Mol. Neurosci. 2018, 11, 387. [Google Scholar] [CrossRef]
- Raber, J.; Duvoisin, R.M. Novel metabotropic glutamate receptor 4 and glutamate receptor 8 therapeutics for the treatment of anxiety. Expert Opin. Investig. Drugs 2015, 24, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Oswald, C.; Marshall, F.H. Applying Structure-Based Drug Design Approaches to Allosteric Modulators of GPCRs. Trends Pharmacol. Sci. 2017, 38, 837–847. [Google Scholar] [CrossRef]
- Koehl, A.; Hu, H.; Feng, D.; Sun, B.; Zhang, Y.; Robertson, M.J.; Chu, M.; Kobilka, T.S.; Laermans, T.; Steyaert, J.; et al. Structural insights into the activation of metabotropic glutamate receptors. Nature 2019, 566, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Kunishima, N.; Shimada, Y.; Tsuji, Y.; Sato, T.; Yamamoto, M.; Kumasaka, T.; Nakanishi, S.; Jingami, H.; Morikawa, K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 2000, 407, 971. [Google Scholar] [CrossRef] [PubMed]
- Doré, A.S.; Okrasa, K.; Patel, J.C.; Serrano-Vega, M.; Bennett, K.; Cooke, R.M.; Errey, J.C.; Jazayeri, A.; Khan, S.; Tehan, B.; et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 2014, 511, 557. [Google Scholar] [CrossRef]
- Wu, H.; Wang, C.; Gregory, K.J.; Han, G.W.; Cho, H.P.; Xia, Y.; Niswender, C.M.; Katritch, V.; Meiler, J.; Cherezov, V.; et al. Structure of a Class C GPCR Metabotropic Glutamate Receptor 1 Bound to an Allosteric Modulator. Science 2014, 344, 58. [Google Scholar] [CrossRef]
- Christopher, J.A.; Aves, S.J.; Bennett, K.A.; Doré, A.S.; Errey, J.C.; Jazayeri, A.; Marshall, F.H.; Okrasa, K.; Serrano-Vega, M.J.; Tehan, B.G.; et al. Fragment and Structure-Based Drug Discovery for a Class C GPCR: Discovery of the mGlu5 Negative Allosteric Modulator HTL14242 (3-Chloro-5-[6-(5-fluoropyridin-2-yl)pyrimidin-4-yl]benzonitrile). J. Med. Chem. 2015, 58, 6653–6664. [Google Scholar] [CrossRef]
- Christopher, J.A.; Orgován, Z.; Congreve, M.; Doré, A.S.; Errey, J.C.; Marshall, F.H.; Mason, J.S.; Okrasa, K.; Rucktooa, P.; Serrano-Vega, M.J.; et al. Structure-Based Optimization Strategies for G Protein-Coupled Receptor (GPCR) Allosteric Modulators: A Case Study from Analyses of New Metabotropic Glutamate Receptor 5 (mGlu5) X-ray Structures. J. Med. Chem. 2018, 62, 207–222. [Google Scholar] [CrossRef]
- Monn, J.A.; Henry, S.S.; Massey, S.M.; Clawson, D.K.; Chen, Q.; Diseroad, B.A.; Bhardwaj, R.M.; Atwell, S.; Lu, F.; Wang, J.; et al. Synthesis and Pharmacological Characterization of C4β-Amide-Substituted 2-Aminobicyclo[3.1.0]hexane-2,6-dicarboxylates. Identification of (1S,2S,4S,5R,6S)-2-Amino-4-[(3-methoxybenzoyl)amino]bicyclo[3.1.0]hexane-2,6-dicarboxylic Acid (LY2794193), a Highly Potent and Selective mGlu3 Receptor Agonist. J. Med. Chem. 2018, 61, 2303–2328. [Google Scholar]
- Lindsley, C.W.; Emmitte, K.A.; Hopkins, C.R.; Bridges, T.M.; Gregory, K.J.; Niswender, C.M.; Conn, P.J. Practical Strategies and Concepts in GPCR Allosteric Modulator Discovery: Recent Advances with Metabotropic Glutamate Receptors. Chem. Rev. 2016, 116, 6707–6741. [Google Scholar] [CrossRef]
- Tresadern, G.; Rombouts, F.J.R.; Oehlrich, D.; Macdonald, G.; Trabanco, A.A. Industrial medicinal chemistry insights: Neuroscience hit generation at Janssen. Drug Discov. Today 2017, 22, 1478–1488. [Google Scholar] [CrossRef]
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martínez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing Pitfalls in Virtual Screening: A Critical Review. J. Chem. Inf. Model. 2012, 52, 867–881. [Google Scholar] [CrossRef]
- Ciordia, M.; Pérez-Benito, L.; Delgado, F.; Trabanco, A.S.A.; Tresadern, G. Application of free energy perturbation for the design of BACE1 inhibitors. J. Chem. Inf. Model. 2016, 56, 1856–1871. [Google Scholar] [CrossRef]
- Keränen, H.; Pérez-Benito, L.; Ciordia, M.; Delgado, F.; Steinbrecher, T.B.; Oehlrich, D.; van Vlijmen, H.W.; Trabanco, A.S.A.; Tresadern, G. Acylguanidine beta secretase 1 inhibitors: A combined experimental and free energy perturbation study. J. Chem. Theory Comput. 2017, 13, 1439–1453. [Google Scholar] [CrossRef]
- Pérez-Benito, L.; Keränen, H.; van Vlijmen, H.; Tresadern, G. Predicting Binding Free Energies of PDE2 Inhibitors. The Difficulties of Protein Conformation. Sci. Rep. 2018, 8, 4883. [Google Scholar] [CrossRef]
- Nicoletti, F.; Bruno, V.; Ngomba, R.T.; Gradini, R.; Battaglia, G. Metabotropic glutamate receptors as drug targets: What’s new? Curr. Opin. Pharmacol. 2015, 20, 89–94. [Google Scholar] [CrossRef]
- Mary, S.; Gomeza, J.; Prézeau, L.; Bockaert, J.; Pin, J.-P. A Cluster of Basic Residues in the Carboxyl-terminal Tail of the Short Metabotropic Glutamate Receptor 1 Variants Impairs Their Coupling to Phospholipase C. J. Biol. Chem. 1998, 273, 425–432. [Google Scholar] [CrossRef]
- Ray, K.; Hauschild, B.C. Cys-140 Is Critical for Metabotropic Glutamate Receptor-1 Dimerization. J. Biol. Chem. 2000, 275, 34245–34251. [Google Scholar] [CrossRef]
- Litschig, S.; Gasparini, F.; Rueegg, D.; Stoehr, N.; Flor, P.J.; Vranesic, I.; Prézeau, L.; Pin, J.-P.; Thomsen, C.; Kuhn, R. CPCCOEt, a Noncompetitive Metabotropic Glutamate Receptor 1 Antagonist, Inhibits Receptor Signaling Without Affecting Glutamate Binding. Mol. Pharmacol. 1999, 55, 453. [Google Scholar]
- Pagano, A.; Rüegg, D.; Litschig, S.; Stoehr, N.; Stierlin, C.; Heinrich, M.; Floersheim, P.; Prezèau, L.; Carroll, F.; Pin, J.-P.; et al. The Non-competitive Antagonists 2-Methyl-6-(phenylethynyl)pyridine and 7-Hydroxyiminocyclopropan[b]chromen-1a-carboxylic Acid Ethyl Ester Interact with Overlapping Binding Pockets in the Transmembrane Region of Group I Metabotropic Glutamate Receptors. J. Biol. Chem. 2000, 275, 33750–33758. [Google Scholar] [CrossRef]
- Malherbe, P.; Kratochwil, N.; Zenner, M.-T.; Piussi, J.; Diener, C.; Kratzeisen, C.; Fischer, C.; Porter, R.H.P. Mutational Analysis and Molecular Modeling of the Binding Pocket of the Metabotropic Glutamate 5 Receptor Negative Modulator 2-Methyl-6-(phenylethynyl)-pyridine. Mol. Pharmacol. 2003, 64, 823. [Google Scholar] [CrossRef]
- Knoflach, F.; Mutel, V.; Jolidon, S.; Kew, J.N.C.; Malherbe, P.; Vieira, E.; Wichmann, J.; Kemp, J.A. Positive allosteric modulators of metabotropic glutamate 1 receptor: Characterization, mechanism of action, and binding site. Proc. Natl. Acad. Sci. USA 2001, 98, 13402. [Google Scholar] [CrossRef]
- Hemstapat, K.; de Paulis, T.; Chen, Y.; Brady, A.E.; Grover, V.K.; Alagille, D.; Tamagnan, G.D.; Conn, P.J. A Novel Class of Positive Allosteric Modulators of Metabotropic Glutamate Receptor Subtype 1 Interact with a Site Distinct from That of Negative Allosteric Modulators. Mol. Pharmacol. 2006, 70, 616. [Google Scholar] [CrossRef]
- Fukuda, J.; Suzuki, G.; Kimura, T.; Nagatomi, Y.; Ito, S.; Kawamoto, H.; Ozaki, S.; Ohta, H. Identification of a novel transmembrane domain involved in the negative modulation of mGluR1 using a newly discovered allosteric mGluR1 antagonist, 3-cyclohexyl-5-fluoro-6-methyl-7-(2-morpholin-4-ylethoxy)-4H-chromen-4-one. Neuropharmacology 2009, 57, 438–445. [Google Scholar] [CrossRef]
- Chen, Y.; Goudet, C.; Pin, J.-P.; Conn, P.J. N-{4-Chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hydroxybenzamide (CPPHA) Acts through a Novel Site as a Positive Allosteric Modulator of Group 1 Metabotropic Glutamate Receptors. Mol. Pharmacol. 2008, 73, 909. [Google Scholar] [CrossRef]
- Suzuki, G.; Kimura, T.; Satow, A.; Kaneko, N.; Fukuda, J.; Hikichi, H.; Sakai, N.; Maehara, S.; Kawagoe-Takaki, H.; Hata, M.; et al. Pharmacological characterization of a new, orally active and potent allosteric metabotropic glutamate receptor 1 antagonist, 4-[1-(2-fluoropyridin-3-yl)-5-methyl-1H-1,2,3-triazol-4-yl]-N-isopropyl-N-methyl-3,6-dihydropyridine-1(2H)-carboxamide (FTIDC). J. Pharmacol. Exp. Ther. 2007, 321, 1144. [Google Scholar] [CrossRef]
- Noeske, T.; Jirgensons, A.; Starchenkovs, I.; Renner, S.; Jaunzeme, I.; Trifanova, D.; Hechenberger, M.; Bauer, T.; Kauss, V.; Parsons, C.G.; et al. Virtual Screening for Selective Allosteric mGluR1 Antagonists and Structure-Activity Relationship Investigations for Coumarine Derivatives. ChemMedChem 2007, 2, 1763–1773. [Google Scholar] [CrossRef]
- Wang, M.; Hampson, D.R. An evaluation of automated in silico ligand docking of amino acid ligands to Family C G-protein coupled receptors. Biorg. Med. Chem. 2006, 14, 2032–2039. [Google Scholar] [CrossRef]
- Vanejevs, M.; Jatzke, C.; Renner, S.; Müller, S.; Hechenberger, M.; Bauer, T.; Klochkova, A.; Pyatkin, I.; Kazyulkin, D.; Aksenova, E.; et al. Positive and Negative Modulation of Group I Metabotropic Glutamate Receptors. J. Med. Chem. 2008, 51, 634–647. [Google Scholar] [CrossRef]
- Harpsøe, K.; Isberg, V.; Tehan, B.G.; Weiss, D.; Arsova, A.; Marshall, F.H.; Bräuner-Osborne, H.; Gloriam, D.E. Selective Negative Allosteric Modulation Of Metabotropic Glutamate Receptors—A Structural Perspective of Ligands and Mutants. Sci. Rep. 2015, 5, 13869. [Google Scholar] [CrossRef]
- Munk, C.; Harpsøe, K.; Hauser, A.S.; Isberg, V.; Gloriam, D.E. Integrating structural and mutagenesis data to elucidate GPCR ligand binding. Curr. Opin. Pharmacol. 2016, 30, 51–58. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, X.; Chen, X.; He, Y.; Qiao, L.; Zhang, Y.; Li, G.; Xiang, Y. Virtual Screening and Molecular Dynamics Study of Potential Negative Allosteric Modulators of mGluR1 from Chinese Herbs. Molecules 2015, 20, 12769–12786. [Google Scholar] [CrossRef]
- Jang, J.W.; Cho, N.-C.; Min, S.-J.; Cho, Y.S.; Park, K.D.; Seo, S.H.; No, K.T.; Pae, A.N. Novel Scaffold Identification of mGlu1 Receptor Negative Allosteric Modulators Using a Hierarchical Virtual Screening Approach. Chem. Biol. Drug Des. 2016, 87, 239–256. [Google Scholar] [CrossRef]
- Lakkaraju, S.K.; Yu, W.; Raman, E.P.; Hershfeld, A.V.; Fang, L.; Deshpande, D.A.; MacKerell, A.D. Mapping Functional Group Free Energy Patterns at Protein Occluded Sites: Nuclear Receptors and G-Protein Coupled Receptors. J. Chem. Inf. Model. 2015, 55, 700–708. [Google Scholar] [CrossRef]
- van Westen, G.J.P.; Gaulton, A.; Overington, J.P. Chemical, Target, and Bioactive Properties of Allosteric Modulation. PLoS Comput. Biol. 2014, 10, e1003559. [Google Scholar] [CrossRef]
- Bai, Q.; Yao, X. Investigation of allosteric modulation mechanism of metabotropic glutamate receptor 1 by molecular dynamics simulations, free energy and weak interaction analysis. Sci. Rep. 2016, 6, 21763. [Google Scholar] [CrossRef]
- Śliwa, P.; Kurczab, R.; Bojarski, A.J. ONIOM and FMO-EDA study of metabotropic glutamate receptor 1: Quantum insights into the allosteric binding site. Int. J. Quantum. Chem. 2018, 118, e25617. [Google Scholar] [CrossRef]
- Borroto-Escuela, D.O.; Fuxe, K. Basimglurant for treatment of major depressive disorder: A novel negative allosteric modulator of metabotropic glutamate receptor 5. Expert Opin. Investig. Drugs 2015, 24, 1247–1260. [Google Scholar]
- Waung, M.W.; Akerman, S.; Wakefield, M.; Keywood, C.; Goadsby, P.J. Metabotropic glutamate receptor 5: A target for migraine therapy. Ann. Clin. Trans. Neurol. 2016, 3, 560–571. [Google Scholar] [CrossRef]
- Scharf, S.H.; Jaeschke, G.; Wettstein, J.G.; Lindemann, L. Metabotropic glutamate receptor 5 as drug target for Fragile X syndrome. Curr. Opin. Pharmacol. 2015, 20, 124–134. [Google Scholar] [CrossRef]
- Mühlemann, A.; Ward, N.A.; Kratochwil, N.; Diener, C.; Fischer, C.; Stucki, A.; Jaeschke, G.; Malherbe, P.; Porter, R.H.P. Determination of key amino acids implicated in the actions of allosteric modulation by 3,3′-difluorobenzaldazine on rat mGlu5 receptors. Eur. J. Pharmacol. 2006, 529, 95–104. [Google Scholar] [CrossRef]
- Chen, Y.; Nong, Y.; Goudet, C.; Hemstapat, K.; de Paulis, T.; Pin, J.-P.; Conn, P.J. Interaction of Novel Positive Allosteric Modulators of Metabotropic Glutamate Receptor 5 with the Negative Allosteric Antagonist Site Is Required for Potentiation of Receptor Responses. Mol. Pharmacol. 2007, 71, 1389. [Google Scholar] [CrossRef]
- Gregory, K.J.; Nguyen, E.D.; Reiff, S.D.; Squire, E.F.; Stauffer, S.R.; Lindsley, C.W.; Meiler, J.; Conn, P.J. Probing the Metabotropic Glutamate Receptor 5 (mGlu5) Positive Allosteric Modulator (PAM) Binding Pocket: Discovery of Point Mutations That Engender a “Molecular Switch” in PAM Pharmacology. Mol. Pharmacol. 2013, 83, 991. [Google Scholar] [CrossRef]
- Gregory, K.J.; Noetzel, M.J.; Rook, J.M.; Vinson, P.N.; Stauffer, S.R.; Rodriguez, A.L.; Emmitte, K.A.; Zhou, Y.; Chun, A.C.; Felts, A.S.; et al. Investigating Metabotropic Glutamate Receptor 5 Allosteric Modulator Cooperativity, Affinity, and Agonism: Enriching Structure-Function Studies and Structure-Activity Relationships. Mol. Pharmacol. 2012, 82, 860. [Google Scholar] [CrossRef]
- Turlington, M.; Noetzel, M.J.; Chun, A.; Zhou, Y.; Gogliotti, R.D.; Nguyen, E.D.; Gregory, K.J.; Vinson, P.N.; Rook, J.M.; Gogi, K.K.; et al. Exploration of Allosteric Agonism Structure–Activity Relationships within an Acetylene Series of Metabotropic Glutamate Receptor 5 (mGlu5) Positive Allosteric Modulators (PAMs): Discovery of 5-((3-Fluorophenyl)ethynyl)-N-(3-methyloxetan-3-yl)picolinamide (ML254). J. Med. Chem. 2013, 56, 7976–7996. [Google Scholar]
- Gregory, K.J.; Nguyen, E.D.; Malosh, C.; Mendenhall, J.L.; Zic, J.Z.; Bates, B.S.; Noetzel, M.J.; Squire, E.F.; Turner, E.M.; Rook, J.M.; et al. Identification of Specific Ligand-Receptor Interactions That Govern Binding and Cooperativity of Diverse Modulators to a Common Metabotropic Glutamate Receptor 5 Allosteric Site. ACS Chem. Neurosci. 2014, 5, 282–295. [Google Scholar] [CrossRef]
- Malherbe, P.; Kratochwil, N.; Mühlemann, A.; Zenner, M.-T.; Fischer, C.; Stahl, M.; Gerber, P.R.; Jaeschke, G.; Porter, R.H.P. Comparison of the binding pockets of two chemically unrelated allosteric antagonists of the mGlu5 receptor and identification of crucial residues involved in the inverse agonism of MPEP. J. Neurochem. 2006, 98, 601–615. [Google Scholar] [CrossRef]
- Noeske, T.; Trifanova, D.; Kauss, V.; Renner, S.; Parsons, C.G.; Schneider, G.; Weil, T. Synergism of virtual screening and medicinal chemistry: Identification and optimization of allosteric antagonists of metabotropic glutamate receptor 1. Biorg. Med. Chem. 2009, 17, 5708–5715. [Google Scholar] [CrossRef]
- Renner, S.; Hechenberger, M.; Noeske, T.; Böcker, A.; Jatzke, C.; Schmuker, M.; Parsons, C.G.; Weil, T.; Schneider, G. Searching for Drug Scaffolds with 3D Pharmacophores and Neural Network Ensembles. Angew. Chem. Int. Ed. 2007, 46, 5336–5339. [Google Scholar] [CrossRef]
- Lowe, E.W.; Ferrebee, A.; Rodriguez, A.L.; Conn, P.J.; Meiler, J. 3D-QSAR CoMFA study of benzoxazepine derivatives as mGluR5 positive allosteric modulators. Bioorg. Med. Chem. Lett. 2010, 20, 5922–5924. [Google Scholar] [CrossRef]
- Mueller, R.; Dawson, E.S.; Meiler, J.; Rodriguez, A.L.; Chauder, B.A.; Bates, B.S.; Felts, A.S.; Lamb, J.P.; Menon, U.N.; Jadhav, S.B.; et al. Discovery of 2-(2-Benzoxazoyl amino)-4-Aryl-5-Cyanopyrimidine as Negative Allosteric Modulators (NAMs) of Metabotropic Glutamate Receptor 5 (mGlu5): From an Artificial Neural Network Virtual Screen to an In Vivo Tool Compound. ChemMedChem 2012, 7, 406–414. [Google Scholar] [CrossRef]
- Kaae, B.H.; Harpsøe, K.; Kvist, T.; Mathiesen, J.M.; Mølck, C.; Gloriam, D.; Jimenez, H.N.; Uberti, M.A.; Nielsen, S.M.; Nielsen, B.; et al. Structure-Activity Relationships for Negative Allosteric mGluR5 Modulators. ChemMedChem 2012, 7, 440–451. [Google Scholar] [CrossRef]
- Dalton, J.A.R.; Gómez-Santacana, X.; Llebaria, A.; Giraldo, J. Computational Analysis of Negative and Positive Allosteric Modulator Binding and Function in Metabotropic Glutamate Receptor 5 (In)Activation. J. Chem. Inf. Model. 2014, 54, 1476–1487. [Google Scholar] [CrossRef]
- Anighoro, A.; Graziani, D.; Bettinelli, I.; Cilia, A.; De Toma, C.; Longhi, M.; Mangiarotti, F.; Menegon, S.; Pirona, L.; Poggesi, E.; et al. Insights into the interaction of negative allosteric modulators with the metabotropic glutamate receptor 5: Discovery and computational modeling of a new series of ligands with nanomolar affinity. Biorg. Med. Chem. 2015, 23, 3040–3058. [Google Scholar] [CrossRef]
- Feng, Z.; Ma, S.; Hu, G.; Xie, X.-Q. Allosteric Binding Site and Activation Mechanism of Class C G-Protein Coupled Receptors: Metabotropic Glutamate Receptor Family. AAPS J. 2015, 17, 737–753. [Google Scholar] [CrossRef]
- Dalton, J.A.R.; Pin, J.-P.; Giraldo, J. Analysis of positive and negative allosteric modulation in metabotropic glutamate receptors 4 and 5 with a dual ligand. Sci. Rep. 2017, 7, 4944. [Google Scholar] [CrossRef]
- Bian, Y.; Feng, Z.; Yang, P.; Xie, X.-Q. Integrated In Silico Fragment-Based Drug Design: Case Study with Allosteric Modulators on Metabotropic Glutamate Receptor 5. AAPS J. 2017, 19, 1235–1248. [Google Scholar] [CrossRef]
- Fu, T.; Zheng, G.; Tu, G.; Yang, F.; Chen, Y.; Yao, X.; Li, X.; Xue, W.; Zhu, F. Exploring the Binding Mechanism of Metabotropic Glutamate Receptor 5 Negative Allosteric Modulators in Clinical Trials by Molecular Dynamics Simulations. ACS Chem. Neurosci. 2018, 9, 1492–1502. [Google Scholar] [CrossRef]
- Vijaya Prabhu, S.; Singh, S.K. E-pharmacophore-based screening of mGluR5 negative allosteric modulators for central nervous system disorder. Comput. Biol. Chem. 2018, 78, 414–423. [Google Scholar] [CrossRef]
- Vijaya Prabhu, S.; Singh, S.K. Atom-based 3D-QSAR, induced fit docking, and molecular dynamics simulations study of thieno[2–b]pyridines negative allosteric modulators of mGluR5. J. Recept. Signal Transduct. 2018, 38, 225–239. [Google Scholar] [CrossRef]
- Llinas del Torrent, C.; Casajuana-Martin, N.; Pardo, L.; Tresadern, G.; Pérez-Benito, L. Mechanisms Underlying Allosteric Molecular Switches of Metabotropic Glutamate Receptor 5. J. Chem. Inf. Model. 2019. [Google Scholar] [CrossRef] [PubMed]
- Trabanco, A.A.; Cid, J.M.; Lavreysen, H.; Macdonald, G.J.; Tresadern, G. Progress in the Developement of Positive Allosteric Modulators of the Metabotropic Glutamate Receptor 2. Curr. Med. Chem. 2011, 18, 47–68. [Google Scholar] [CrossRef] [PubMed]
- Trabanco, A.A.; Cid, J.M. mGluR2 positive allosteric modulators: A patent review (2009–present). Expert Opin. Ther. Patents 2013, 23, 629–647. [Google Scholar] [CrossRef]
- Trabanco, A.A.; Tresadern, G.; Macdonald, G.J.; Vega, J.A.; de Lucas, A.I.; Matesanz, E.; García, A.; Linares, M.L.; Alonso de Diego, S.A.; Alonso, J.M.; et al. Imidazo[1,2-a]pyridines: Orally Active Positive Allosteric Modulators of the Metabotropic Glutamate 2 Receptor. J. Med. Chem. 2012, 55, 2688–2701. [Google Scholar] [CrossRef]
- Cid, J.M.; Duvey, G.; Cluzeau, P.; Nhem, V.; Macary, K.; Raux, A.; Poirier, N.; Muller, J.; Boléa, C.; Finn, T.; et al. Discovery of 1,5-Disubstituted Pyridones: A New Class of Positive Allosteric Modulators of the Metabotropic Glutamate 2 Receptor. ACS Chem. Neurosci. 2010, 1, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Cid, J.M.; Tresadern, G.; Duvey, G.; Lütjens, R.; Finn, T.; Rocher, J.-P.; Poli, S.; Vega, J.A.; de Lucas, A.I.; Matesanz, E.; et al. Discovery of 1-Butyl-3-chloro-4-(4-phenyl-1-piperidinyl)-(1H)-pyridone (JNJ-40411813): A Novel Positive Allosteric Modulator of the Metabotropic Glutamate 2 Receptor. J. Med. Chem. 2014, 57, 6495–6512. [Google Scholar] [CrossRef] [PubMed]
- Cid, J.M.; Duvey, G.; Tresadern, G.; Nhem, V.; Furnari, R.; Cluzeau, P.; Vega, J.A.; de Lucas, A.I.; Matesanz, E.; Alonso, J.M.; et al. Discovery of 1,4-Disubstituted 3-Cyano-2-pyridones: A New Class of Positive Allosteric Modulators of the Metabotropic Glutamate 2 Receptor. J. Med. Chem. 2012, 55, 2388–2405. [Google Scholar] [CrossRef] [PubMed]
- Lundström, L.; Bissantz, C.; Beck, J.; Wettstein, J.G.; Woltering, T.J.; Wichmann, J.; Gatti, S. Structural determinants of allosteric antagonism at metabotropic glutamate receptor 2: Mechanistic studies with new potent negative allosteric modulators. Br. J. Pharmacol. 2011, 164, 521–537. [Google Scholar] [CrossRef]
- Lundström, L.; Kuhn, B.; Beck, J.; Borroni, E.; Wettstein, J.G.; Woltering, T.J.; Gatti, S. Mutagenesis and Molecular Modeling of the Orthosteric Binding Site of the mGlu2 Receptor Determining Interactions of the Group II Receptor Antagonist 3H-HYDIA. ChemMedChem 2009, 4, 1086–1094. [Google Scholar] [CrossRef]
- Farinha, A.; Lavreysen, H.; Peeters, L.; Russo, B.; Masure, S.; Trabanco, A.A.; Cid, J.; Tresadern, G. Molecular determinants of positive allosteric modulation of the human metabotropic glutamate receptor 2. Br. J. Pharmacol. 2015, 172, 2383–2396. [Google Scholar] [CrossRef]
- Pérez-Benito, L.; Doornbos, M.L.J.; Cordomí, A.; Peeters, L.; Lavreysen, H.; Pardo, L.; Tresadern, G. Molecular Switches of Allosteric Modulation of the Metabotropic Glutamate 2 Receptor. Structure 2017, 25, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Doornbos, M.L.J.; Wang, X.; Vermond, S.C.; Peeters, L.; Pérez-Benito, L.; Trabanco, A.A.; Lavreysen, H.; Cid, J.M.; Heitman, L.H.; Tresadern, G.; et al. Covalent Allosteric Probe for the Metabotropic Glutamate Receptor 2: Design, Synthesis, and Pharmacological Characterization. J. Med. Chem. 2018, 62, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Doornbos, M.L.J.; Pérez-Benito, L.; Tresadern, G.; Mulder-Krieger, T.; Biesmans, I.; Trabanco, A.A.; Cid, J.M.; Lavreysen, H.; Ijzerman, A.P.; Heitman, L.H. Molecular mechanism of positive allosteric modulation of the metabotropic glutamate receptor 2 by JNJ-46281222. Br. J. Pharmacol. 2015, 173, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Cid, J.M.; Tresadern, G.; Vega, J.A.; de Lucas, A.I.; del Cerro, A.; Matesanz, E.; Linares, M.L.; García, A.; Iturrino, L.; Pérez-Benito, L.; et al. Discovery of 8-Trifluoromethyl-3-cyclopropylmethyl-7-[(4-(2,4-difluorophenyl)-1-piperazinyl)methyl]-1,2,4-triazolo[4,3-a]pyridine (JNJ-46356479), a Selective and Orally Bioavailable mGlu2 Receptor Positive Allosteric Modulator (PAM). J. Med. Chem. 2016, 59, 8495–8507. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Guadix, A.E.; Costantino, G. Molecular Dynamics Simulation of the Heterodimeric mGluR2/5HT2A Complex. An Atomistic Resolution Study of a Potential New Target in Psychiatric Conditions. J. Chem. Inf. Model. 2009, 49, 1602–1616. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Costantino, G.; de Fabritiis, G.; Pastor, M.; Selent, J. Membrane-Sensitive Conformational States of Helix 8 in the Metabotropic Glu2 Receptor, a Class C GPCR. PLoS ONE 2012, 7, e42023. [Google Scholar] [CrossRef]
- Xue, L.; Rovira, X.; Scholler, P.; Zhao, H.; Liu, J.; Pin, J.-P.; Rondard, P. Major ligand-induced rearrangement of the heptahelical domain interface in a GPCR dimer. Nat. Chem. Biol. 2014, 11, 134. [Google Scholar] [CrossRef]
- Tresadern, G.; Cid, J.M.; Macdonald, G.J.; Vega, J.A.; de Lucas, A.I.; García, A.; Matesanz, E.; Linares, M.L.; Oehlrich, D.; Lavreysen, H.; et al. Scaffold hopping from pyridones to imidazo[1,2-a]pyridines. New positive allosteric modulators of metabotropic glutamate 2 receptor. Bioorg. Med. Chem. Lett. 2010, 20, 175–179. [Google Scholar] [CrossRef]
- Cid, J.M.; Tresadern, G.; Vega, J.A.; de Lucas, A.I.; Matesanz, E.; Iturrino, L.; Linares, M.L.; Garcia, A.; Andrés, J.I.; Macdonald, G.J.; et al. Discovery of 3-Cyclopropylmethyl-7-(4-phenylpiperidin-1-yl)-8-trifluoromethyl[1,2,4]triazolo[4,3-a]pyridine (JNJ-42153605): A Positive Allosteric Modulator of the Metabotropic Glutamate 2 Receptor. J. Med. Chem. 2012, 55, 8770–8789. [Google Scholar] [CrossRef]
- Ahnaou, A.; Lavreysen, H.; Tresadern, G.; Cid, J.M.; Drinkenburg, W.H. mGlu2 Receptor Agonism, but Not Positive Allosteric Modulation, Elicits Rapid Tolerance towards Their Primary Efficacy on Sleep Measures in Rats. PLoS ONE 2015, 10, e0144017. [Google Scholar] [CrossRef]
- Szabó, G.; Túrós, G.I.; Kolok, S.; Vastag, M.; Sánta, Z.; Dékány, M.; Lévay, G.I.; Greiner, I.; Natsumi, M.; Tatsuya, W.; et al. Fragment Based Optimization of Metabotropic Glutamate Receptor 2 (mGluR2) Positive Allosteric Modulators in the Absence of Structural Information. J. Med. Chem. 2019, 62, 234–246. [Google Scholar] [CrossRef]
- Tresadern, G.; Cid, J.-M.; Trabanco, A.A. QSAR design of triazolopyridine mGlu2 receptor positive allosteric modulators. J. Mol. Graph. Model. 2014, 53, 82–91. [Google Scholar] [CrossRef]
- Harrison, P.J.; Lyon, L.; Sartorius, L.J.; Burnet, P.; Lane, T.A. Review: The group II metabotropic glutamate receptor 3 (mGluR3, mGlu3, GRM3): Expression, function and involvement in schizophrenia. J. Psychopharmacol. 2008, 22, 308–322. [Google Scholar] [CrossRef]
- Maksymetz, J.; Moran, S.P.; Conn, P.J. Targeting metabotropic glutamate receptors for novel treatments of schizophrenia. Mol. Brain 2017, 10, 15. [Google Scholar] [CrossRef]
- Yao, Y.; Pattabiraman, N.; Michne, W.F.; Huang, X.-P.; Hampson, D.R. Molecular modeling and mutagenesis of the ligand-binding pocket of the mGlu3 subtype of metabotropic glutamate receptor. J. Neurochem. 2003, 86, 947–957. [Google Scholar] [CrossRef]
- DiRaddo, J.O.; Miller, E.J.; Bowman-Dalley, C.; Wroblewska, B.; Javidnia, M.; Grajkowska, E.; Wolfe, B.B.; Liotta, D.C.; Wroblewski, J.T. Chloride is an Agonist of Group II and III Metabotropic Glutamate Receptors. Mol. Pharmacol. 2015, 88, 450–459. [Google Scholar] [CrossRef]
- Tora, A.S.; Rovira, X.; Cao, A.-M.; Cabayé, A.; Olofsson, L.; Malhaire, F.; Scholler, P.; Baik, H.; Van Eeckhaut, A.; Smolders, I.; et al. Chloride ions stabilize the glutamate-induced active state of the metabotropic glutamate receptor 3. Neuropharmacology 2018, 140, 275–286. [Google Scholar] [CrossRef]
- Doornbos, M.L.J.; Van der Linden, I.; Vereyken, L.; Tresadern, G.; Ijzerman, A.P.; Lavreysen, H.; Heitman, L.H. Constitutive activity of the metabotropic glutamate receptor 2 explored with a whole-cell label-free biosensor. Biochem. Pharmacol. 2018, 152, 201–210. [Google Scholar] [CrossRef]
- Monn, J.A.; Valli, M.J.; Massey, S.M.; Wright, R.A.; Salhoff, C.R.; Johnson, B.G.; Howe, T.; Alt, C.A.; Rhodes, G.A.; Robey, R.L.; et al. Design, Synthesis, and Pharmacological Characterization of (+)-2-Aminobicyclo[3.1.0]hexane-2,6-dicarboxylic Acid (LY354740): A Potent, Selective, and Orally Active Group 2 Metabotropic Glutamate Receptor Agonist Possessing Anticonvulsant and Anxiolytic Properties. J. Med. Chem. 1997, 40, 528–537. [Google Scholar]
- Monn, J.A.; Massey, S.M.; Valli, M.J.; Henry, S.S.; Stephenson, G.A.; Bures, M.; Hérin, M.; Catlow, J.; Giera, D.; Wright, R.A.; et al. Synthesis and Metabotropic Glutamate Receptor Activity of S-Oxidized Variants of (−)-4-Amino-2-thiabicyclo-[3.1.0]hexane-4,6-dicarboxylate: Identification of Potent, Selective, and Orally Bioavailable Agonists for mGlu2/3 Receptors. J. Med. Chem. 2007, 50, 233–240. [Google Scholar] [CrossRef]
- Monn, J.A.; Prieto, L.; Taboada, L.; Pedregal, C.; Hao, J.; Reinhard, M.R.; Henry, S.S.; Goldsmith, P.J.; Beadle, C.D.; Walton, L.; et al. Synthesis and Pharmacological Characterization of C4-Disubstituted Analogs of 1S,2S,5R,6S-2-Aminobicyclo[3.1.0]hexane-2,6-dicarboxylate: Identification of a Potent, Selective Metabotropic Glutamate Receptor Agonist and Determination of Agonist-Bound Human mGlu2 and mGlu3 Amino Terminal Domain Structures. J. Med. Chem. 2015, 58, 1776–1794. [Google Scholar] [PubMed]
- Hopkins, C.R.; Lindsley, C.W.; Niswender, C.M. mGluR4-positive allosteric modulation as potential treatment for Parkinson’s disease. Future Med. Chem. 2009, 1, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Selvam, C.; Oueslati, N.; Lemasson, I.A.; Brabet, I.; Rigault, D.; Courtiol, T.; Cesarini, S.; Triballeau, N.; Bertrand, H.-O.; Goudet, C.; et al. A Virtual Screening Hit Reveals New Possibilities for Developing Group III Metabotropic Glutamate Receptor Agonists. J. Med. Chem. 2010, 53, 2797–2813. [Google Scholar] [CrossRef] [PubMed]
- Rovira, X.; Malhaire, F.; Scholler, P.; Rodrigo, J.; Gonzalez-Bulnes, P.; Llebaria, A.; Pin, J.-P.; Giraldo, J.; Goudet, C. Overlapping binding sites drive allosteric agonism and positive cooperativity in type 4 metabotropic glutamate receptors. FASEB J. 2014, 29, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Villa, H.; Trabanco, A.A. Progress toward allosteric ligands of metabotropic glutamate 7 (mGlu7) receptor: 2008–present. MedChemComm 2019, 10, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Reed, C.W.; Yohn, S.E.; Washecheck, J.P.; Roenfanz, H.F.; Quitalig, M.C.; Luscombe, V.B.; Jenkins, M.T.; Rodriguez, A.L.; Engers, D.W.; Blobaum, A.L.; et al. Discovery of an Orally Bioavailable and Central Nervous System (CNS) Penetrant mGlu7 Negative Allosteric Modulator (NAM) in Vivo Tool Compound: N-(2-(1H-1,2,4-triazol-1-yl)-5-(trifluoromethoxy)phenyl)-4-(cyclopropylmethoxy)-3-methoxybenzamide (VU6012962). J. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tresadern, G.; Trabanco, A.A.; Pérez-Benito, L.; Overington, J.P.; van Vlijmen, H.W.T.; van Westen, G.J.P. Identification of Allosteric Modulators of Metabotropic Glutamate 7 Receptor Using Proteochemometric Modeling. J. Chem. Inf. Model. 2017, 57, 2976–2985. [Google Scholar] [CrossRef]
- Cid, J.M.; Lavreysen, H.; Tresadern, G.; Pérez-Benito, L.; Tovar, F.; Fontana, A.; Trabanco, A.A. Computationally Guided Identification of Allosteric Agonists of the Metabotropic Glutamate 7 Receptor. ACS Chem. Neurosci. 2018. [Google Scholar] [CrossRef]
- Cleva, R.M.; Olive, M.F. Metabotropic glutamate receptors and drug addiction. Wiley Interdiscip.Rev. Membr. Transp. Signal. 2012, 1, 281–295. [Google Scholar] [CrossRef]
- Duvoisin, R.M.; Pfankuch, T.; Wilson, J.M.; Grabell, J.; Chhajlani, V.; Brown, D.G.; Johnson, E.; Raber, J. Acute pharmacological modulation of mGluR8 reduces measures of anxiety. Behav. Brain Res. 2010, 212, 168–173. [Google Scholar] [CrossRef]
- Chen, Q.; Ho, J.D.; Ashok, S.; Vargas, M.C.; Wang, J.; Atwell, S.; Bures, M.; Schkeryantz, J.M.; Monn, J.A.; Hao, J. Structural Basis for (S)-3,4-Dicarboxyphenylglycine (DCPG) As a Potent and Subtype Selective Agonist of the mGlu8 Receptor. J. Med. Chem. 2018, 61, 10040–10052. [Google Scholar] [CrossRef]
- Bessis, A.-S.; Rondard, P.; Gaven, F.; Brabet, I.; Triballeau, N.; Prézeau, L.; Acher, F.; Pin, J.-P. Closure of the Venus flytrap module of mGlu8 receptor and the activation process: Insights from mutations converting antagonists into agonists. Proc. Natl. Acad. Sci. USA 2002, 99, 11097. [Google Scholar] [CrossRef]
- Schkeryantz, J.M.; Chen, Q.; Ho, J.D.; Atwell, S.; Zhang, A.; Vargas, M.C.; Wang, J.; Monn, J.A.; Hao, J. Determination of L-AP4-bound human mGlu8 receptor amino terminal domain structure and the molecular basis for L-AP4′s group III mGlu receptor functional potency and selectivity. Bioorg. Med. Chem. Lett. 2018, 28, 612–617. [Google Scholar] [CrossRef]
- Lenselink, E.B.; Louvel, J.; Forti, A.F.; van Veldhoven, J.P.D.; de Vries, H.; Mulder-Krieger, T.; McRobb, F.M.; Negri, A.; Goose, J.; Abel, R.; et al. Predicting Binding Affinities for GPCR Ligands Using Free-Energy Perturbation. ACS Omega 2016, 1, 293–304. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | PDB ID | Method | Resolution | Description | Reference |
|---|---|---|---|---|---|
| mGlu1 | 4OR2 | X-ray diffraction | 2.8 Å | Crystal structure of 7TM domain of human mGlu1 bound to FITM (NAM). Soluble cytochrome b562 present for stabilization. | [17] |
| mGlu5 | 4OO9 | X-ray diffraction | 2.6 Å | Structure of the human 7TM domain of mGlu5 bound to mavoglurant (NAM). Lysozyme used for stabilization. | [16] |
| 5CGC | X-ray diffraction | 3.1 Å | Structure of human 7TM domain of mGlu5 bound to 3-chloro-4-fluoro-5-[6-(1H-pyrazol-1-yl)pyrimidin-4-yl]benzonitrile (NAM). Endolysin present in the structure used for stabilization. | [18] | |
| 5CGD | X-ray diffraction | 2.6 Å | Structure of human 7TM of mGlu5 bound to HTL14242 (NAM). Endolysin present in the structure used for stabilization. | [18] | |
| 6FFH | X-ray diffraction | 2.7 Å | Crystal structure of 7TM domain of human mGlu5 bound to fenobam (NAM). Endolysin present in the structure for stabilization. | [19] | |
| 6FFI | X-ray diffraction | 2.2 Å | Crystal structure of 7TM domain of human mGlu5 bound to M-MPEP (NAM). Endolysin present for stabilization. | [19] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Llinas del Torrent, C.; Pérez-Benito, L.; Tresadern, G. Computational Drug Design Applied to the Study of Metabotropic Glutamate Receptors. Molecules 2019, 24, 1098. https://doi.org/10.3390/molecules24061098
Llinas del Torrent C, Pérez-Benito L, Tresadern G. Computational Drug Design Applied to the Study of Metabotropic Glutamate Receptors. Molecules. 2019; 24(6):1098. https://doi.org/10.3390/molecules24061098
Chicago/Turabian StyleLlinas del Torrent, Claudia, Laura Pérez-Benito, and Gary Tresadern. 2019. "Computational Drug Design Applied to the Study of Metabotropic Glutamate Receptors" Molecules 24, no. 6: 1098. https://doi.org/10.3390/molecules24061098
APA StyleLlinas del Torrent, C., Pérez-Benito, L., & Tresadern, G. (2019). Computational Drug Design Applied to the Study of Metabotropic Glutamate Receptors. Molecules, 24(6), 1098. https://doi.org/10.3390/molecules24061098