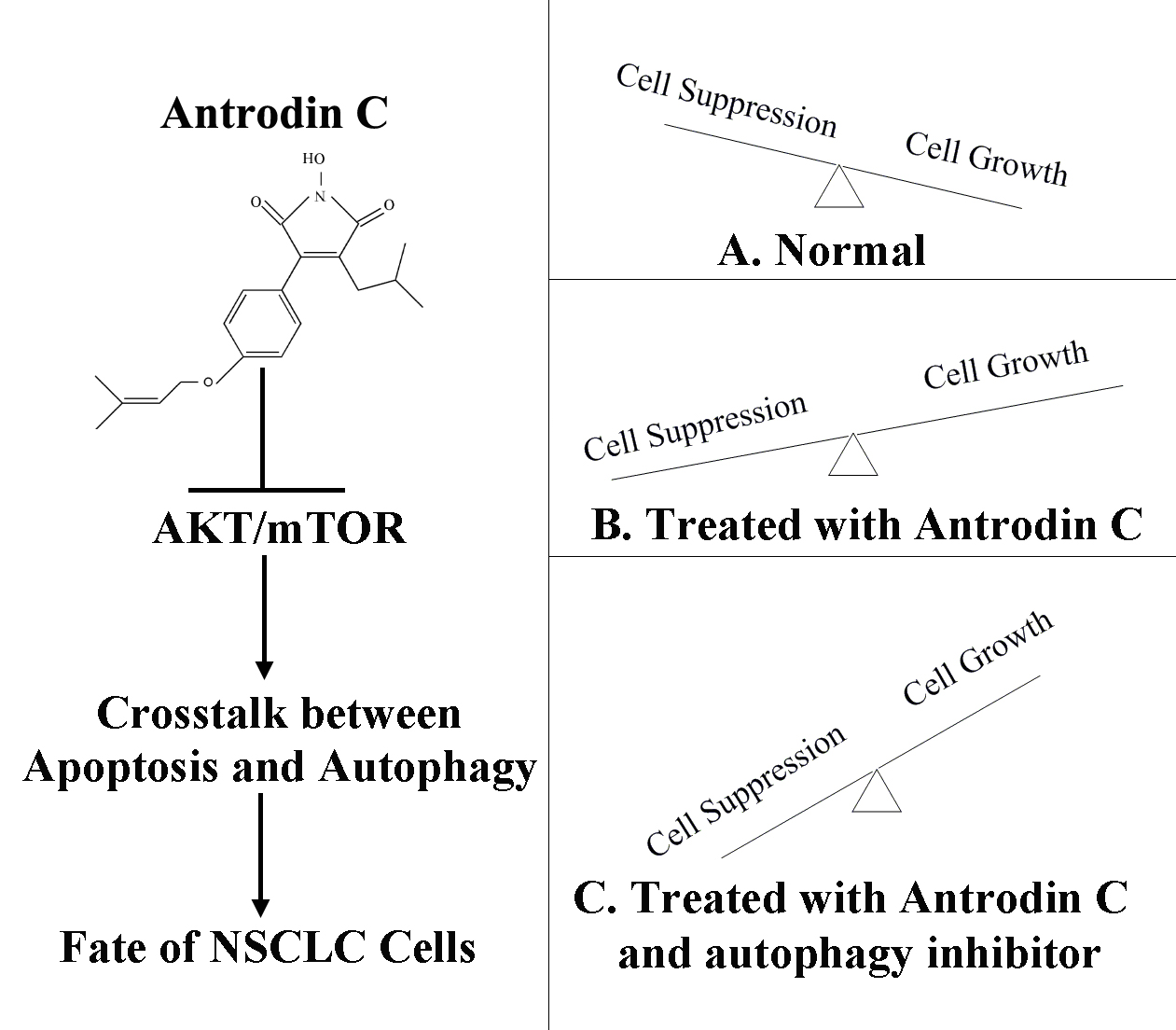
Antrodin C, an NADPH Dependent Metabolism, Encourages Crosstalk between Autophagy and Apoptosis in Lung Carcinoma Cells by Use of an AMPK Inhibition-Independent Blockade of the Akt/mTOR Pathway


 ,
,
Abstract
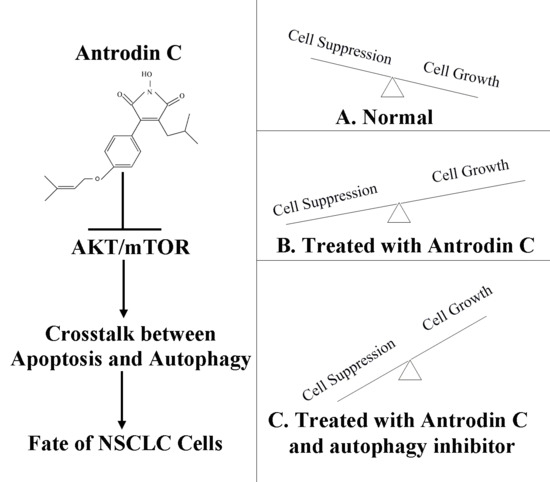
:
1. Introduction
2. Results
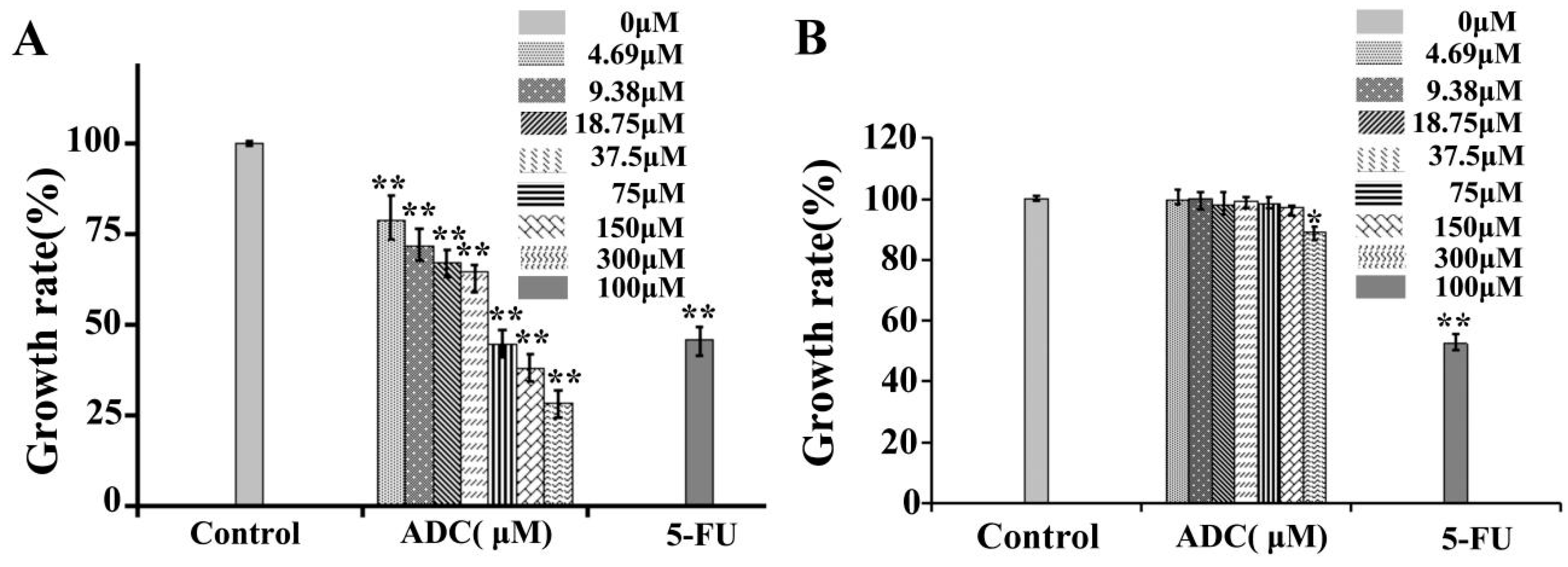
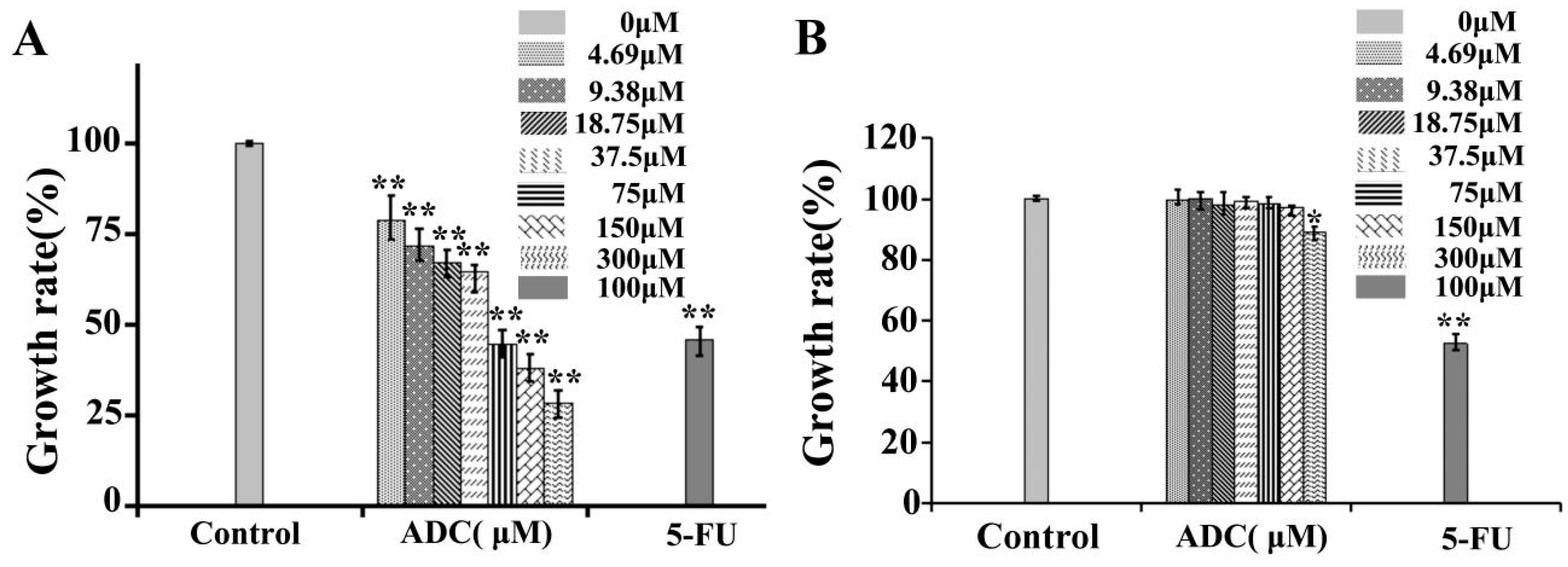
2.1. Effects of ADC In Vitro Cell Proliferation of SPCA-1 and BEAS-2B
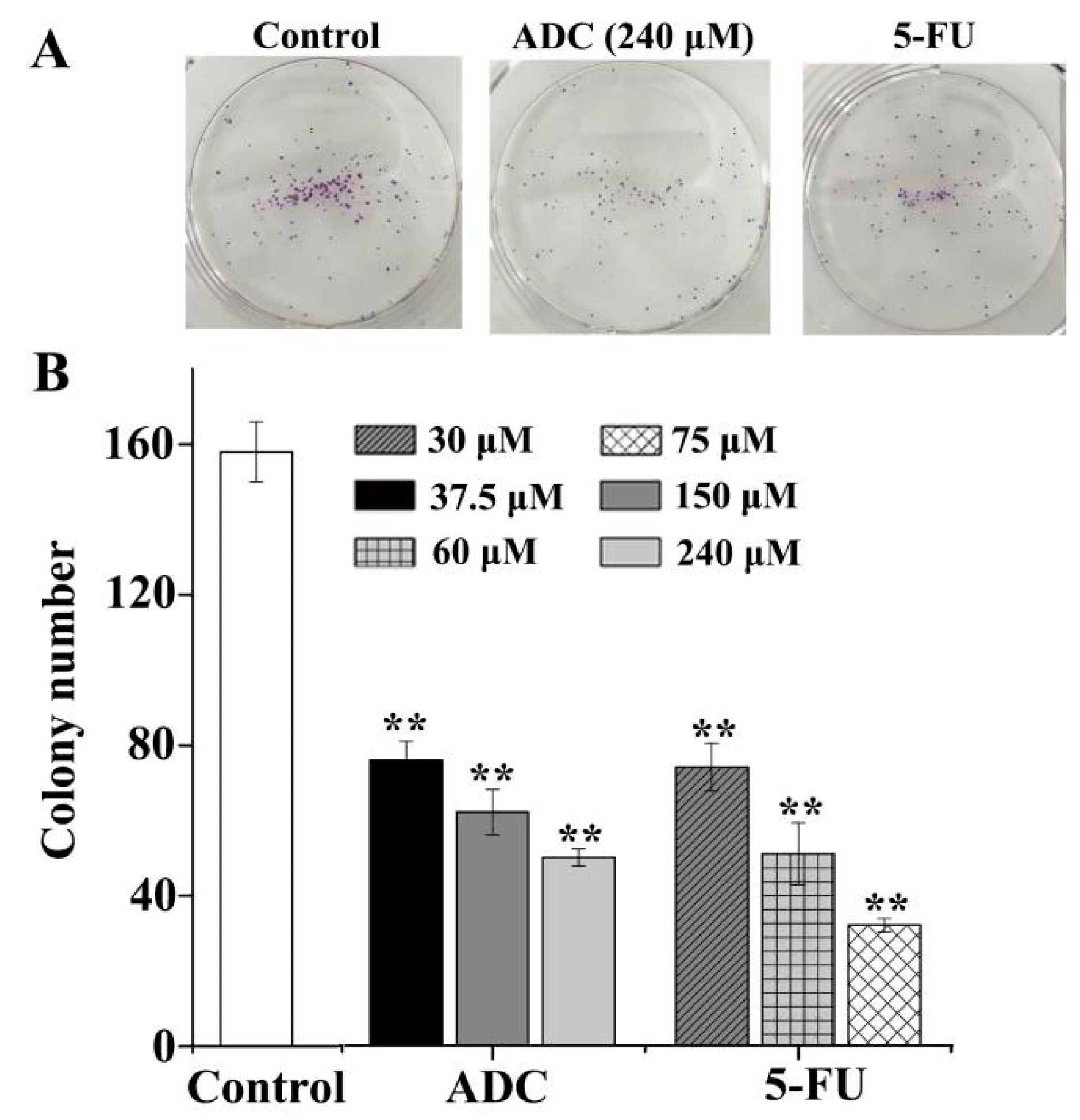
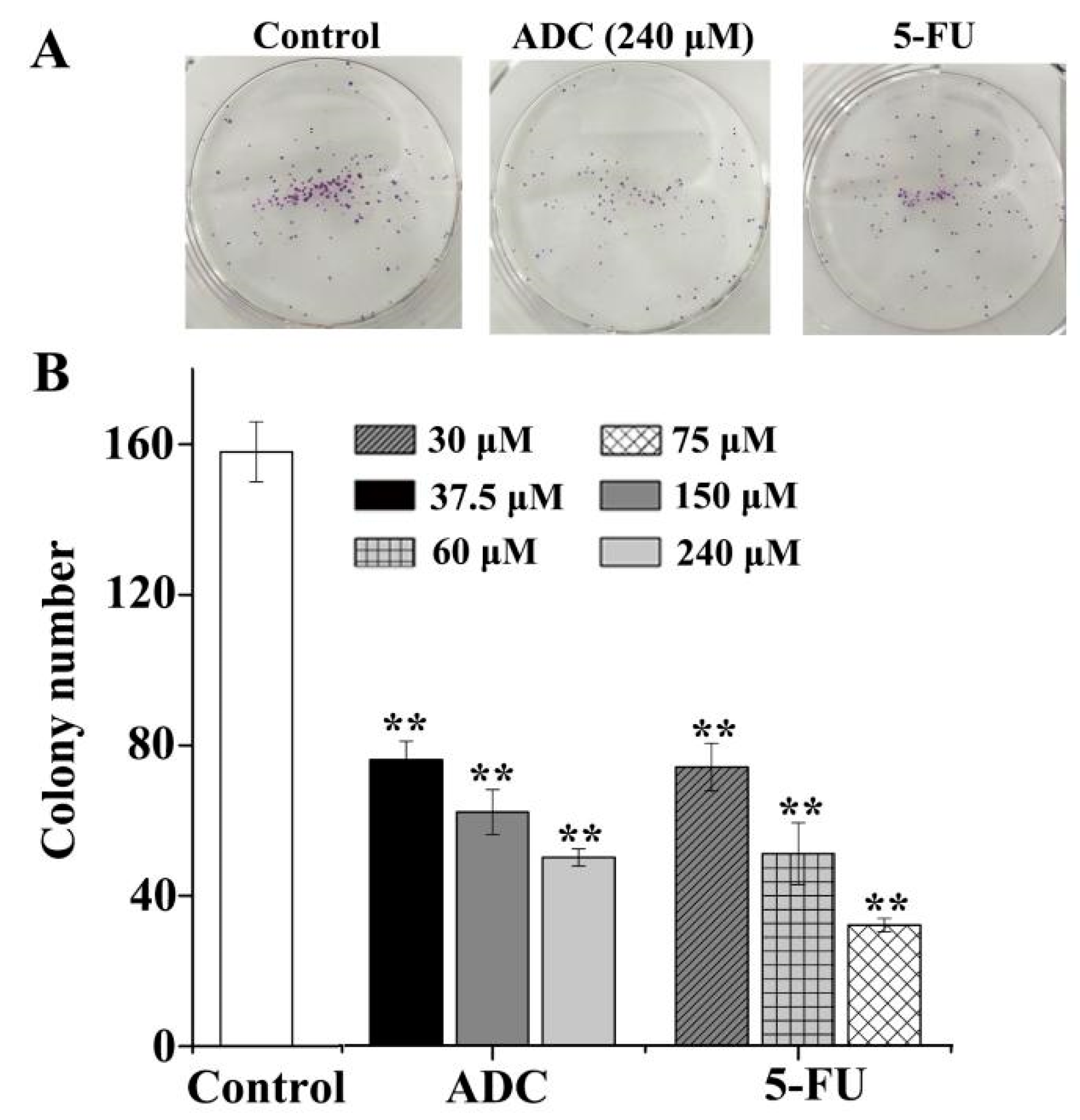
2.2. Effects of ADC In Vitro on the Colony Forming Ability of SPCA-1 Cells
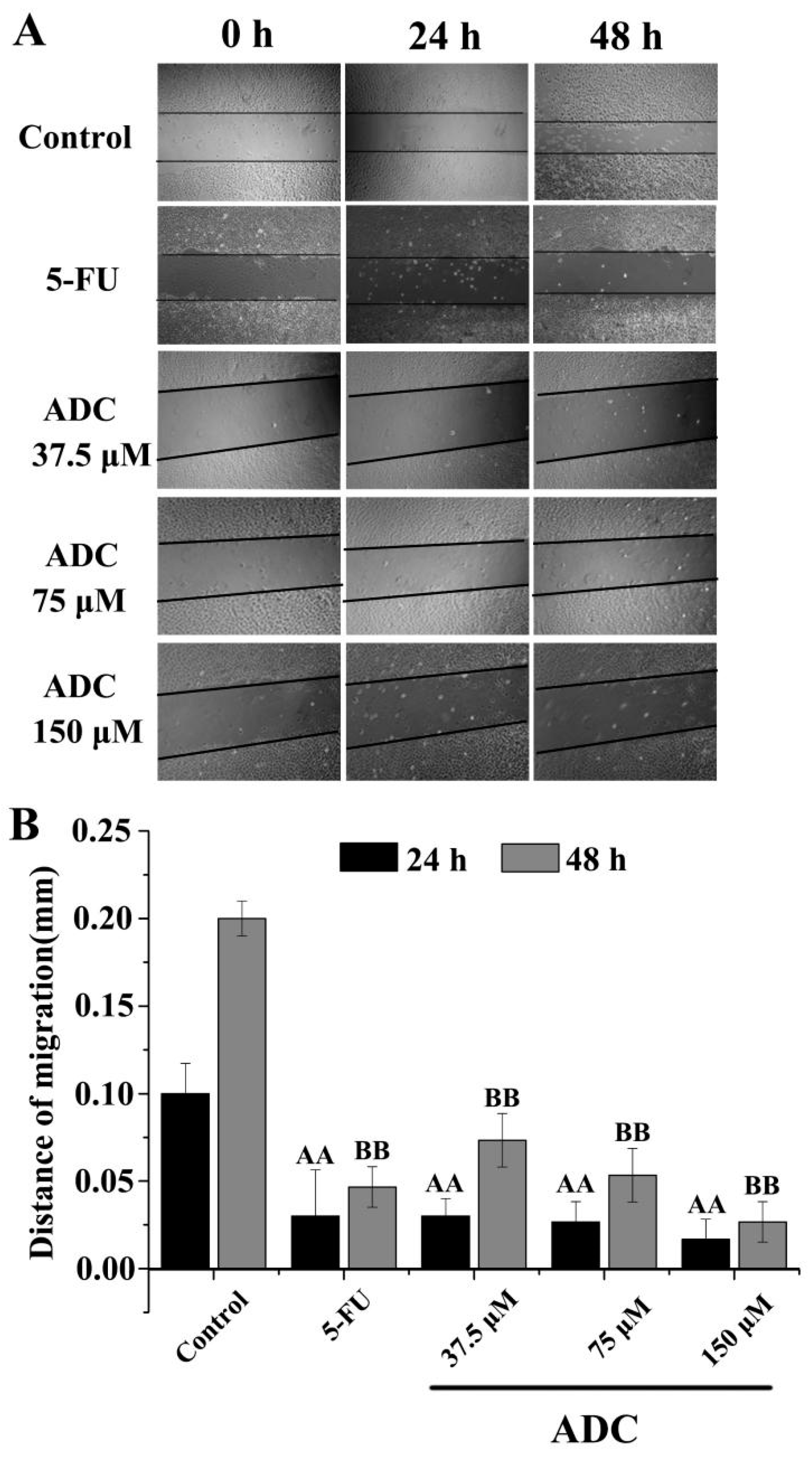
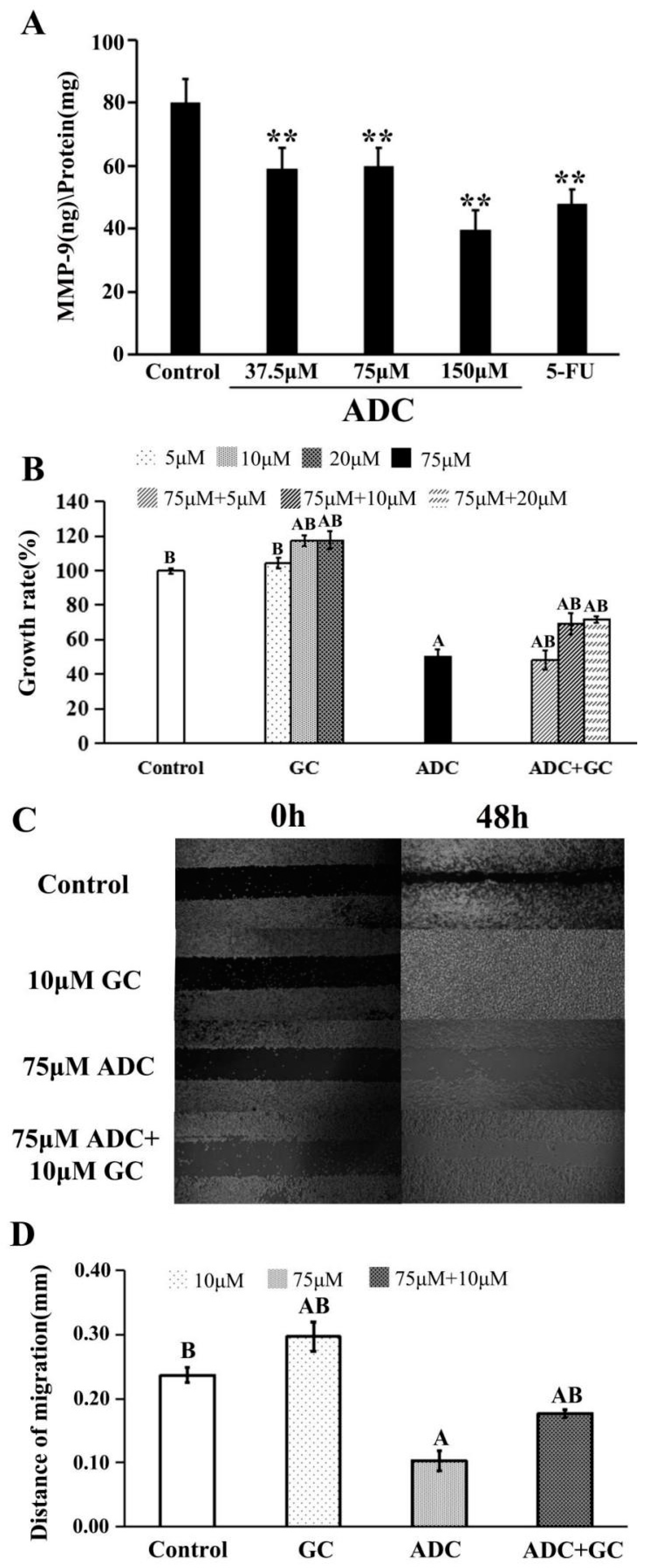
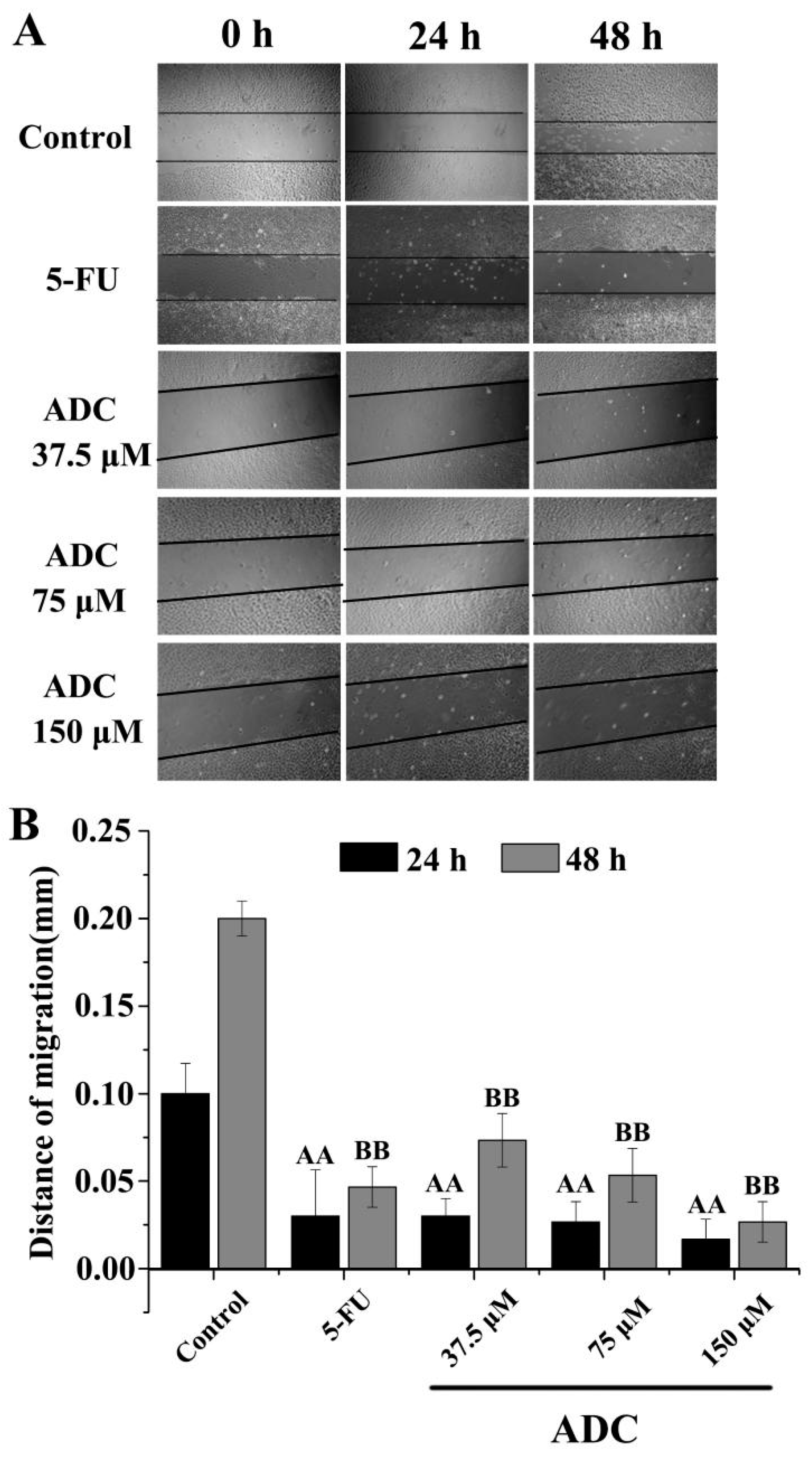
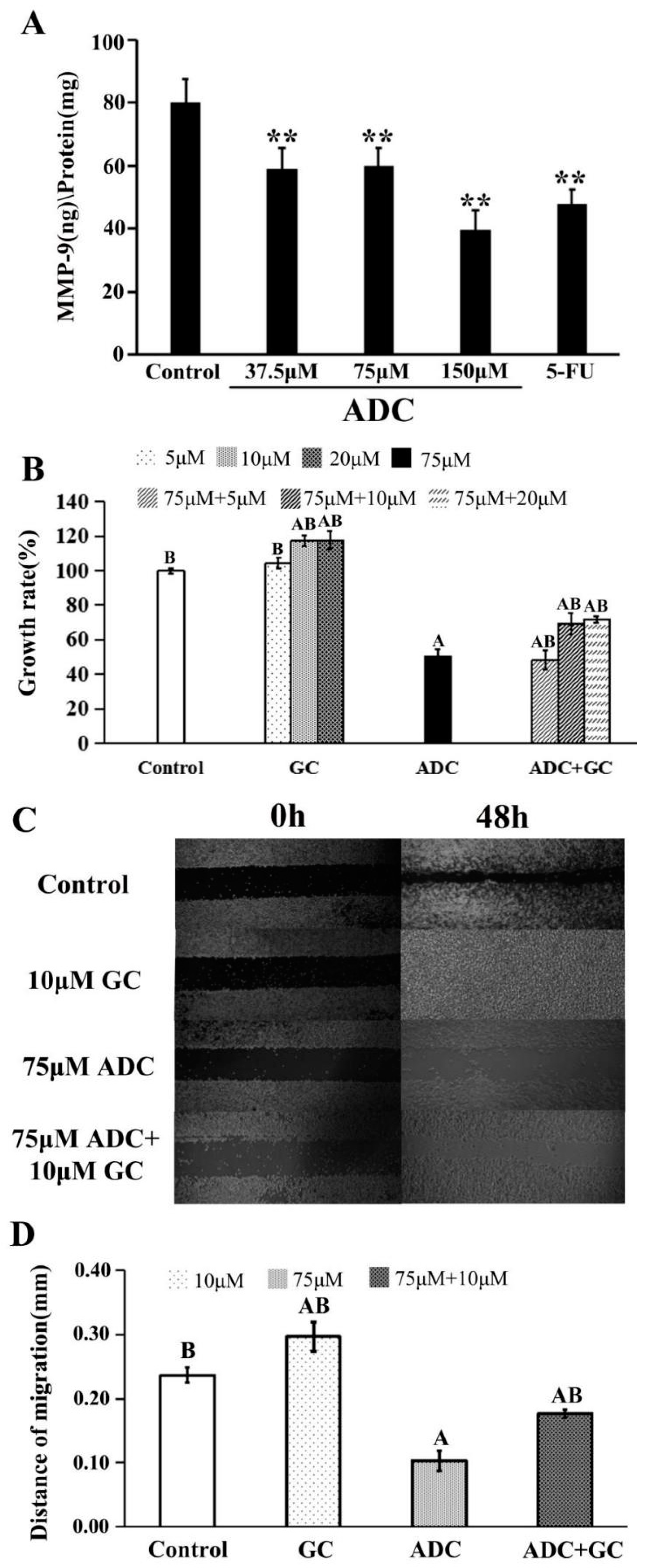
2.3. Effects of ADC In Vitro on Cell Migration of SPCA-1 Cells
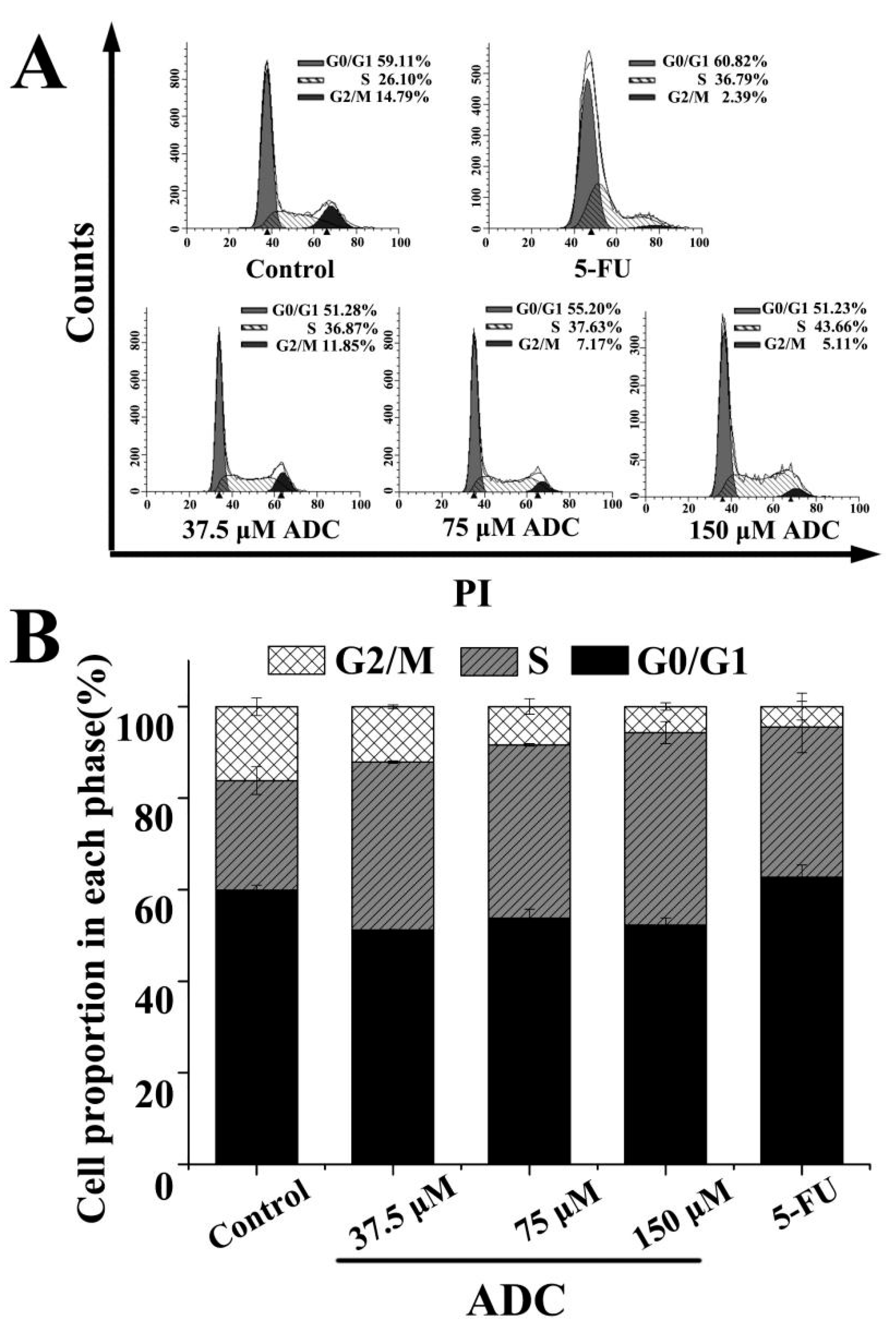
2.4. Influence of ADC on Cell Cycle of SPCA-1 Cells
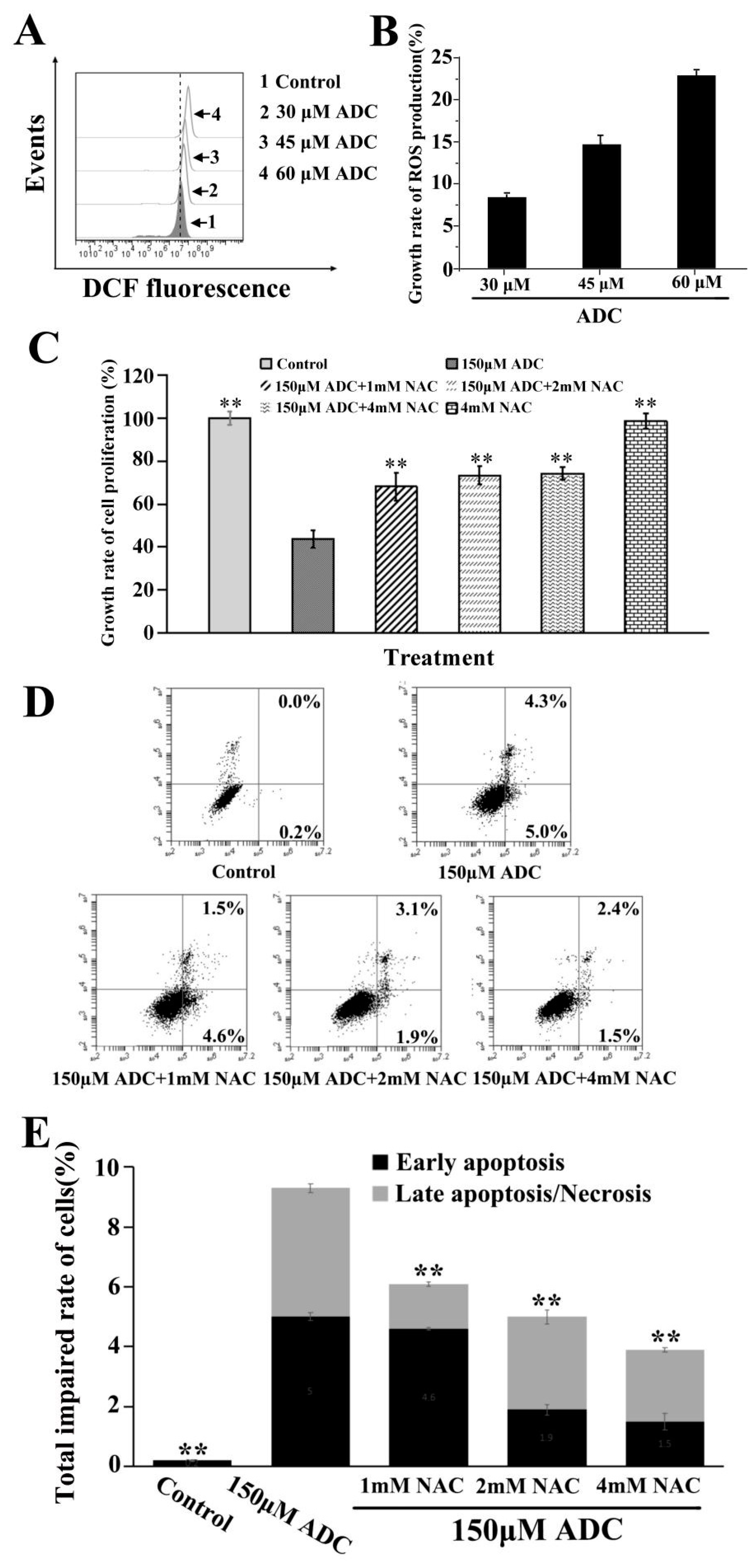
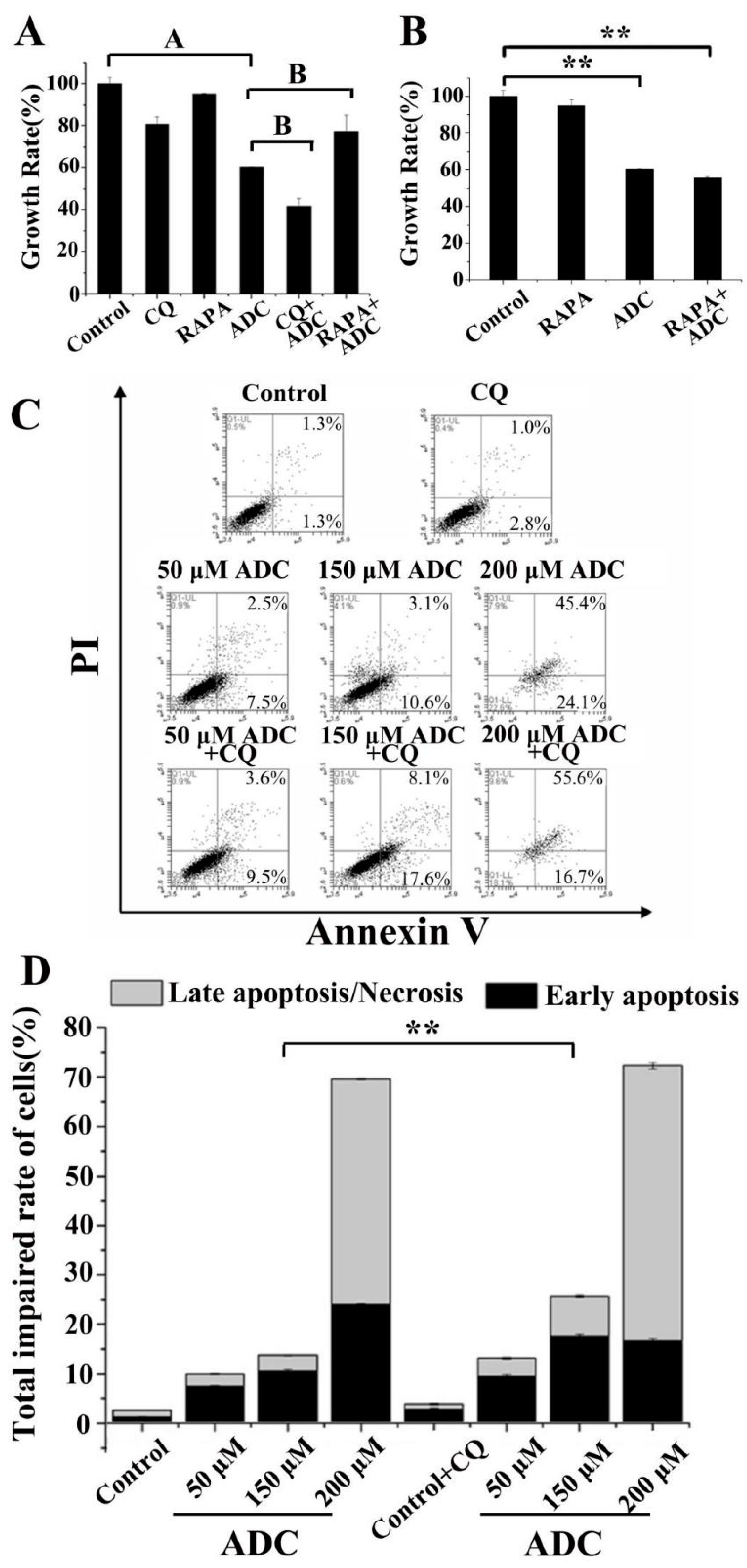
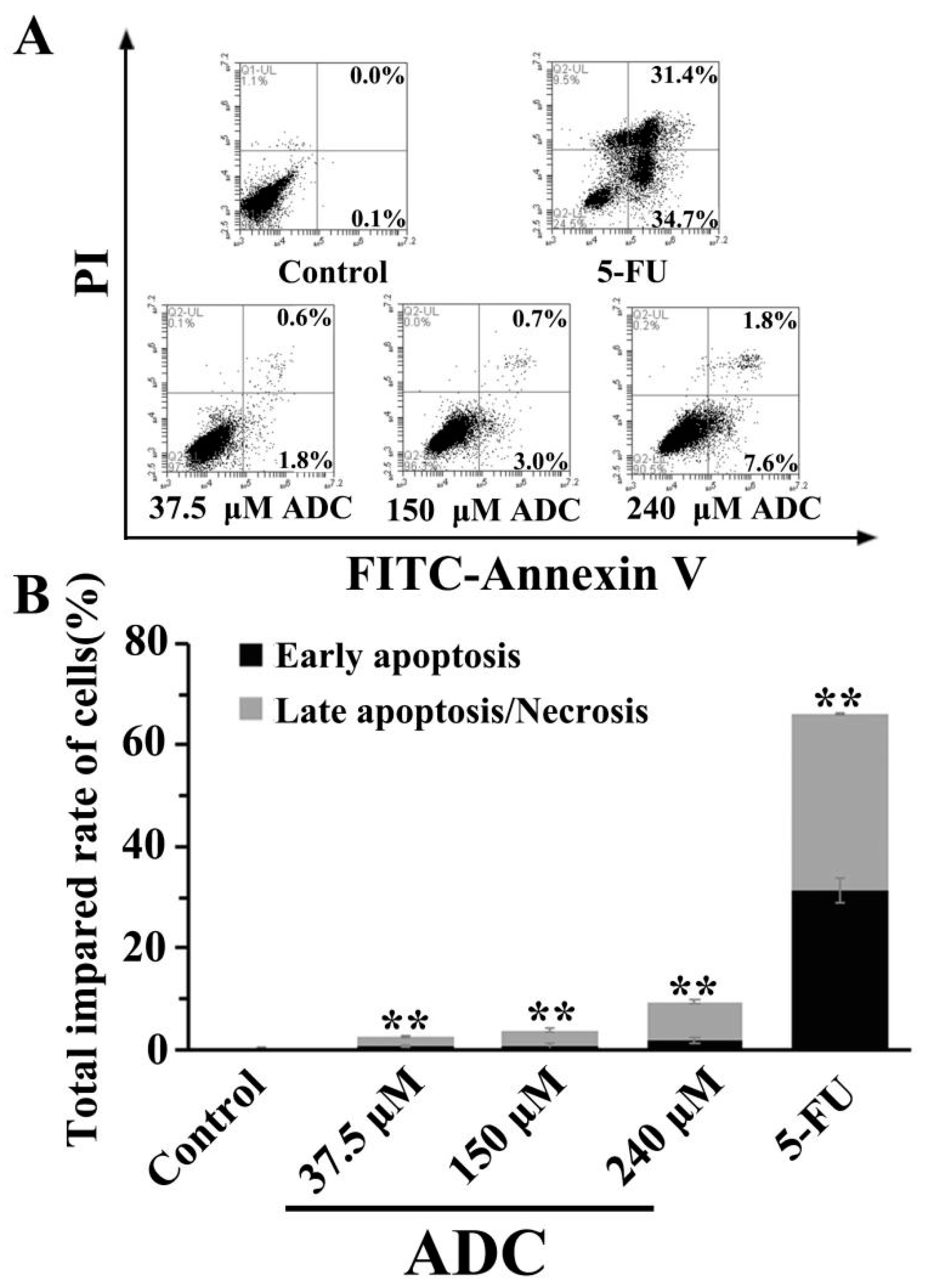
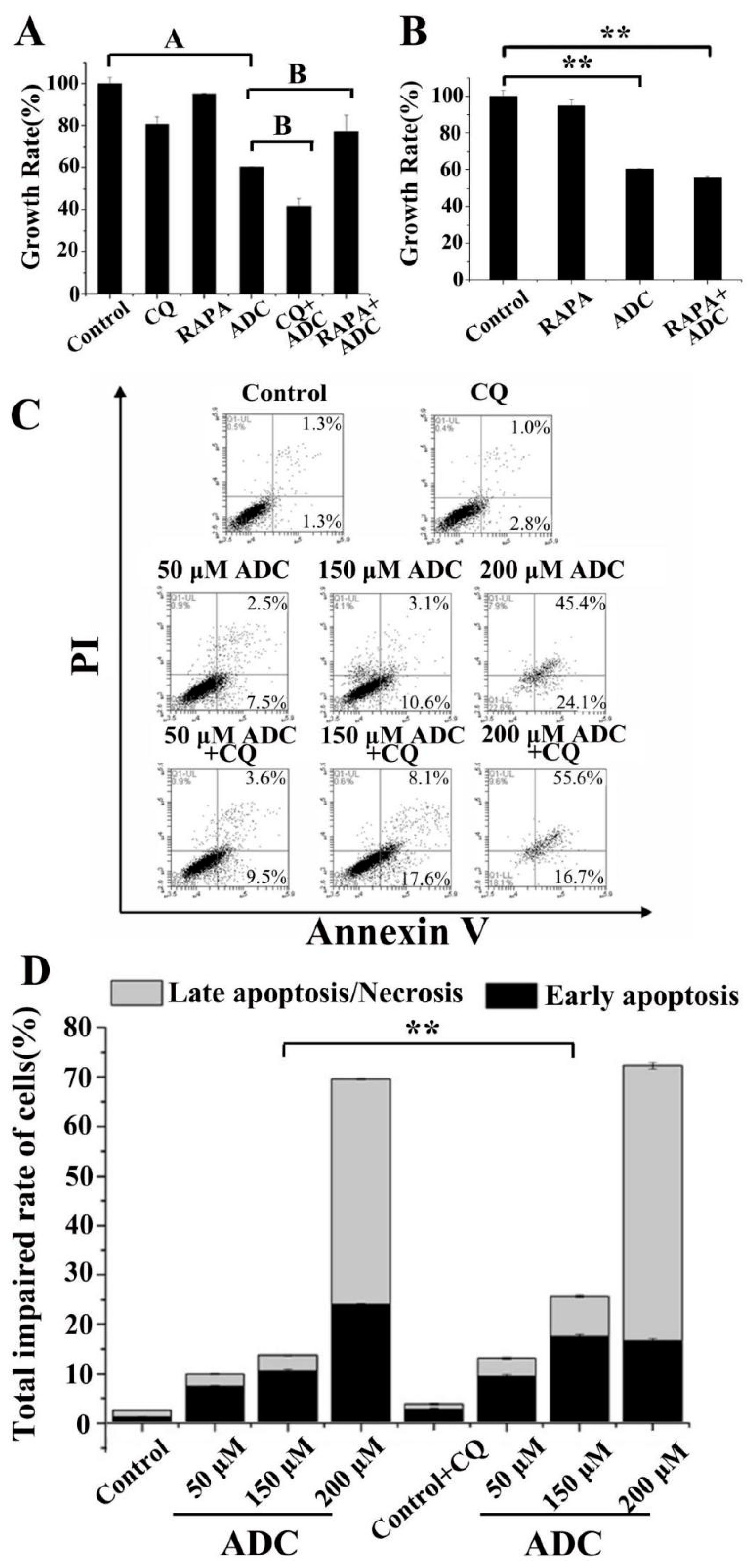
2.5. Effects of ADC on SPCA-1 Cell Apoptosis
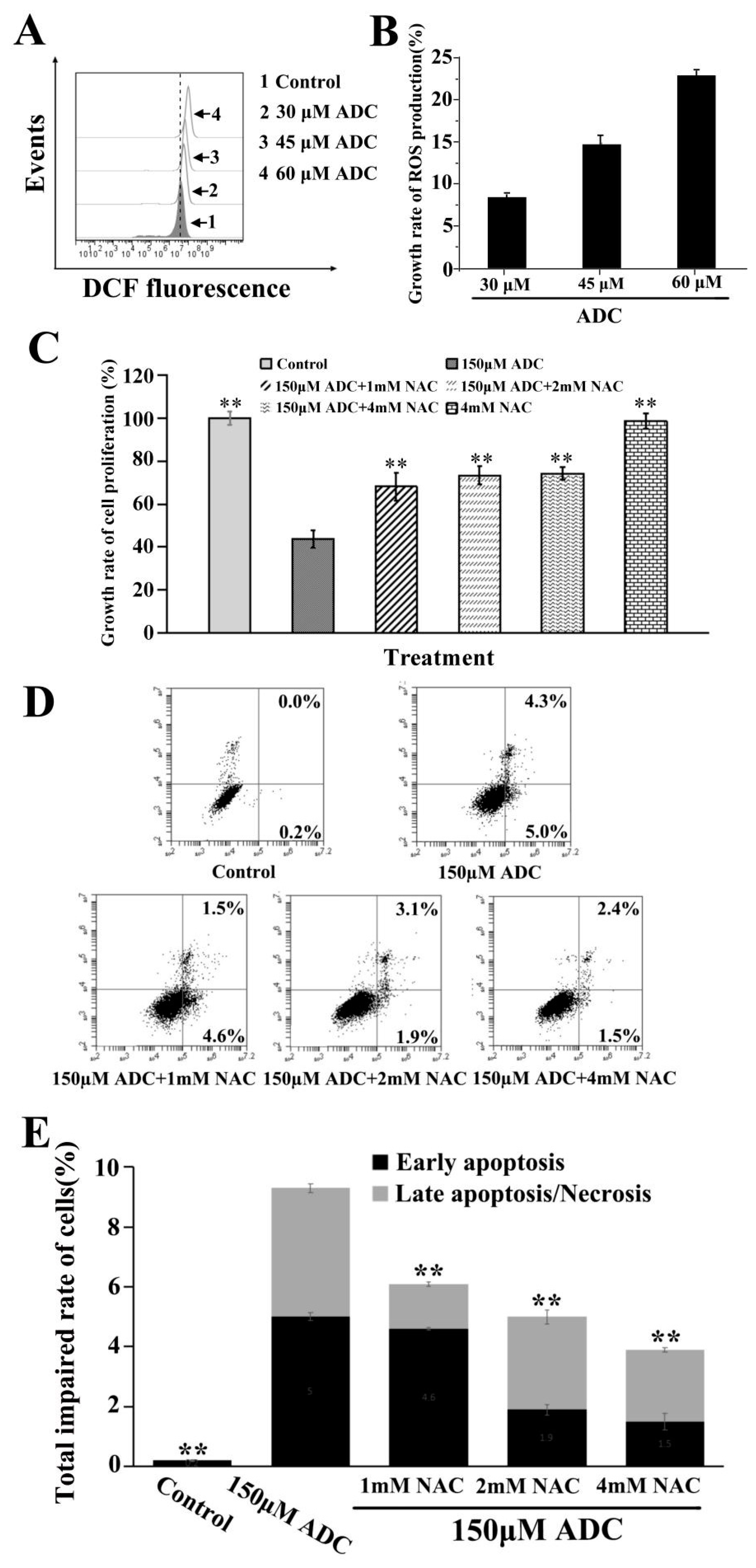
2.6. Effects of ADC on ROS Released by SPCA-1 Cells
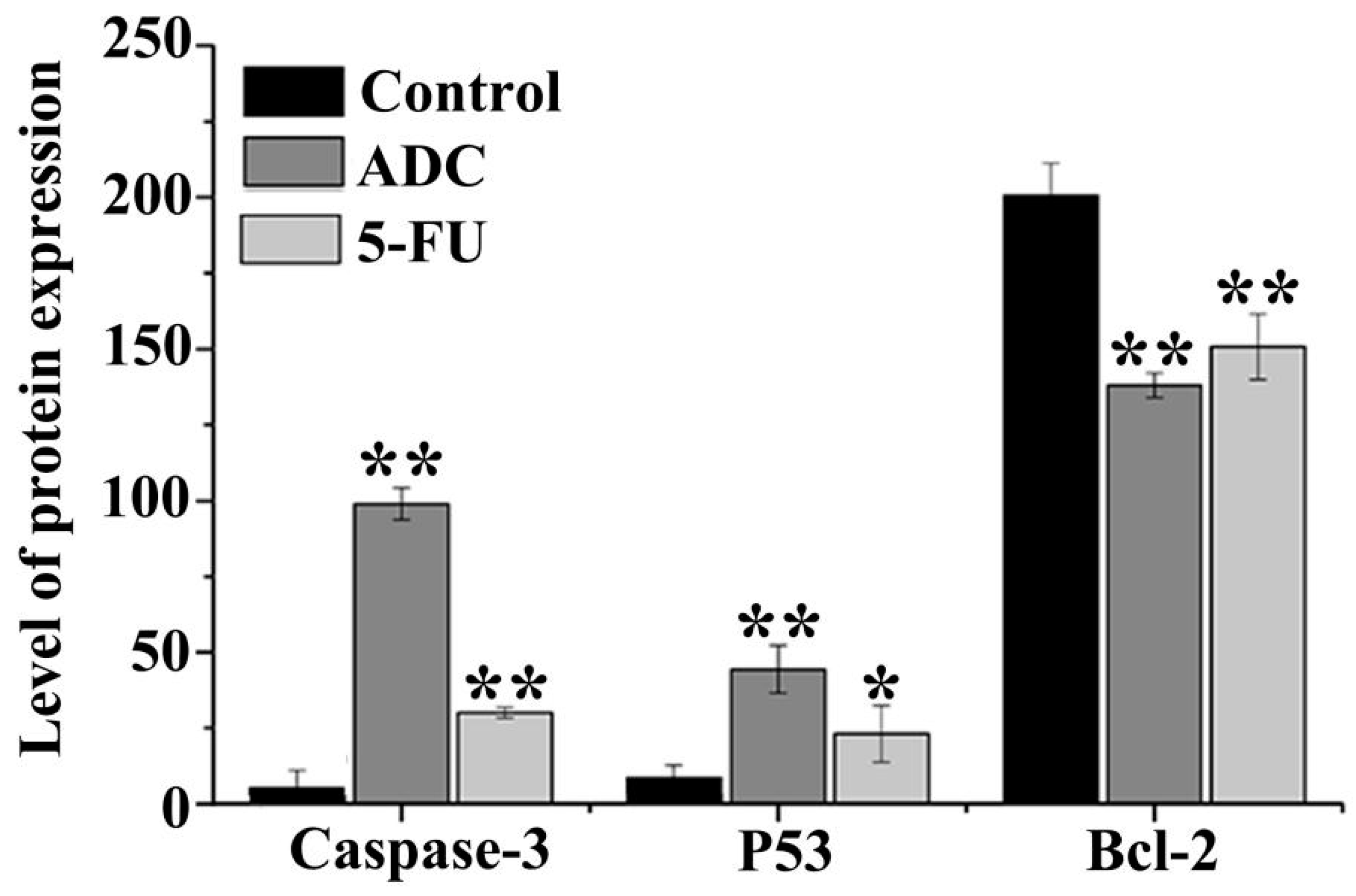
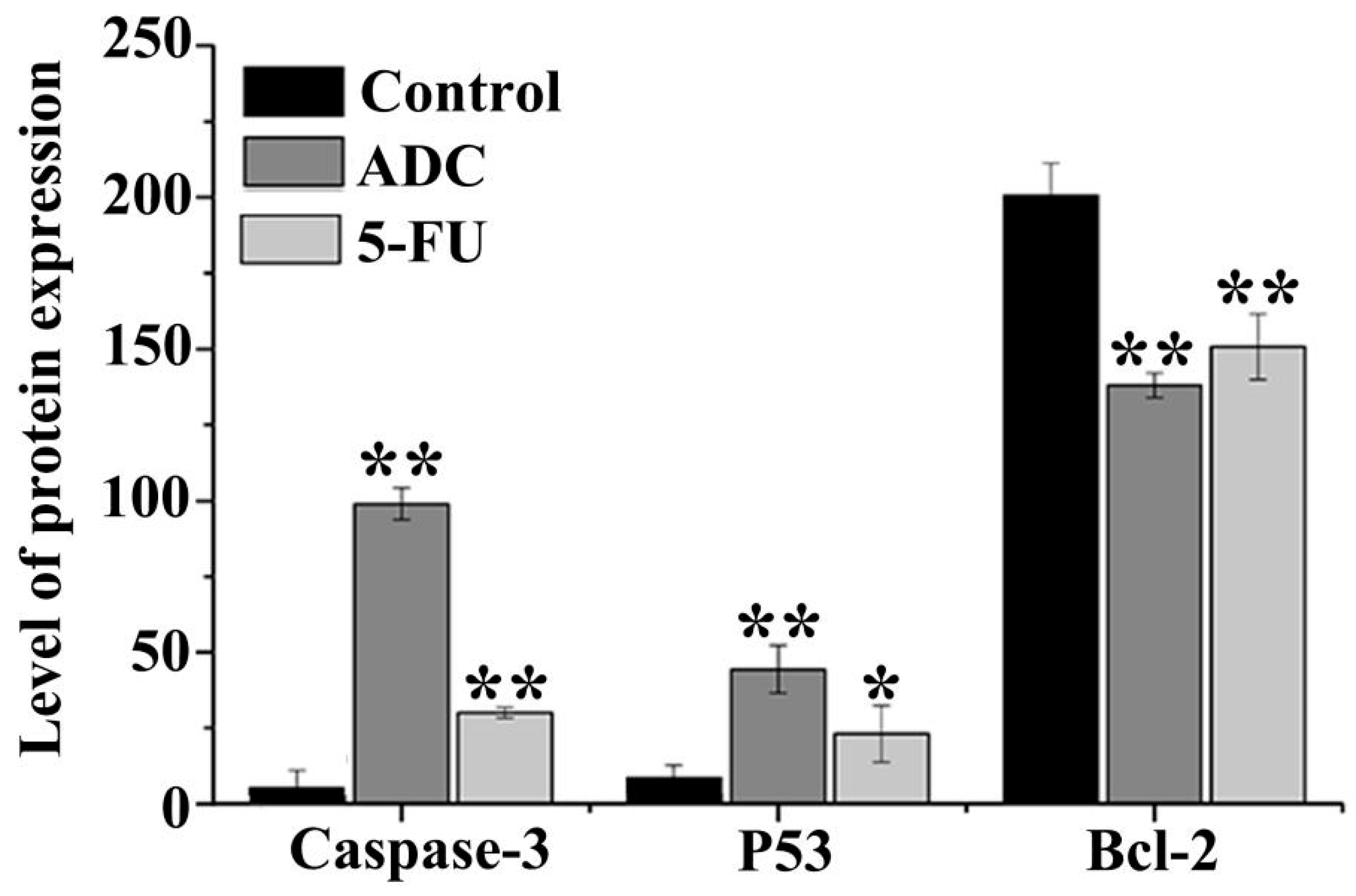
2.7. Effects of ADC on Activation of Caspase-3, P53, and Bcl-2
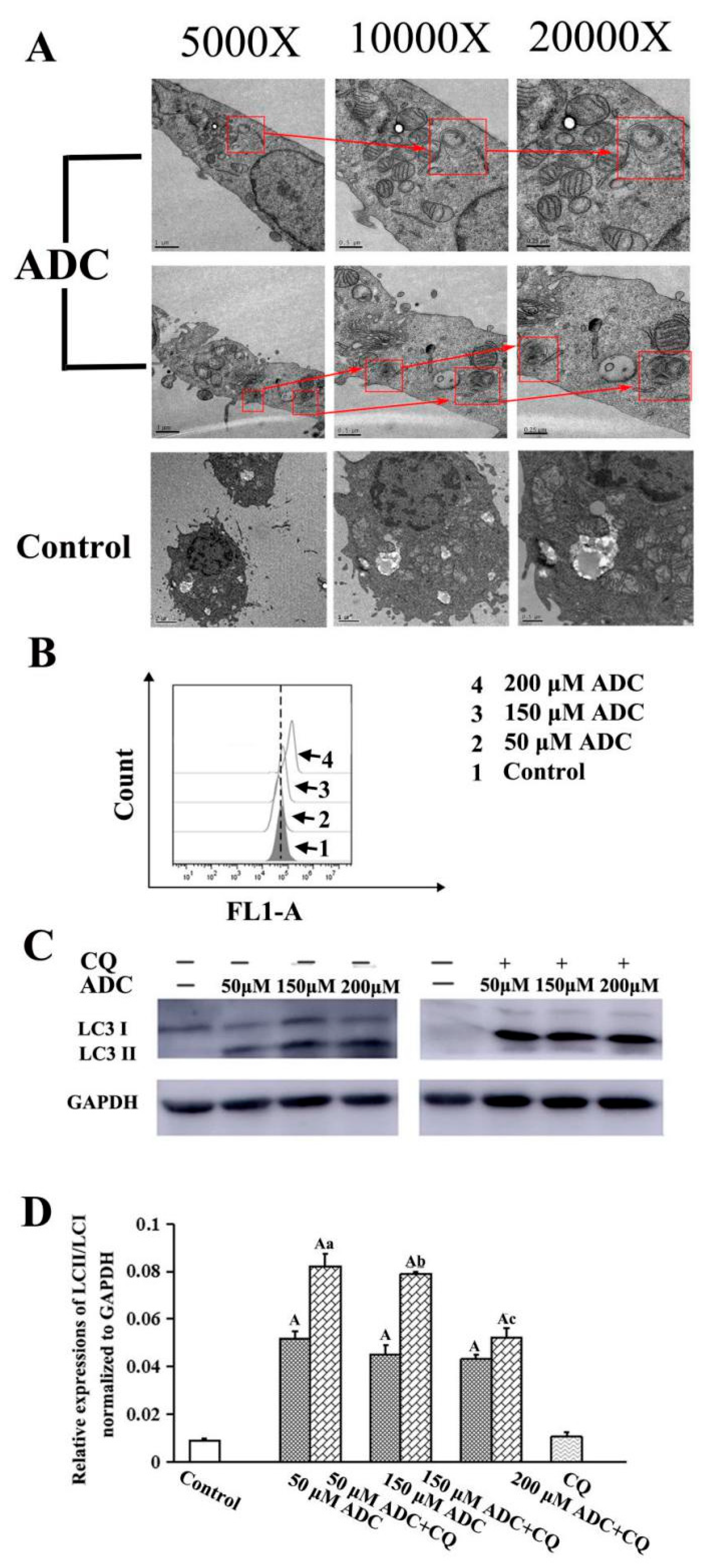
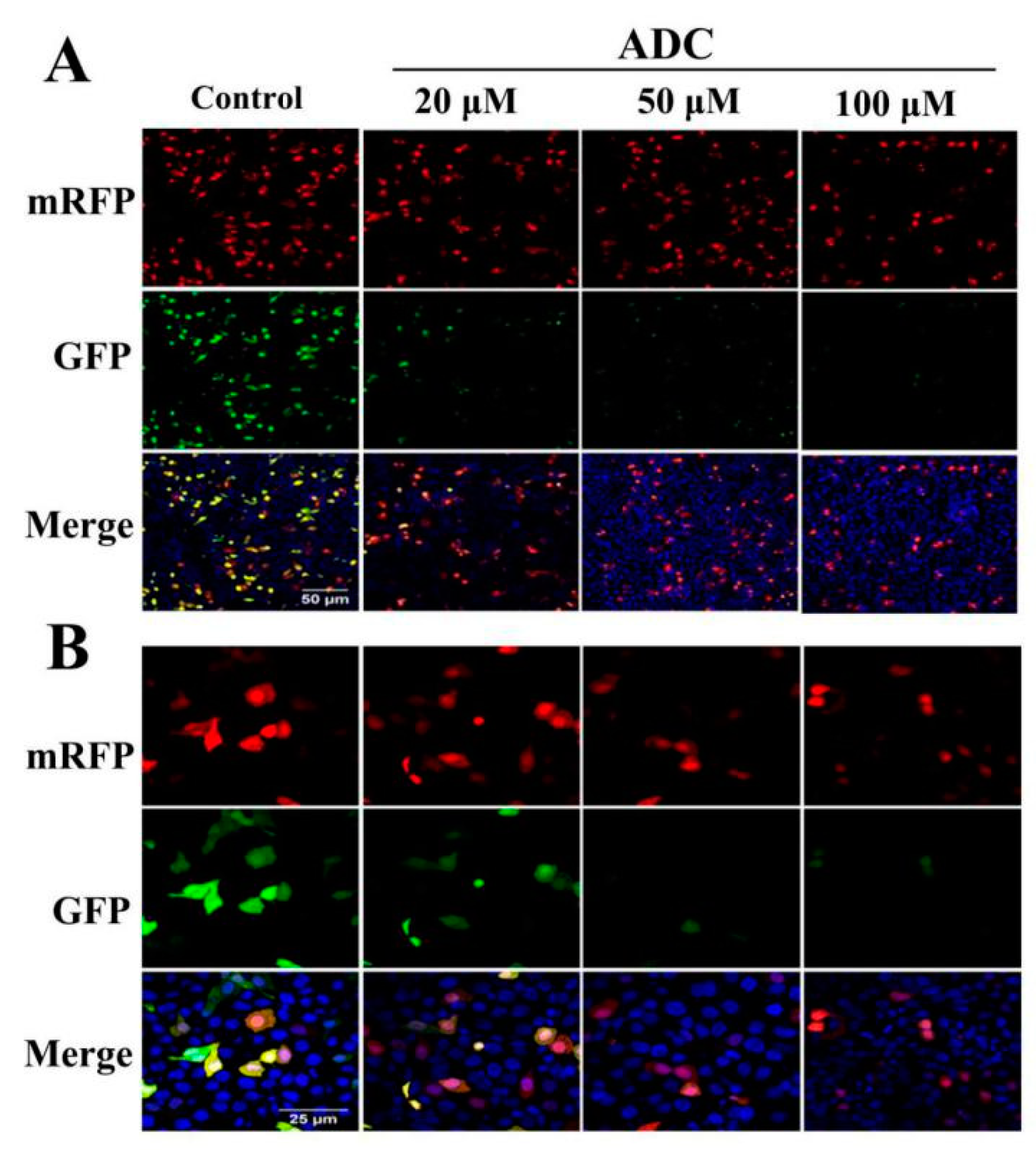
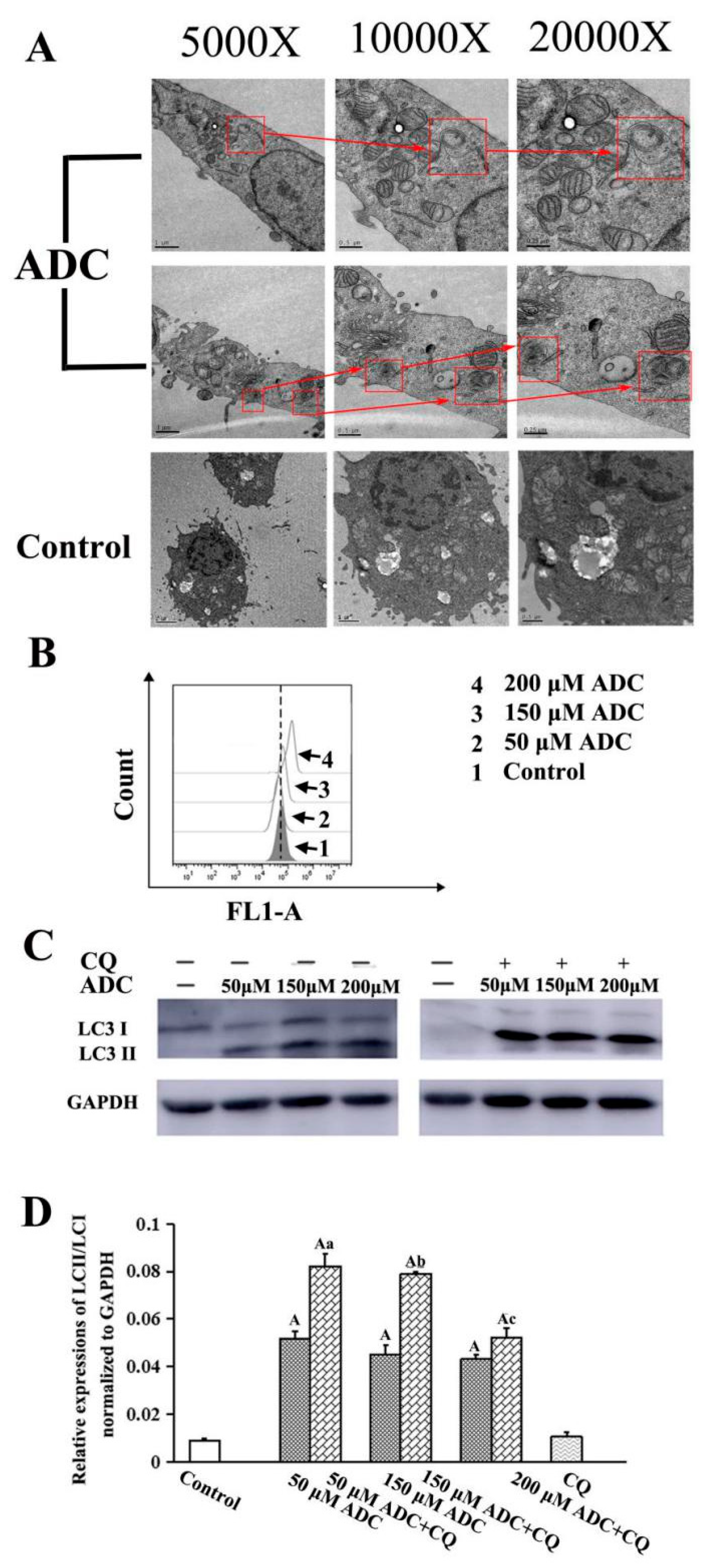
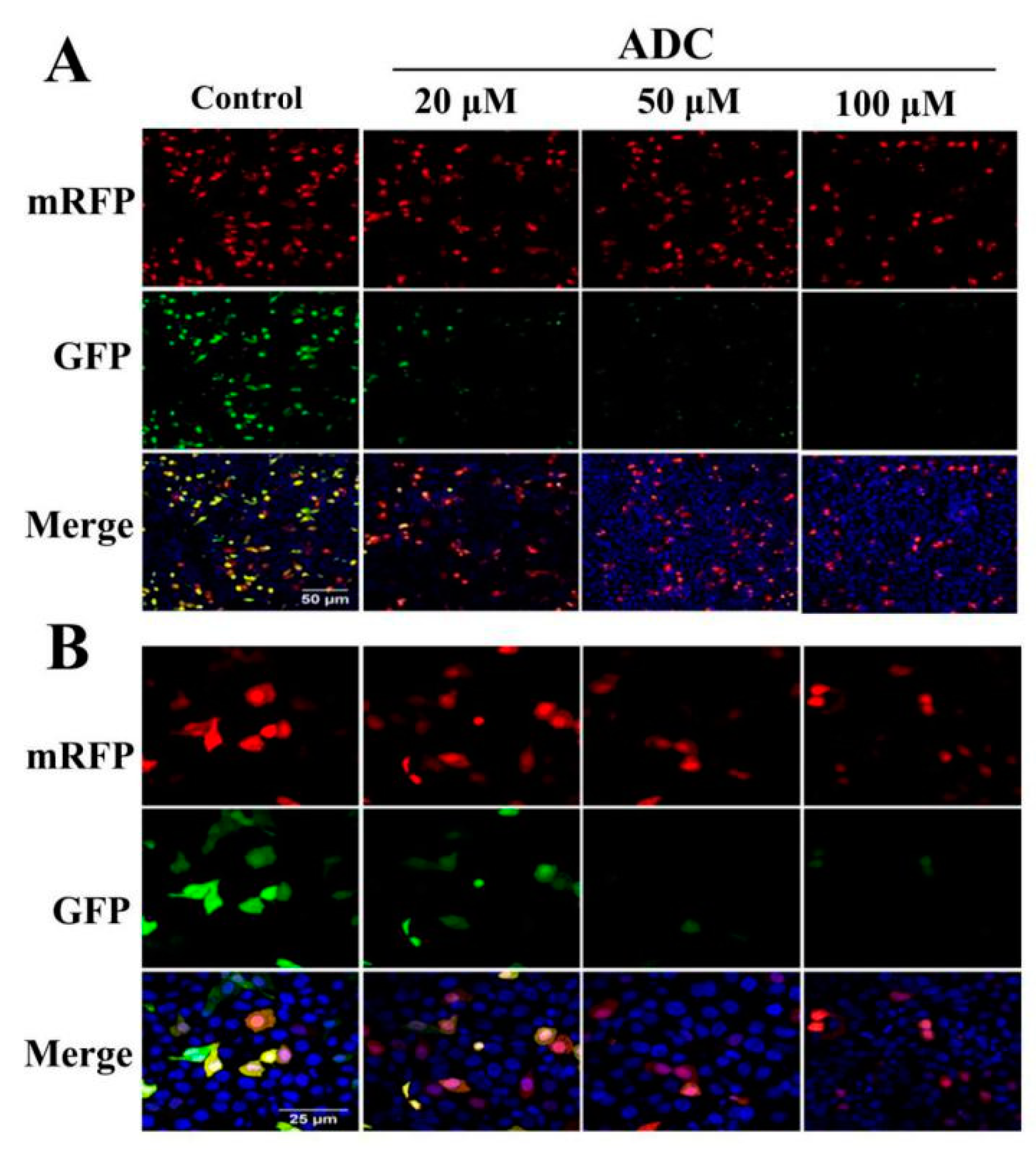
2.8. Effects of ADC on Autophagy Activation in SPCA-1 Cells
2.9. Protective Effects of Autophagy on ADC-Induced Apoptotic SPCA-1 Cell Death
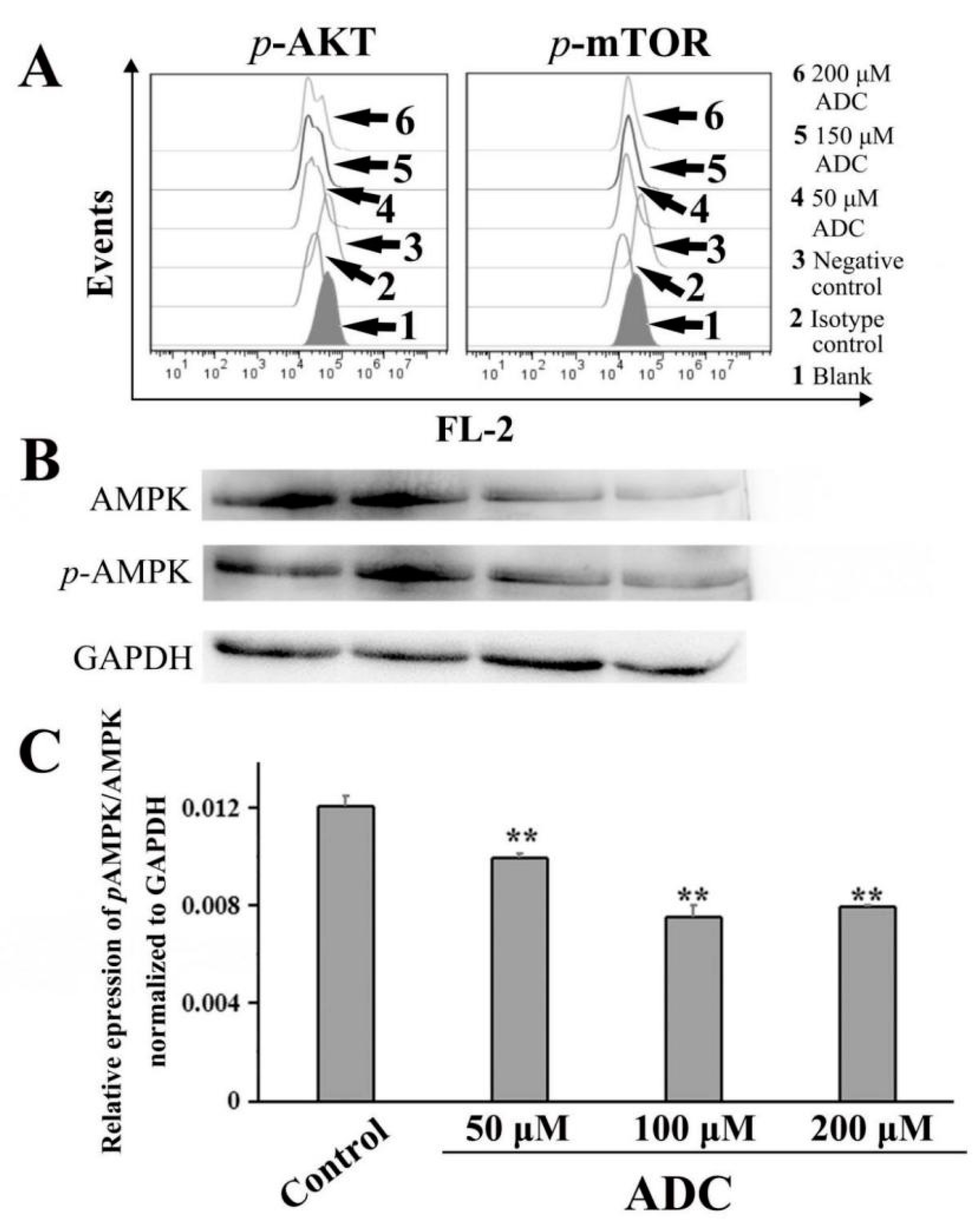
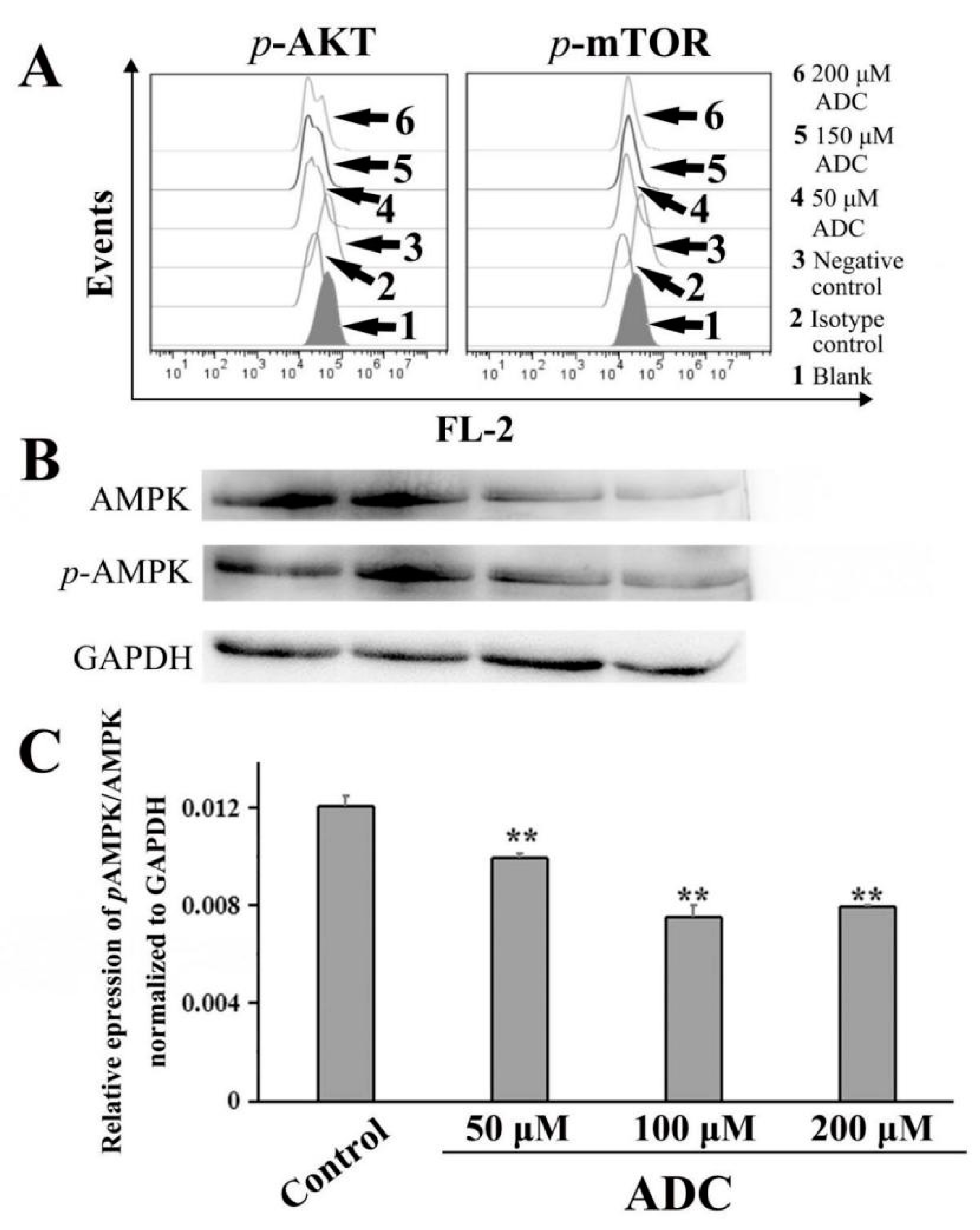
2.10. ADC Downregulated the AKT-mTOR Pathway and AMP-Activated Protein Kinase (AMPK) Pathway
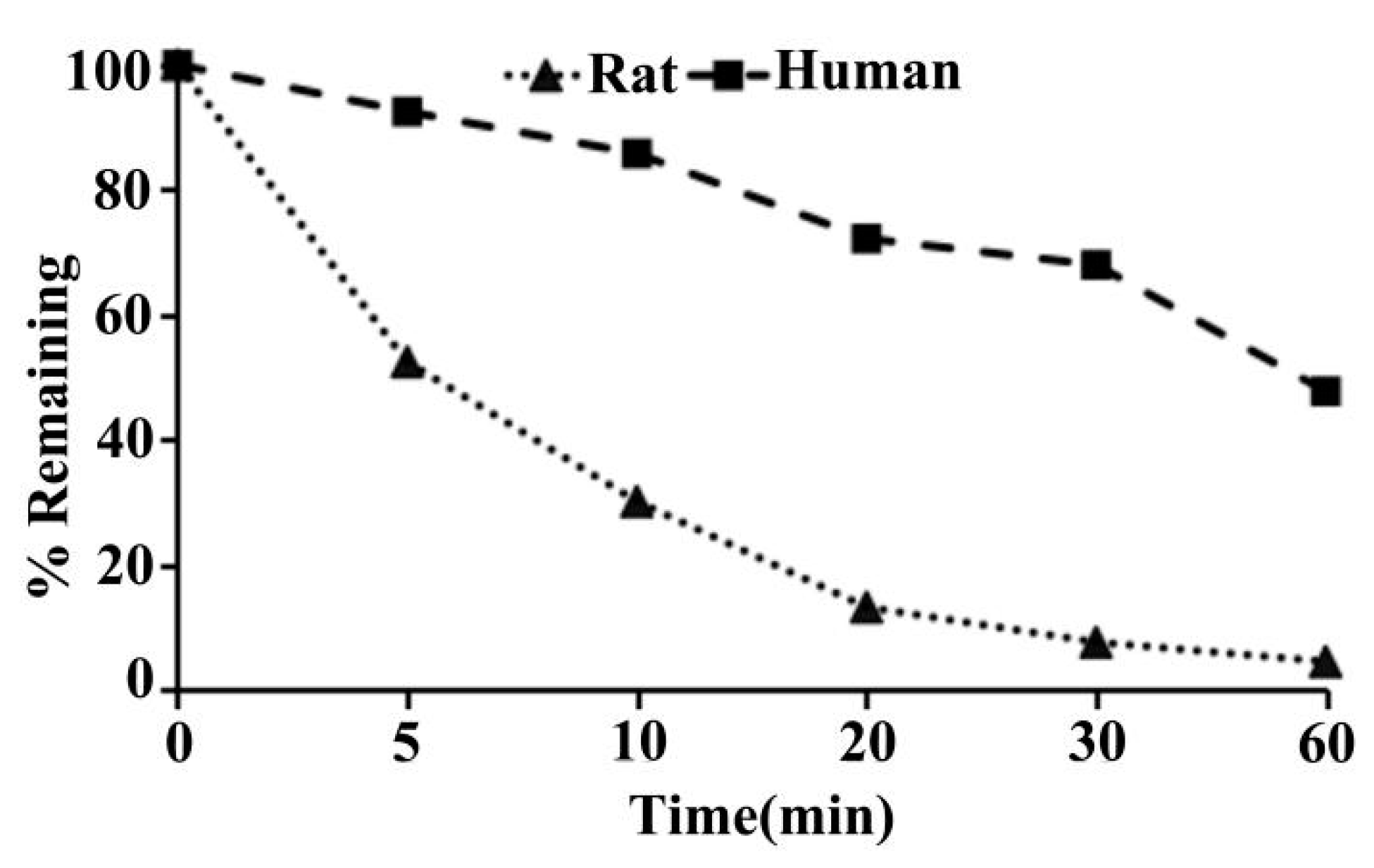
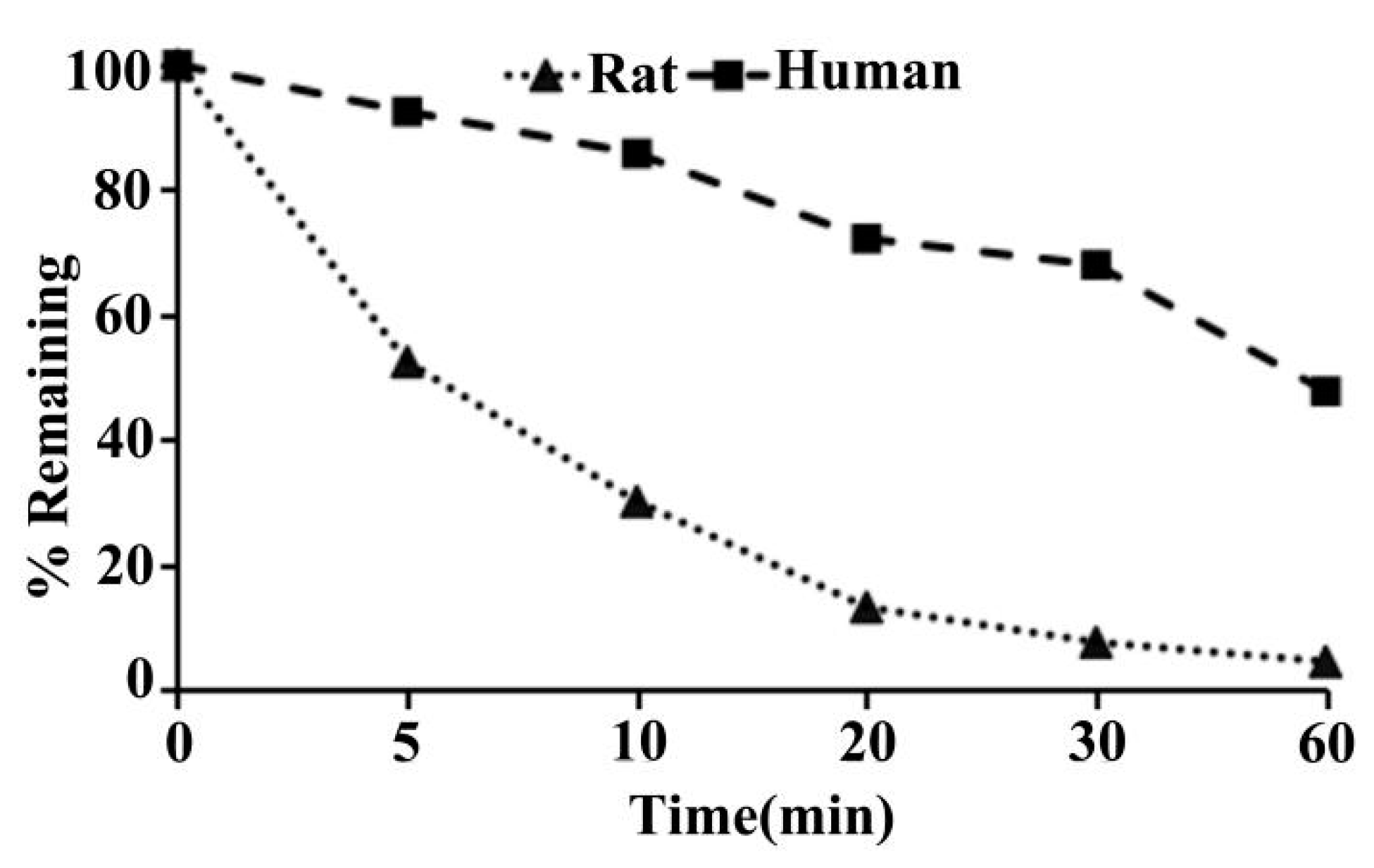
2.11. Metabolic Stability of ADC in SD Rat and Human Liver Microsomes
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Lines and Cell Culture
4.3. Cell Viability Assay
4.4. Clone Formation Assay
4.5. Wound-Healing Assay
4.6. Cell Cycle Analysis
4.7. Cell Apoptosis Detection
4.8. ROS Detection
4.9. ELISA Assay
4.10. Transmission Electron Microscopy Analysis
4.11. Flow Cytometric Analysis of Autophagy with Cyto-ID Staining
4.12. Western Blotting Analysis
4.13. Live-Cell Imaging for Autophagic Flux
4.14. FACS/Phosflow
4.15. Role of Autophagy
4.16. In Vitro ADC Metabolism
4.17. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Ikari, A.; Sato, T.; Watanabe, R.; Yamazaki, Y.; Sugatani, J. Increase in claudin-2 expression by an EGFR/MEK/ERK/c-Fos pathway in lung adenocarcinoma A549 cells. Biochim. Biophys. Acta 2012, 1823, 1110–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.P.; Deep, G.; Chittezhath, M.; Kaur, M.; Dwyer-Nield, L.D.; Malkinson, A.M.; Agarwal, R. Effect of silibinin on the growth and progression of primary lung tumors in mice. J. Natl. Cancer Inst. 2006, 98, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; He, S.M.; He, Z.X.; Li, M.; Yang, Y.; Pang, J.X.; Zhang, X.; Chow, K.; Zhou, Q.; Duan, W.; et al. Plumbagin induces apoptotic and autophagic cell death through inhibition of the PI3K/Akt/mTOR pathway in human non-small cell lung cancer cells. Cancer Lett. 2014, 344, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.L.; Lin, Z.B. Effects of Ganoderma lucidum polysaccharides on proliferation and cytotoxicity of cytokine-induced killer cells. Acta Pharmacol. Sin. 2005, 26, 1130–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.C.; Du, YC.; Chuu, J.J.; Hwang, S.L.; Hsieh, P.C.; Hung, C.S.; Chang, F.R.; Wu, Y.C. Active extracts of wild fruiting bodies of Antrodia camphorata (EEAC) induce leukemia HL 60 cells apoptosis partially through histone hypoacetylation and synergistically promote anticancer effect of trichostatin A. Arch. Toxicol. 2009, 83, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Kong, Z.L.; Chang, J.S.; Chang, K.L.B. Antiproliferative effect of Antrodia camphorata polysaccharides encapsulated in chitosan–silica nanoparticles strongly depends on the metabolic activity type of the cell line. J. Nanopart. Res. 2013, 15, 1–13. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.; Kim, S.L.; Choi, J.W.; Seo, J.Y.; Choi, D.J.; Park, Y.I. Corn silk maysin induces apoptotic cell death in PC-3 prostate cancer cells via mitochondria-dependent pathway. Life Sci. 2014, 119, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Miao, L.; Lv, C.; Sun, H.; Wei, S.; Wang, B.; Huang, C.; Jiao, B. Wentilactone B induces G2/M phase arrest and apoptosis via the Ras/Raf/MAPK signaling pathway in human hepatoma SMMC-7721 cells. Cell Death Dis. 2013, 4, e657. [Google Scholar] [CrossRef]
- Ryter, S.W.; Choi, A.M. Autophagy in lung disease pathogenesis and therapeutics. Redox Biol. 2015, 4, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.S. Autophagy and cancer. Exp. Mol. Med. 2012, 44, 109–120. [Google Scholar] [CrossRef]
- Maes, H.; Rubio, N.; Garg, A.D.; Agostinis, P. Autophagy: Shaping the tumor microenvironment and therapeutic response. Trends Mol. Med. 2013, 19, 428–446. [Google Scholar] [CrossRef] [PubMed]
- Gozuacik, D.; Kimchi, A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004, 23, 2891–2906. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.H.; Ryvarden, L.; Chang, T.T. Antrodia camphorata (“niu-chang-chih”), new combination of a medicinal fungus in Taiwan. Bot. Bull. Acad. Sin. Taipei 1997, 38, 273–275. [Google Scholar]
- Lee, K.H.; Morris-Natschke, S.L.; Yang, X.; Huang, R.; Zhou, T.; Wu, S.F.; Shi, Q.; Itokawa, H. Recent progress of research on medicinal mushrooms, foods, and other herbal products used in traditional Chinese medicine. J. Tradit. Complement. Med. 2012, 2, 84–95. [Google Scholar] [CrossRef]
- Cha, W.S.; Ding, J.L.; Choi, D.B. Comparative evaluation of antioxidant, nitrite scavenging, and antitumor effects of Antrodia camphorata extract. Biotechnol. Bioprocess Eng. 2009, 14, 232–237. [Google Scholar] [CrossRef]
- Phuong, D.T.; Ma, C.M.; Hattori, M.; Jin, J.S. Inhibitory effects of antrodins A-E from Antrodia cinnamomea and their metabolites on hepatitis C virus protease. Phytother. Res. 2010, 23, 582–584. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C.; El-Shazly, M.; Wu, T.Y.; Du, Y.C.; Chang, T.T.; Chen, C.F.; Hsu, Y.M.; Lai, K.H.; Chiu, C.P.; Chang, F.R.; et al. Recent research and development of Antrodia cinnamomea. Pharmacol. Ther. 2013, 139, 124–156. [Google Scholar] [CrossRef] [PubMed]
- Villaume, M.T.; Sella, E.; Saul, G.; Borzilleri, R.; Fargnoli, J.; Johnston, K.A.; Zhang, H.; Fereshteh, M.P.; Dhar, T.G.M.; Baran, P.S. Antroquinonol A: Scalable Synthesis and PreclinicalBiology of aPhase 2 Drug Candidate. ACS Cent. Sci. 2016, 2, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Baranczewski, P.; Stańczak, A.; Sundberg, K.; Svensson, R.; Wallin, A.; Jansson, J.; Garberg, P.; Postlind, H. Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol. Rep. 2006, 58, 453–472. [Google Scholar] [PubMed]
- Di, L.; Kerns, E.H.; Carter, G.T. Drug-like property concepts in pharmaceutical design. Curr. Pharm. Des. 2009, 15, 2184–2194. [Google Scholar] [CrossRef] [PubMed]
- Healt, J.P. Epithelial cell migration in the intestine. Cell Biol. Int. 1996, 20, 139–146. [Google Scholar]
- Tsubouchi, S. Theoretical implications for cell migration through the crypt and the villus of labelling studies conducted at each position within the crypt. Cell Tissue Kinet. 1983, 16, 441–456. [Google Scholar] [PubMed]
- Meineke, F.A.; Potten, C.S.; Loeffler, M. Cell migration and organization in the intestinal crypt using a lattice-free model. Cell Prolif. 2001, 34, 253–266. [Google Scholar] [CrossRef]
- Pin, C.; Watson, A.J.; Carding, S.R. Modelling the Spatio-Temporal Cell Dynamics Reveals Novel Insights on Cell Differentiation and Proliferation in the Small Intestinal Crypt. PLoS ONE 2012, 7, e37115. [Google Scholar] [CrossRef] [PubMed]
- Dunn, S.J.; Näthke, I.S.; Osborne, J.M. Computational models reveal a passive mechanism for cell migration in the crypt. PLoS ONE 2013, 8, e80516. [Google Scholar] [CrossRef] [PubMed]
- Mirams, G.R.; Fletcher, A.G.; Maini, P.K.; Byrne, H.M. A theoretical investigation of the effect of proliferation and adhesion on monoclonal conversion in the colonic crypt. J. Theor. Biol. 2012, 312, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Osborne, J.M.; Walter, A.; Kershaw, S.K.; Mirams, G.R.; Fletcher, A.G.; Pathmanathan, P.; Gavaghan, D.; Jensen, O.E.; Maini, P.K.; Byrne, H.M. A hybrid approach to multi-scale modelling of cancer. Philos. Trans. A Math. Phys. Eng Sci. 2010, 368, 5013–5028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Leeuwen, I.M.; Mirams, G.R.; Walter, A.; Fletcher, A.; Murray, P.; Osborne, J.; Varma, S.; Young, S.J.; Cooper, J.; Doyle, B.; et al. An integrative computational model for intestinal tissue renewal. Cell Prolif. 2009, 42, 617–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vihinen, P.; Kähäri, V.M. Matrix metalloproteinases in cancer: Prognostic markers and therapeutic targets. Int. J. Cancer. 2002, 99, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, A.K.; Singh, M.; Bharti, A.C.; Asotra, K.; Sundaram, S.; Mehrotra, R. Genetic polymorphism of matrix metalloproteinases and their inhibitors in potentially malignant lesions of the head and neck. J. Biomed. Sci. 2010, 17, 10. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New Functions for the Matrix Metalloproteinases in Cancer Progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.C.; Kim, H.G.; Lee, S.A.; Lim, S.; Park, E.H.; Kim, S.J.; Lim, C.J. Genipin-induced apoptosis in hepatoma cells is mediated by reactive oxygen species/c-Jun NH-terminal kinase-dependent activation of mitochondrial pathway. Biochem. Pharmacol. 2005, 70, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Rathore, S.; Datta, G.; Kaur, I.; Malhotra, P.; Mohmmed, A. Disruption of cellular homeostasis induces organelle stress and triggers apoptosis like cell-death pathways in malaria parasite. Cell Death Dis. 2015, 6, e1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Sun, S.; Li, N.; Ho, A.S.; Kiang, K.M.; Zhang, X.; Cheng, Y.S.; Poon, M.W.; Lee, D.; Pu, J.K. Rutin increases the cytotoxicity of temozolomide in glioblastoma via autophagy inhibition. J. Neurooncol. 2015, 132, 1–8. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, J.; Wu, Y.F.; Lou, J.; Mao, Y.Y.; Shen, H.H.; Chen, Z.H. mTOR and autophagy in regulation of acute lung injury: A review and perspective. Microb. Infect. 2014, 16, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.T.; Tai, C.J.; Su, C.H.; Chang, F.M.; Choong, C.Y.; Wang, C.K.; Tai, C.J. The Ethanolic Extract of Taiwanofungus camphoratus (Antrodia camphorata) Induces Cell Cycle Arrest and Enhances Cytotoxicity of Cisplatin and Doxorubicin on Human Hepatocellular Carcinoma Cells. Biomed. Res. Int. 2017, 2015, 415269. [Google Scholar]
- Chen, L.Y.; Sheu, M.T.; Liu, D.Z.; Liao, C.K.; Ho, H.O.; Kao, W.Y.; Ho, Y.S.; Lee, W.S.; Su, C.H. Pretreatment with an ethanolic extract of Taiwanofungus camphoratus (Antrodia camphorata) enhances the cytotoxic effects of amphotericin B. J. Agric. Food Chem. 2011, 59, 11255–11263. [Google Scholar] [CrossRef]
- Kumar, V.B.; Yuan, T.C.; Liou, J.W.; Yang, C.J.; Sung, P.J.; Weng, C.F. Antroquinonol inhibits NSCLC proliferation by altering PI3K/mTOR proteins and miRNA expression profiles. Mutat. Res. 2011, 707, 42–52. [Google Scholar] [CrossRef]
- Kawabe, T. G2 checkpoint abrogators as anticancer drugs. Mol. Cancer Ther. 2004, 3, 513–519. [Google Scholar] [PubMed]
- Ujiki, M.B.; Ding, X.Z.; Salabat, M.R.; Bentrem, D.J.; Golkar, L.; Milam, B.; Talamonti, M.S.; Bell, R.H.; Iwamura, T.; Adrian, T.E. Apigenin inhibits pancreatic cancer cell proliferation through G2/M cell cycle arrest. Mol. Cancer 2006, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zheng, M.; Chen, F.; Zhu, Y.; Yong, W.; Lin, H.; Sun, Y.; Han, X. Gefitinib Inhibits the Proliferation of Pancreatic Cancer Cells via Cell Cycle Arrest. Anat. Rec. 2010, 292, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Y.; Hail, N.; Lotan, R. Apoptosis as a novel target for cancer chemoprevention. J. Natl. Cancer Inst. 2004, 96, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.R.; Min, K.J.; Woo, S.M.; Choe, M.; Choi, K.S.; Lee, Y.K.; Yoon, G.; Kwon, T.K. Inhibition of Cathepsin S Induces Mitochondrial ROS That Sensitizes TRAIL-Mediated Apoptosis Through p53-Mediated Downregulation of Bcl-2 and c-FLIP. Antioxid. Redox Signal. 2017, 27, 215–233. [Google Scholar] [CrossRef]
- Guo, L.; Tan, K.; Wang, H.; Zhang, X. Pterostilbene inhibits hepatocellular carcinoma through p53/SOD2/ROS-mediated mitochondrial apoptosis. Oncol. Rep. 2016, 36, 3233–3240. [Google Scholar] [CrossRef]
- Zhou, M.; Shen, S.; Zhao, X.; Gong, X. Luteoloside induces G0/G1 arrest and pro-death autophagy through the ROS-mediated AKT/mTOR/p70S6K signalling pathway in human non-small cell lung cancer cell lines. Biochem. Biophys. Res. Commun. 2017, 494, 263–269. [Google Scholar] [CrossRef]
- Song, Y.; Kong, L.; Sun, B.; Gao, L.; Chu, P.; Ahsan, A.; Qaed, E.; Lin, Y.; Peng, J.; Ma, X.; et al. Induction of autophagy by an oleanolic acid derivative, SZC017, promotes ROS-dependent apoptosis through Akt and JAK2/STAT3 signaling pathway in human lung cancer cells. Cell Biol. Int. 2017, 41, 1367–1378. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Li, X.B.; Hou, S.G.; Sun, Y.; Shi, Y.R.; Lin, S.S. Cedrol induces autophagy and apoptotic cell death in A549 non-small cell lung carcinoma cells through the P13K/Akt signaling pathway, the loss of mitochondrial transmembrane potential and the generation of ROS. Int. J. Mol. Med. 2016, 38, 291–299. [Google Scholar] [CrossRef]
- Senthil, K.K.J.; Gokila, V.M.; Wang, S.Y. Activation of Nrf2-mediated anti-oxidant genes by antrodin C prevents hyperglycemia-inducedsenescence and apoptosis in human endothelial cells. Oncotarget 2017, 8, 96568–96587. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Shimizu, S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 2007, 12, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gao, Y.; Rankin, G.O.; Rojanasakul, Y.; Cutler, S.J.; Tu, Y.; Chen, Y.C. Chaetoglobosin K induces apoptosis and G2 cell cycle arrest through p53-dependent pathway in cisplatin-resistant ovarian cancer cells. Cancer Lett. 2015, 356, 418–433. [Google Scholar] [CrossRef]
- Jang, J.H.; Kim, Y.J.; Kim, H.; Kim, S.C.; Cho, J.H. Buforin IIb induces endoplasmic reticulum stress-mediated apoptosis in HeLa cells. Peptides 2015, 69, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, J.K.; Letai, A. Control of mitochondrial apoptosis by the Bcl-2 family. J. Cell. Sci. 2009, 122, 437–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okoshi, R.; Ozaki, T.; Yamamoto, H.; Ando, K.; Koida, N.; Ono, S.; Koda, T.; Kamijo, T.; Nakagawara, A.; Kizaki, H. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J. Biol. Chem. 2008, 283, 3979–3987. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, C. Inhibition of autophagy enhances synergistic effects of Salidroside and anti-tumor agents against colorectal cancer. Redox Biol. 2015, 4, 215–225. [Google Scholar]
- Komatsu, M.; Kominami, E.; Tanaka, K. Autophagy and neurodegeneration. Autophagy 2006, 2, 315–317. [Google Scholar] [CrossRef]
- Jiang, X.; Overholtzer, M.; Thompson, C.B. Autophagy in cellular metabolism and cancer. J. Clin. Investig. 2015, 125, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liang, X.; Chang, H.; Shu, F.; Wu, Y.; Zhang, T.; Fu, Y.; Zhang, Q.; Zhu, J.D.; Mi, M. Ampelopsin-induced autophagy protects breast cancer cells from apoptosis through Akt-mTOR pathway via endoplasmic reticulum stress. Cancer Sci. 2014, 105, 1279–1287. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.; Huang, W.; Liu, X.; Liu, X.; Chen, N.; Xu, Q.; Hu, Y.; Song, W.; Zhou, J. IMPDH2 promotes colorectal cancer progression through activation of the PI3K/AKT/mTOR and PI3K/AKT/FOXO1 signaling pathways. J. Exp. Clin. Cancer Res. 2018, 37, 304. [Google Scholar] [CrossRef]
- Paul, D.; Bargale, A.B.; Rapole, S.; Shetty, P.K.; Santra, M.K. Protein Phosphatase 1 Regulatory Subunit SDS22 Inhibits Breast Cancer Cell Tumorigenesis by Functioning as a Negative Regulator of the AKT Signaling Pathway. Neoplasia 2019, 21, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Shankar, S.; Srivastava, R.K. Rottlerin induces autophagy and apoptosis in prostate cancer stem cells via PI3K/Akt/mTOR signaling pathway. Cancer Lett. 2014, 343, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Kumar, D.; Shankar, S.; Srivastava, R.K. Rottlerin induces autophagy which leads to apoptotic cell death through inhibition of PI3K/Akt/mTOR pathway in human pancreatic cancer stem cells. Biochem. Pharmacol. 2012, 84, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.; Jeong, S.; Jing, K.; Shin, S.; Kim, S.; Heo, J.Y.; Kweon, G.R.; Park, S.K.; Wu, T.; Park, J.I.; et al. Docosahexaenoic Acid Induces Cell Death in Human Non-Small Cell Lung Cancer Cells by Repressing mTOR via AMPK Activation and PI3K/Akt Inhibition. Biomed. Res. Int. 2015, 2015, 239764. [Google Scholar] [CrossRef] [PubMed]
- Vucicevic, L.; Misirkic, M.; Janjetovic, K.; Vilimanovich, U.; Sudar, E.; Isenovic, E.; Prica, M.; Harhaji-Trajkovic, L.; Kravic-Stevovic, T.; Bumbasirevic, V.; et al. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy 2011, 7, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papandreou, I.; Lim, A.L.; Laderoute, K.; Denko, N.C. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008, 15, 1572–1581. [Google Scholar] [CrossRef] [Green Version]
- Filomeni, G.; Desideri, E.; Cardaci, S.; Graziani, I.; Piccirillo, S.; Rotilio, G.; Ciriolo, M.R. Carcinoma cells activate AMP-activated protein kinase-dependent autophagy as survival response to kaempferol-mediated energetic impairment. Autophagy 2010, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Law, B.Y.; Wang, M.; Ma, D.L.; Al-Mousa, F.; Michelangeli, F.; Cheng, S.H.; Ng, M.H.; To, K.F.; Mok, A.Y.; Ko, R.Y.; et al. Alisol B, a novel inhibitor of the sarcoplasmic/endoplasmic reticulum Ca (2+) ATPase pump, induces autophagy, endoplasmic reticulum stress, and apoptosis. Mol. Cancer Ther. 2010, 9, 718–730. [Google Scholar] [CrossRef]
- Xu, Z.X.; Liang, J.; Haridas, V.; Gaikwad, A.; Connolly, F.P.; Mills, G.B.; Gutterman, J.U. A plant triterpenoid, avicin D, induces autophagy by activation of AMP-activated protein kinase. Cell Death Differ. 2007, 14, 1948–1957. [Google Scholar] [CrossRef] [Green Version]
- Di, L.; Kerns, E.H.; Ma, X.J.; Huang, Y.; Carter, G.T. Applications of high throughput microsomal stability assay in drug discovery. Comb. Chem. High Throughput Screen 2008, 11, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Schyman, P.; Wallqvist, A. Critically Assessing the Predictive Power of QSAR Models for Human Liver Microsomal Stability. J. Chem. Inf. Model. 2015, 55, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Nyhan, M.J.; O’Donovan, T.R.; Boersma, A.W.; Wiemer, E.A.; McKenna, S.L. MiR-193b promotes autophagy and non-apoptotic cell death in oesophageal cancer cells. BMC Cancer 2016, 16, 101. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | T1/2 (min) | CLint(mic) (μL/min/mg protein) | CLint(liver) (mL/min/kg) |
|---|---|---|---|
| SD rat | 7.5 | 185.8 | 334.4 |
| Human | 54.1 | 25.6 | 23.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Bai, X.; Zhang, H.; Zhang, J.; Wu, Y.; Tang, C.; Liu, Y.; Yang, Y.; Liu, Z.; Jia, W.; et al. Antrodin C, an NADPH Dependent Metabolism, Encourages Crosstalk between Autophagy and Apoptosis in Lung Carcinoma Cells by Use of an AMPK Inhibition-Independent Blockade of the Akt/mTOR Pathway. Molecules 2019, 24, 993. https://doi.org/10.3390/molecules24050993
Yang H, Bai X, Zhang H, Zhang J, Wu Y, Tang C, Liu Y, Yang Y, Liu Z, Jia W, et al. Antrodin C, an NADPH Dependent Metabolism, Encourages Crosstalk between Autophagy and Apoptosis in Lung Carcinoma Cells by Use of an AMPK Inhibition-Independent Blockade of the Akt/mTOR Pathway. Molecules. 2019; 24(5):993. https://doi.org/10.3390/molecules24050993
Chicago/Turabian StyleYang, Hairui, Xu Bai, Henan Zhang, Jingsong Zhang, Yingying Wu, Chuanhong Tang, Yanfang Liu, Yan Yang, Zhendong Liu, Wei Jia, and et al. 2019. "Antrodin C, an NADPH Dependent Metabolism, Encourages Crosstalk between Autophagy and Apoptosis in Lung Carcinoma Cells by Use of an AMPK Inhibition-Independent Blockade of the Akt/mTOR Pathway" Molecules 24, no. 5: 993. https://doi.org/10.3390/molecules24050993
APA StyleYang, H., Bai, X., Zhang, H., Zhang, J., Wu, Y., Tang, C., Liu, Y., Yang, Y., Liu, Z., Jia, W., & Wang, W. (2019). Antrodin C, an NADPH Dependent Metabolism, Encourages Crosstalk between Autophagy and Apoptosis in Lung Carcinoma Cells by Use of an AMPK Inhibition-Independent Blockade of the Akt/mTOR Pathway. Molecules, 24(5), 993. https://doi.org/10.3390/molecules24050993