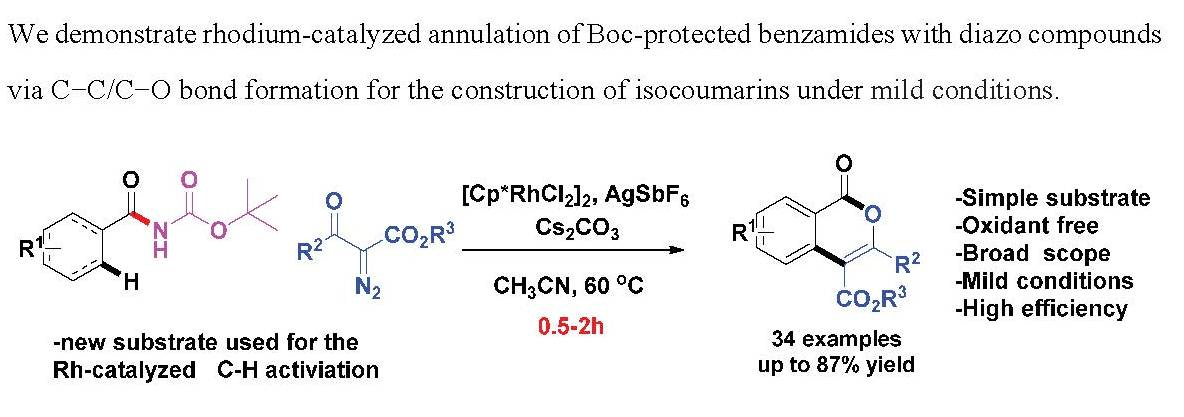
Rh(III)-Catalyzed Annulation of Boc-Protected Benzamides with Diazo Compounds: Approach to Isocoumarins


Abstract

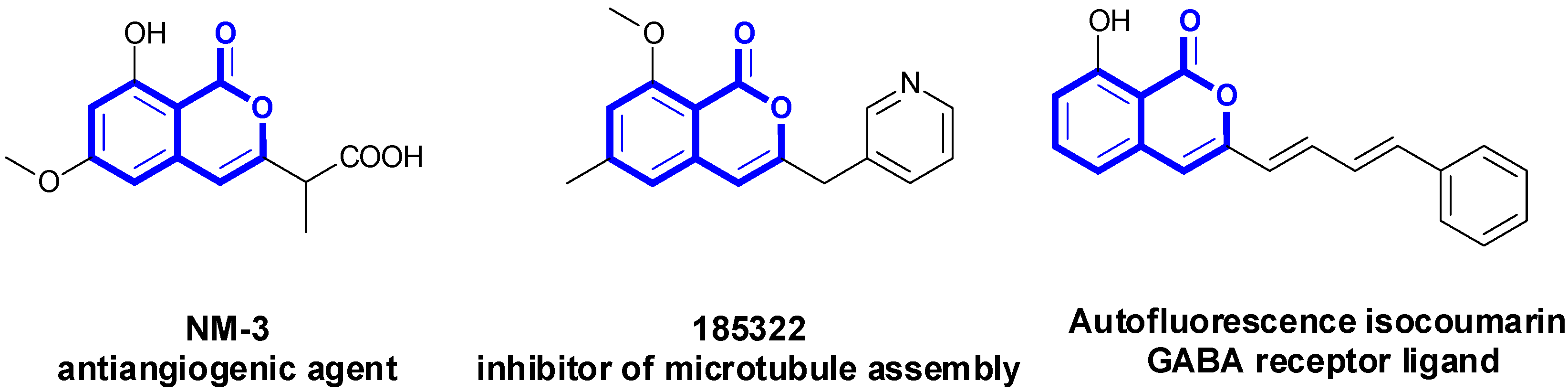
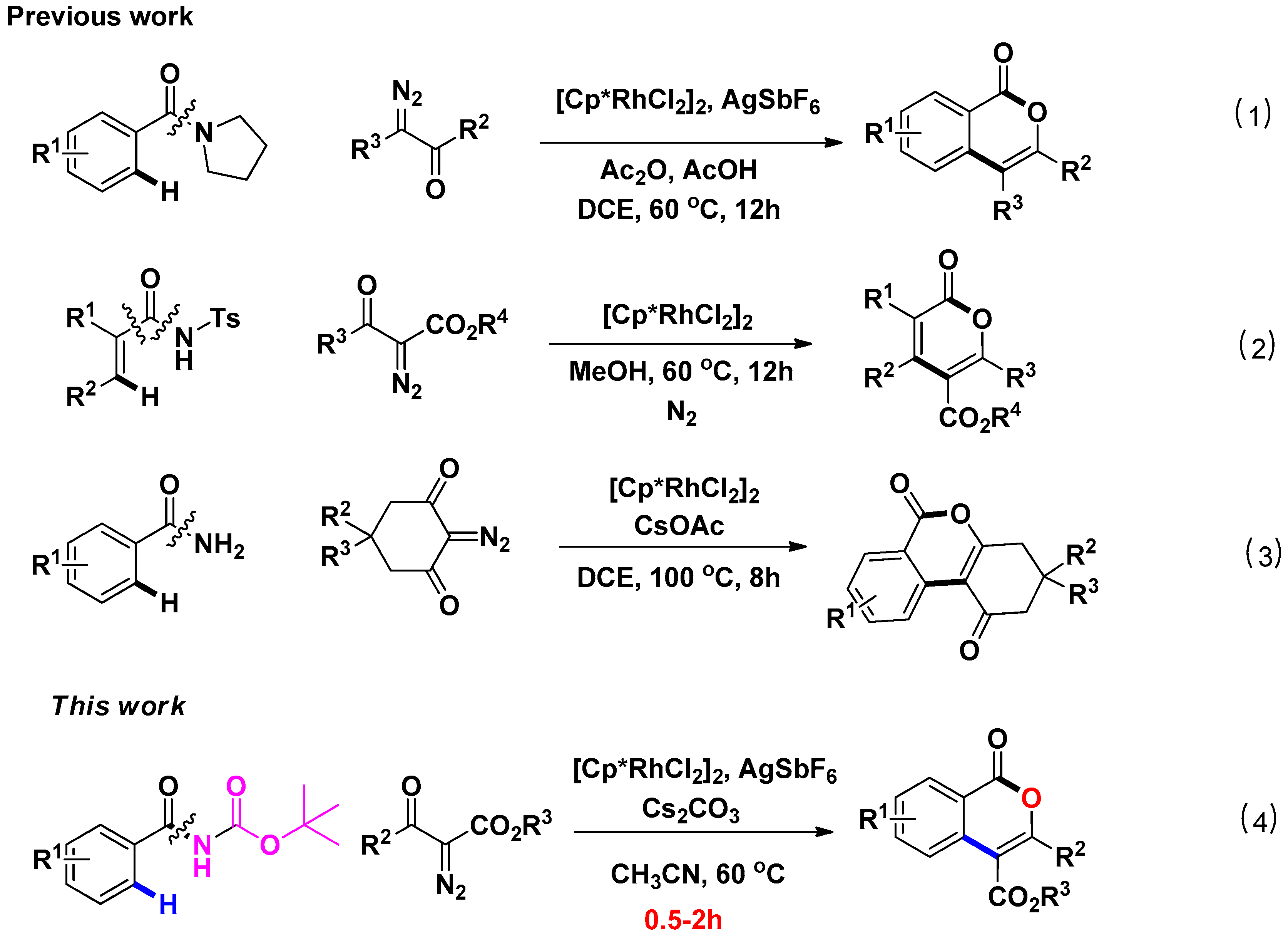
1. Introduction
2. Results and Discussion
2.1. Optimization of Reaction Conditions for Synthesis of Ethyl 3-Methyl-1-oxo-1H-Isochromene-4-Carboxylate 3aa
2.2. Substrate Scope for the Boc-Protected Benzamides
2.3. Substrate Scope for the Diazo Compounds
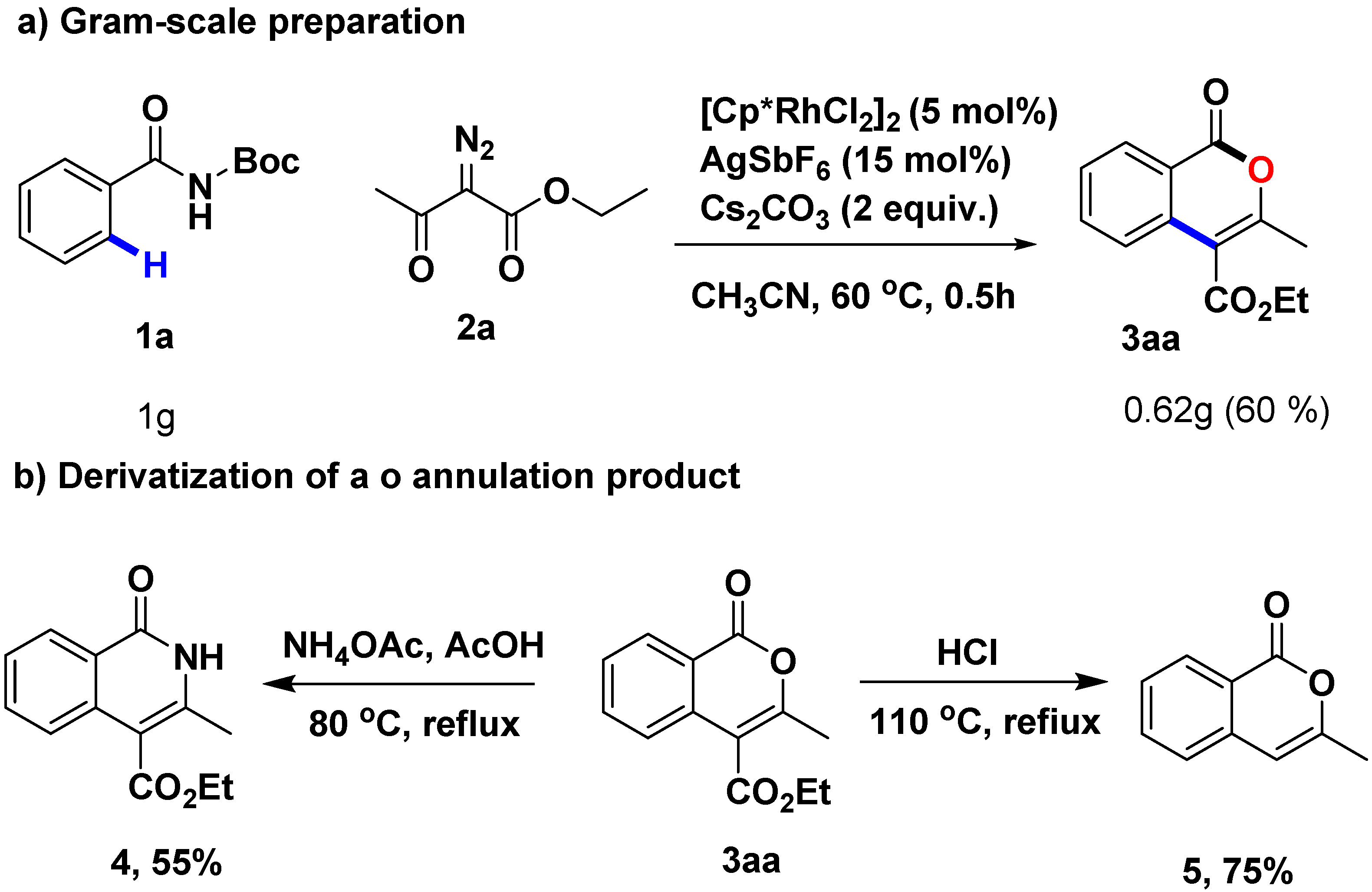
2.4. Gram-Scale Preparation and Derivatization of the Annulation Product
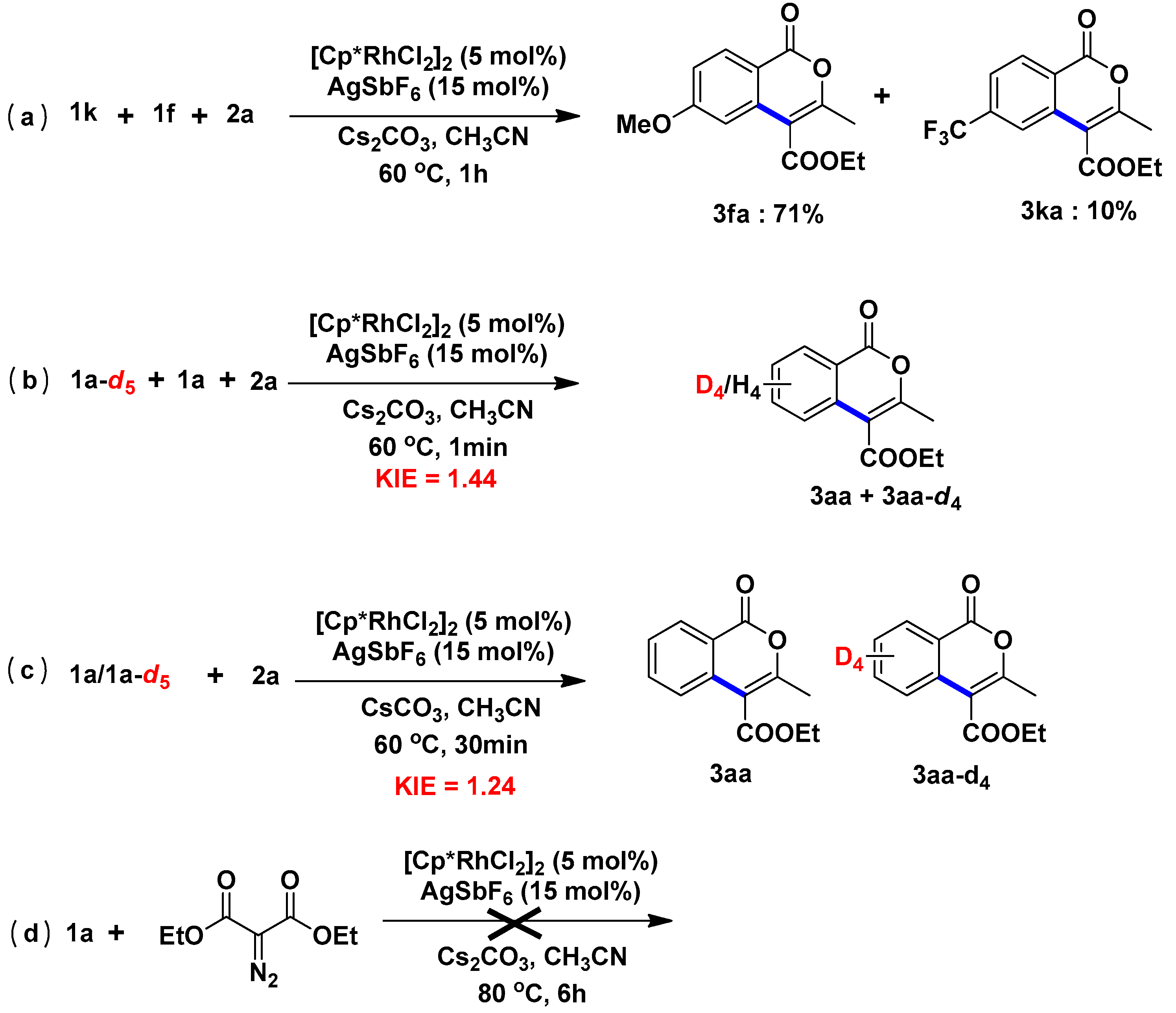
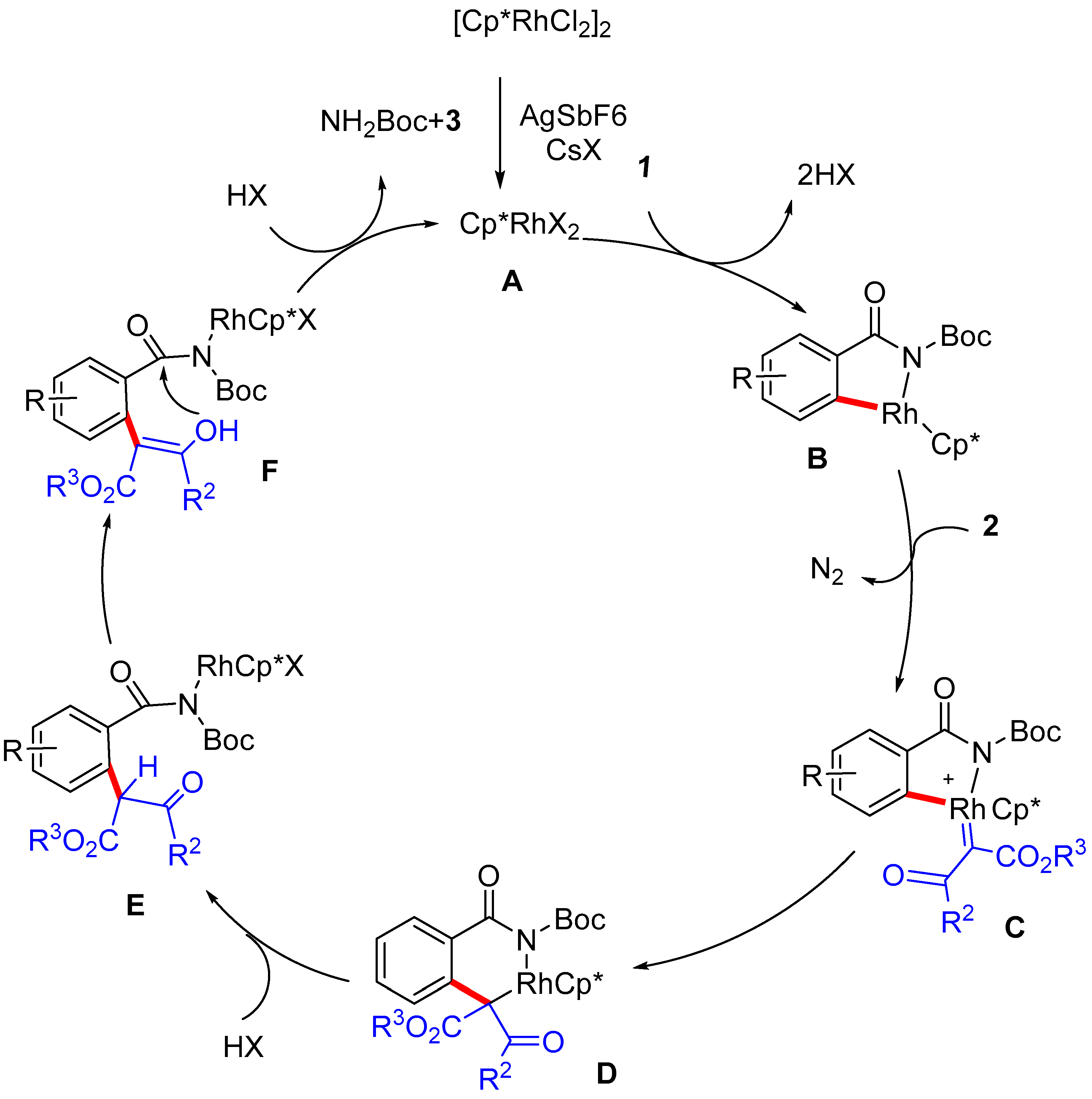
2.5. Mechanism
3. Materials and Methods
3.1. Chemistry
3.2. Experimental Part Method
3.2.1. General Procedure for the Synthesis of Isocoumarins and α-Pyrones
3.2.2. Procedure for the Synthesis of Diazo Substrates
3.2.3. Procedure for the Synthesis of Benzoylcarbamate Derivatives [37]
3.2.4. Characterization of the Products
3.2.5. Derivatization Experiments
3.2.6. Compete Experiments and Mechanistic Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Saeed, A. Isocoumarins, miraculous natural products blessed with diverse pharmacological activities. Eur. J. Med. Chem. 2016, 116, 290–317. [Google Scholar] [CrossRef] [PubMed]
- Agata, N.; Nogi, H.; Bamberg, M.; Milhollen, M.; Pu, M.; Weitman, S.; Kharbanda, S.; Kufe, D. The angiogenesis inhibitor NM-3 is active against human NSCLC xenografts alone and in combination with docetaxel. Cancer Chemoth. Pharm. 2005, 56, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Agata, N.; Kharbanda, S.; Avigan, D.; Kufe, D. A novel isocoumarin derivative induces mitotic phase arrest and apoptosis of human multiple myeloma cells. Cancer Chemoth. Pharm. 2007, 59, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Ozoe, Y.; Kuriyama, T.; Tachibana, Y.; Harimaya, K.; Takahashi, N.; Yaguchi, T.; Suzuki, E.; Imamura, K.-i.; Oyama, K. Isocoumarin Derivative as a Novel GABA Receptor Ligand from Neosartorya quadricincta. J. Pest. Sci. 2004, 29, 328–331. [Google Scholar] [CrossRef]
- Larock, R.C.; Doty, M.J.; Han, X. Synthesis of Isocoumarins and α-Pyrones via Palladium-Catalyzed Annulation of Internal Alkynes. J. Org. Chem. 1999, 64, 8770–8779. [Google Scholar] [CrossRef] [PubMed]
- Rayabarapu, D.K.; Shukla, P.; Cheng, C.-H. Cyclization of Oxa-Bicyclic Alkenes with β-Iodo-(Z)-propenoates and o-Iodobenzoate Catalyzed by Nickel Complexes: A Simple Efficient Route to Annulated Coumarins. Org. Lett. 2003, 5, 4903–4906. [Google Scholar] [CrossRef] [PubMed]
- Cherry, K.; Parrain, J.-L.; Thibonnet, J.; Duchêne, A.; Abarbri, M. Synthesis of Isocoumarins and α-Pyrones via Tandem Stille Reaction/Heterocyclization. J. Org. Chem. 2005, 70, 6669–6675. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Lu, Y.; Liu, S.; Liu, J.; Deng, G.J. Efficient One-Pot Synthesis of Dibenzopyranones via a Decarboxylative Cross-Coupling and Lactonization Sequence. Adv. Synth. Catal. 2011, 353, 2604–2608. [Google Scholar] [CrossRef]
- Panda, N.; Mishra, P.; Mattan, I. Synthesis of Isocoumarins via Silver(I)-Mediated Annulation of Enol Esters. J. Org. Chem. 2016, 81, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Ueura, K.; Satoh, T.; Miura, M. An Efficient Waste-Free Oxidative Coupling via Regioselective C−H Bond Cleavage: Rh/Cu-Catalyzed Reaction of Benzoic Acids with Alkynes and Acrylates under Air. Org. Lett. 2007, 9, 1407–1409. [Google Scholar] [CrossRef] [PubMed]
- Mochida, S.; Hirano, K.; Satoh, T.; Miura, M. Synthesis of Functionalized α-Pyrone and Butenolide Derivatives by Rhodium-Catalyzed Oxidative Coupling of Substituted Acrylic Acids with Alkynes and Alkenes. J. Org. Chem. 2009, 74, 6295–6298. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Pospech, J.; Graczyk, K.; Rauch, K. Versatile Synthesis of Isocoumarins and α-Pyrones by Ruthenium-Catalyzed Oxidative C-H/O–H Bond Cleavages. Org. Lett. 2012, 14, 930–933. [Google Scholar] [CrossRef] [PubMed]
- Frasco, D.A.; Lilly, C.P.; Boyle, P.D.; Ison, E.A. Cp*IrIII-Catalyzed Oxidative Coupling of Benzoic Acids with Alkynes. ACS Catal. 2013, 3, 2421–2429. [Google Scholar] [CrossRef]
- Warratz, S.; Kornhaass, C.; Cajaraville, A.; Niepotter, B.; Stalke, D.; Ackermann, L. Ruthenium(II)-catalyzed C-H activation/alkyne annulation by weak coordination with O2 as the sole oxidant. Angew Chem. Int. Ed. Engl. 2015, 54, 5513–5517. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-G.; Gao, H.; Zhang, S.-S.; Li, Q.; Wang, H. N–O Bond as External Oxidant in Group 9 Cp*M(III)-Catalyzed Oxidative C-H Coupling Reactions. ACS Catal. 2017, 7, 5078–5086. [Google Scholar] [CrossRef]
- Qiu, Y.; Tian, C.; Massignan, L.; Rogge, T.; Ackermann, L. Electrooxidative Ruthenium-Catalyzed C-H/O-H Annulation by Weak O-Coordination. Angew Chem. Int. Ed. Engl. 2018, 57, 5818–5822. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.A.; Bergman, R.G.; Ellman, J.A. Rhodium-Catalyzed C−C Bond Formation via Heteroatom-Directed C−H Bond Activation. Chem. Rev. 2010, 110, 624–655. [Google Scholar] [CrossRef] [PubMed]
- Wencel-Delord, J.; Dröge, T.; Liu, F.; Glorius, F. Towards mild metal-catalyzed C-H bond activation. Chem. Soc. Rev. 2011, 40, 4740–4761. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Wang, F.; Li, X. C-C, C-O and C-N bond formation via rhodium(iii)-catalyzed oxidative C-H activation. Chem. Soc. Rev. 2012, 41, 3651–3678. [Google Scholar] [CrossRef] [PubMed]
- Peneau, A.; Guillou, C.; Chabaud, L. Recent Advances in [Cp*MIII] (M = Co, Rh, Ir)-Catalyzed Intramolecular Annulation Through C-H Activation. Eur. J. Org. Chem. 2018, 2018, 5777–5794. [Google Scholar] [CrossRef]
- Adams, J.; Spero, D.M. Rhodium (II) catalyzed reactions of diazo-carbonyl compounds. Tetrahedron. 1991, 47, 1765–1808. [Google Scholar] [CrossRef]
- Padwa, A.; Austin, D.J. Ligand Effects on the Chemoselectivity of Transition Metal Catalyzed Reactions of α-Diazo Carbonyl Compounds. Angew Chem. Int. Ed. Engl. 1994, 33, 1797–1815. [Google Scholar] [CrossRef]
- Ye, T.; McKervey, M.A. Organic Synthesis with .alpha.-Diazo Carbonyl Compounds. Chem. Rev. 1994, 94, 1091–1160. [Google Scholar] [CrossRef]
- Maas, G. Ruthenium-catalysed carbenoid cyclopropanation reactions with diazo compounds. Chem. Soc. Rev. 2004, 33, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J. Recent development of reactions with α-diazocarbonyl compounds as nucleophiles. Chem. Commun. (Camb). 2009, 5350–5361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J. Recent Developments in Pd-Catalyzed Reactions of Diazo Compounds. Eur. J. Org. Chem. 2011, 2011, 1015–1026. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhang, Y.; Wang, J. Diazo Compounds and N-Tosylhydrazones: Novel Cross-Coupling Partners in Transition-Metal-Catalyzed Reactions. Acc. Chem. Res. 2013, 46, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Hyster, T.K.; Ruhl, K.E.; Rovis, T. A Coupling of Benzamides and Donor/Acceptor Diazo Compounds To Form γ-Lactams via Rh(III)-Catalyzed C-H Activation. J. Am. Chem. Soc. 2013, 135, 5364–5367. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.-W.; Man, K.-Y.; Chan, W.-W.; Zhou, Z.; Yu, W.-Y. Rhodium(iii)-catalyzed formal oxidative [4 + 1] cycloaddition of benzohydroxamic acids and α-diazoesters. A facile synthesis of functionalized benzolactams. Org. Biomol. Chem. 2014, 12, 4112–4116. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Cramer, N. Asymmetric Synthesis of Isoindolones by Chiral Cyclopentadienyl-Rhodium(III)-Catalyzed C–H Functionalizations. Angew Chem. Int. Ed. 2014, 53, 7896–7899. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Yu, K.; Wang, B. Regioselective synthesis of multisubstituted isoquinolones and pyridones via Rh(iii)-catalyzed annulation reactions. Chem. Commun. (Camb). 2015, 51, 17277–17280. [Google Scholar] [CrossRef] [PubMed]
- Phatake, R.S.; Patel, P.; Ramana, C.V. Ir(III)-Catalyzed Carbenoid Functionalization of Benzamides: Synthesis of N-Methoxyisoquinolinediones and N-Methoxyisoquinolinones. Org. Lett. 2016, 18, 2828–2831. [Google Scholar] [CrossRef] [PubMed]
- Li, X.G.; Sun, M.; Liu, K.; Jin, Q.; Liu, P.N. Rh(iii)-catalyzed C-H activation/cyclization of benzamides and diazo compounds to form isocoumarins and α-pyrones. Chem. Commun. (Camb). 2015, 51, 2380–2383. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, D.; Wan, Y.; Ma, C. Rhodium-catalyzed tunable oxidative cyclization toward the selective synthesis of α-pyrones and furans. Chem. Commun. (Camb). 2016, 52, 1661–1664. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Han, G.; Zuo, Y.; Shang, Y. Rh(III)-catalyzed C-H activation of primary benzamides and tandem cyclization with cyclic 2-diazo-1,3-diketones for the synthesis of isocoumarins. Tetrahedron. 2018, 74, 7082–7088. [Google Scholar] [CrossRef]
- Sun, P.; Gao, S.; Yang, C.; Guo, S.; Lin, A.; Yao, H. Controllable Rh(III)-Catalyzed Annulation between Salicylaldehydes and Diazo Compounds: Divergent Synthesis of Chromones and Benzofurans. Org. Lett. 2016, 18, 6464–6467. [Google Scholar] [CrossRef] [PubMed]
- Kanbayashi, N.; Takenaka, K.; Okamura, T.A.; Onitsuka, K. Asymmetric auto-tandem catalysis with a planar-chiral ruthenium complex: sequential allylic amidation and atom-transfer radical cyclization. Angew Chem. Int. Ed. Engl. 2013, 52, 4897–4901. [Google Scholar] [CrossRef] [PubMed]
- Evans, V.; Mahon, M.F.; Webster, R.L. A mild, copper-catalysed amide deprotection strategy: use of tert-butyl as a protecting group. Tetrahedron. 2014, 70, 7593–7597. [Google Scholar] [CrossRef]
- Tanaka, K.-I.; Yoshifuji, S.; Nitta, Y. A New Method for the Synthesis of Amides from Amines: Ruthenium Tetroxide Oxidation of N-Protected Alkylamines. Chem. Pharm. Bull. 1988, 36, 3125–3129. [Google Scholar] [CrossRef]
- Meng, G.; Lei, P.; Szostak, M. A General Method for Two-Step Transamidation of Secondary Amides Using Commercially Available, Air- and Moisture-Stable Palladium/NHC (N-Heterocyclic Carbene) Complexes. Org. Lett. 2017, 19, 2158–2161. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, S.; Trudell, M.L. Novel chromium(VI) catalyzed oxidation of N-alkylamides to imides with periodic acid. Chem. Commun. (Camb). 2004, 1668–1669. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Shi, S.; Lalancette, R.; Szostak, R.; Szostak, M. Reversible Twisting of Primary Amides via Ground State N-C(O) Destabilization: Highly Twisted Rotationally Inverted Acyclic Amides. J. Am. Chem. Soc. 2018, 140, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; de Almeida, A.M.; de Almeida, M.V.; Lindhardt, A.T.; Skrydstrup, T. Synthesis of acyl carbamates via four component Pd-catalyzed carbonylative coupling of aryl halides, potassium cyanate, and alcohols. Org. Lett. 2015, 17, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Furber, M.; Alcaraz, L.; Luckhurst, C.; Bahl, A.; Beaton, H.; Bowers, K.; Collington, J.; Denton, R.; Donald, D.; Kinchin, E.; et al. Discovery and evolution of phenoxypiperidine hydroxyamide dual CCR3/H1 antagonists. Part II: Optimising in vivo clearance. Bioorg. Med. Chem. Lett. 2012, 22, 7707–7710. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sun, P.; Zhang, K.; Yang, T.; Yao, H.; Lin, A. Rh(III)-Catalyzed Redox-Neutral Annulation of Primary Benzamides with Diazo Compounds: Approach to Isoquinolinones. J. Org. Chem. 2016, 81, 2166–2173. [Google Scholar] [CrossRef] [PubMed]
- Perveen, S.; Zhao, Z.; Zhang, G.; Liu, J.; Anwar, M.; Fang, X. Enantioselective Synthesis of 1,2-Dihydronaphthalenes via Oxidative N-Heterocyclic Carbene Catalysis. Org. Lett. 2017, 19, 2470–2473. [Google Scholar] [CrossRef] [PubMed]
- Weerasinghe, M.S.; Karlson, S.T.; Lu, Y.; Wheeler, K.A. Crystal Photodimerization Reactions of Spatially Engineered Isocoumarin Assemblies. Cryst. Growth Des. 2016, 16, 1781–1785. [Google Scholar] [CrossRef]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | R1 | R2 | Base | Addtive | Solvent | 3aa Yield (%) |
|---|---|---|---|---|---|---|
| 1 | H | Acetyl | NaOAc | - | DCE | nr |
| 2 | Me | Boc | NaOAc | - | DCE | nr |
| 3 | Boc | Boc | NaOAc | - | DCE | nr |
| 4 | H | CO2Me | NaOAc | - | DCE | nr |
| 5 | H | CO2ipr | NaOAc | - | DCE | trace |
| 6 | H | Boc | NaOAc | - | DCE | 19 |
| 7 | H | Boc | NaOAc | Ag2O | DCE | nr |
| 8 | H | Boc | NaOAc | AgOAc | DCE | nr |
| 9 | H | Boc | NaOAc | Cu(OAc)2 | DCE | 23 |
| 10 | H | Boc | NaOAc | AgSbF6 | DCE | 33 |
| 11 | H | Boc | NaOAc | AgSbF6 | CH3CN | 47 |
| 12 | H | Boc | NaOAc | AgSbF6 | MeOH | trace |
| 13 | H | Boc | Na2CO3 | AgSbF6 | CH3CN | 67 |
| 14 | H | Boc | Cs2CO3 | AgSbF6 | CH3CN | 80 |
| 15 | H | Boc | Na2Opiv | AgSbF6 | CH3CN | 30 |
| 16 b | H | Boc | NaOAc | - | DCE | nr |
| 17 c | H | Boc | NaOAc | - | DCE | 14 |
| 18 d | H | Boc | NaOAc | - | DCE | 16 |
| 19 e | H | Boc | Cs2CO3 | AgSbF6 | CH3CN | 78 |
| 20 f | H | Boc | Cs2CO3 | AgSbF6 | CH3CN | 54 |
| 21 g | H | Boc | Cs2CO3 | AgSbF6 | CH3CN | 79 |
| 22 h | H | Boc | Cs2CO3 | AgSbF6 | CH3CN | 65 |
| 23 i | H | Boc | Cs2CO3 | AgSbF6 | CH3CN | 78 |

 |  |  |  |
| 3aa, 80% | 3bab, 87% | 3ca, 68% | 3da, 61% |
 |  |  |  |
| 3ea, 79% | 3fab, 81% | 3ga, 72% | 3ga |
 |  |  |  |
| 3ha, 48% | 3ia, 61% | 3ja, 51% | 3kac, 22% |
 |  |  |  |
| 3la, trace | 3ma, 69% | 3na, 75% | 3oa, 61% |
 |  |  |  |
| 3pa, 60% | 3qa, 48% | 3ra, 65% | 3sa, 53% |
 |  |  | |
| 3ta, 82% | 3ua, 51% | 3va, 68% |

 |  |  |  |
| 3ab, 79% | 3acb, 69% | 3adb, 61% | 3aeb, 51% |
 |  |  |  |
| 3af, 68% | 3ag, 61% | 3ah, 77% | 3ai, 45% |
 |  |  |  |
| 3aj, 61% | 3ak, 53% | 3al, 50% | 3amc, 25% |
 |  | ||
| 3an, 70% | 3ao, 63% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, G.; Li, C.; Liu, H. Rh(III)-Catalyzed Annulation of Boc-Protected Benzamides with Diazo Compounds: Approach to Isocoumarins. Molecules 2019, 24, 937. https://doi.org/10.3390/molecules24050937
Dong G, Li C, Liu H. Rh(III)-Catalyzed Annulation of Boc-Protected Benzamides with Diazo Compounds: Approach to Isocoumarins. Molecules. 2019; 24(5):937. https://doi.org/10.3390/molecules24050937
Chicago/Turabian StyleDong, Guangyu, Chunpu Li, and Hong Liu. 2019. "Rh(III)-Catalyzed Annulation of Boc-Protected Benzamides with Diazo Compounds: Approach to Isocoumarins" Molecules 24, no. 5: 937. https://doi.org/10.3390/molecules24050937
APA StyleDong, G., Li, C., & Liu, H. (2019). Rh(III)-Catalyzed Annulation of Boc-Protected Benzamides with Diazo Compounds: Approach to Isocoumarins. Molecules, 24(5), 937. https://doi.org/10.3390/molecules24050937