First-Principles Study of AlPO4-H3, a Hydrated Aluminophosphate Zeotype Containing Two Different Types of Adsorbed Water Molecules


Abstract
:1. Introduction
2. Results
2.1. Method Validation
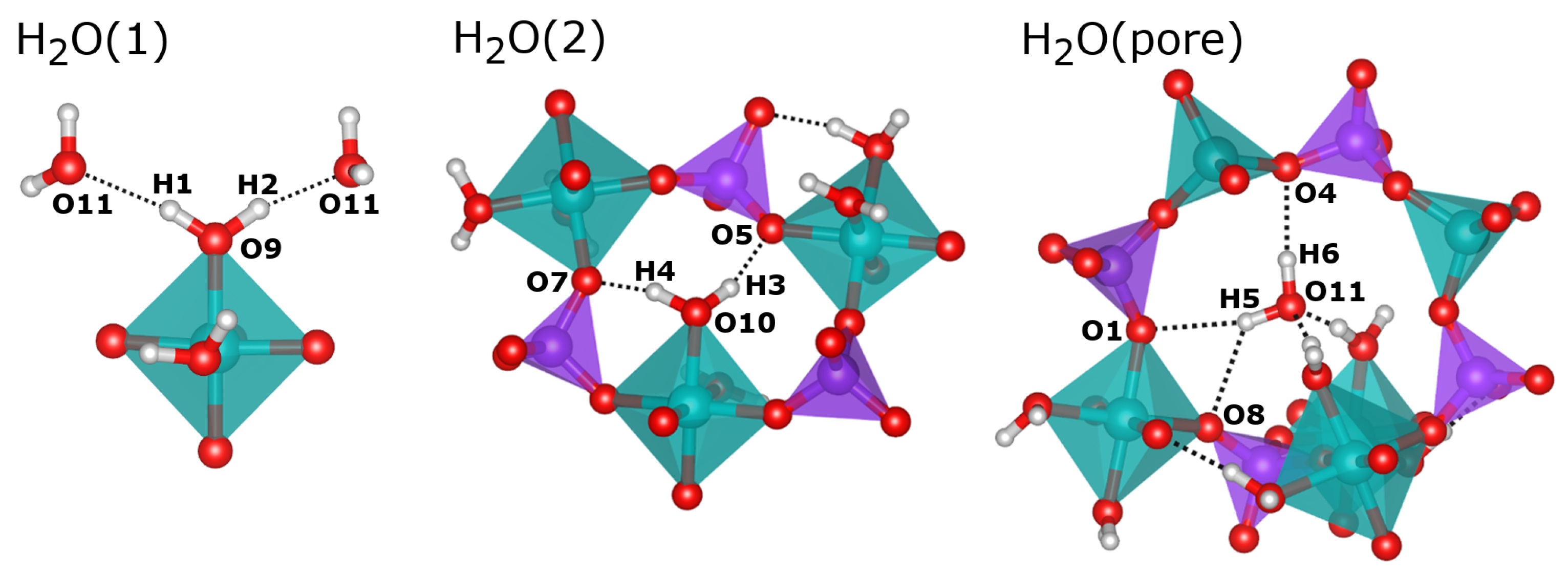
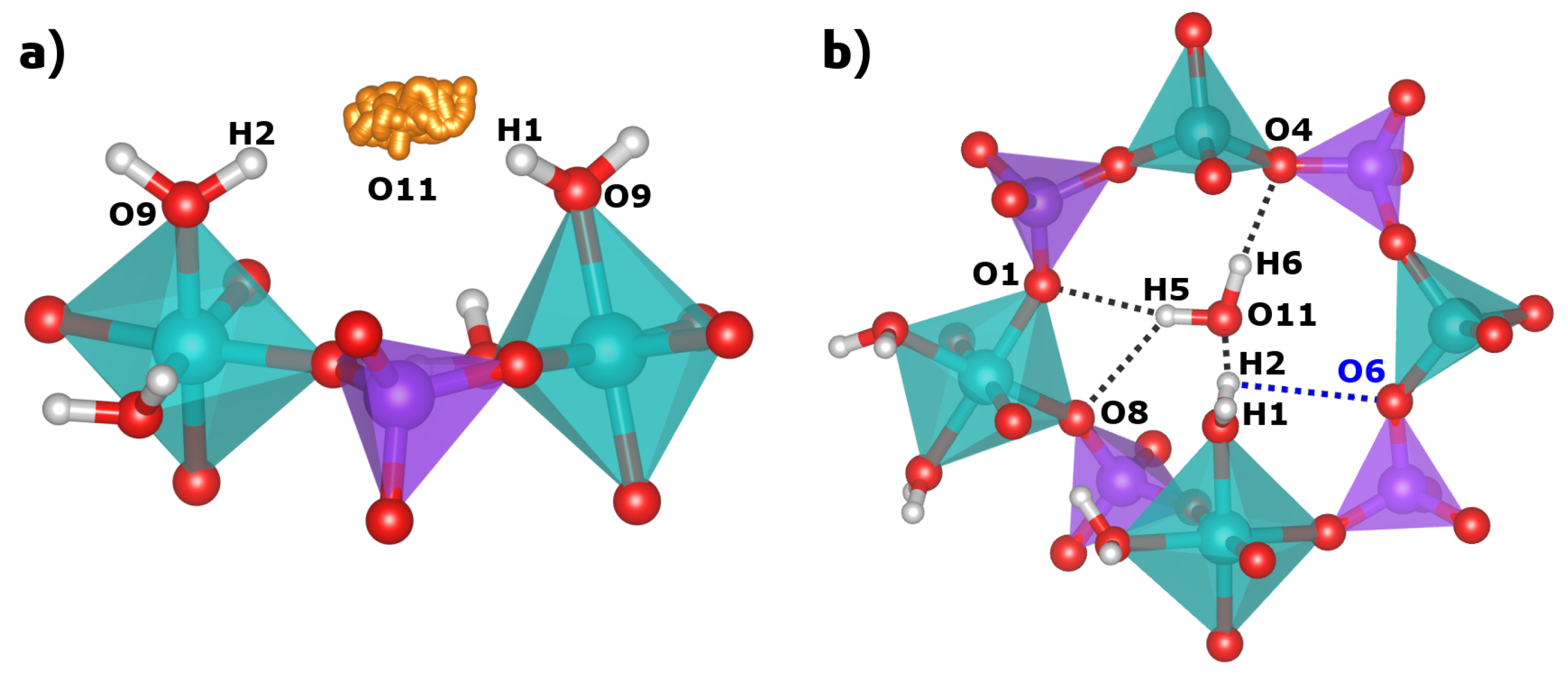
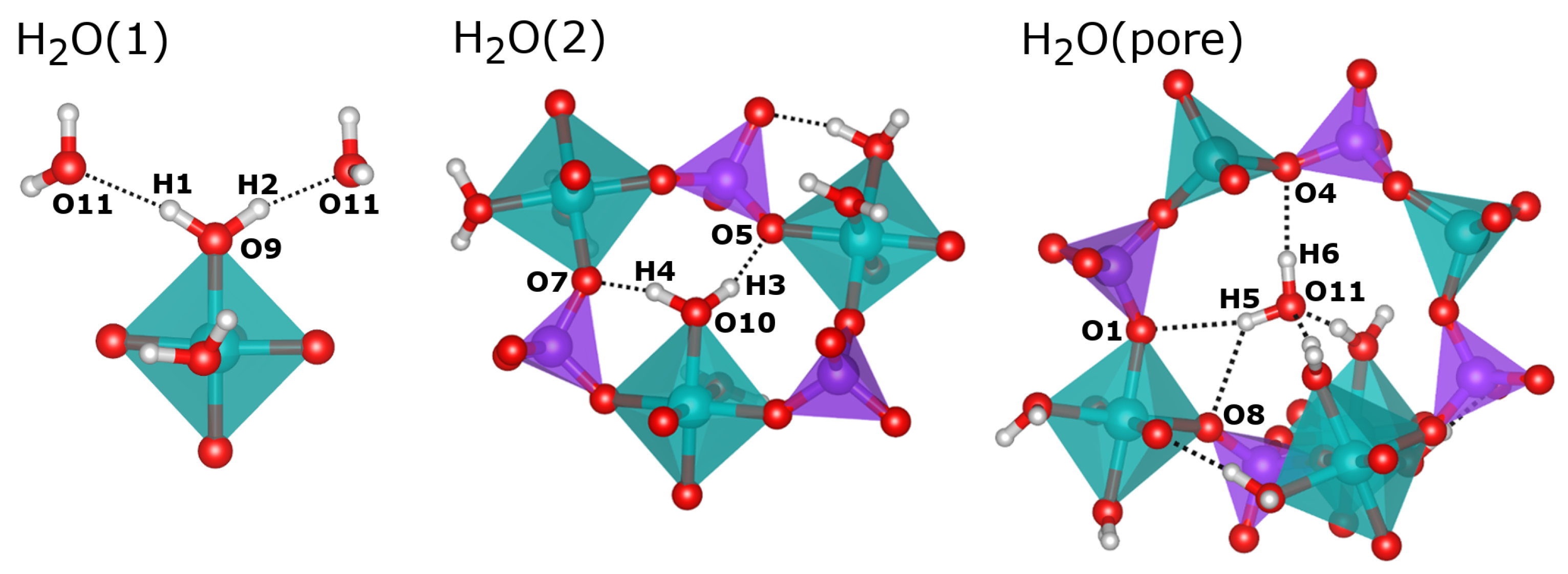
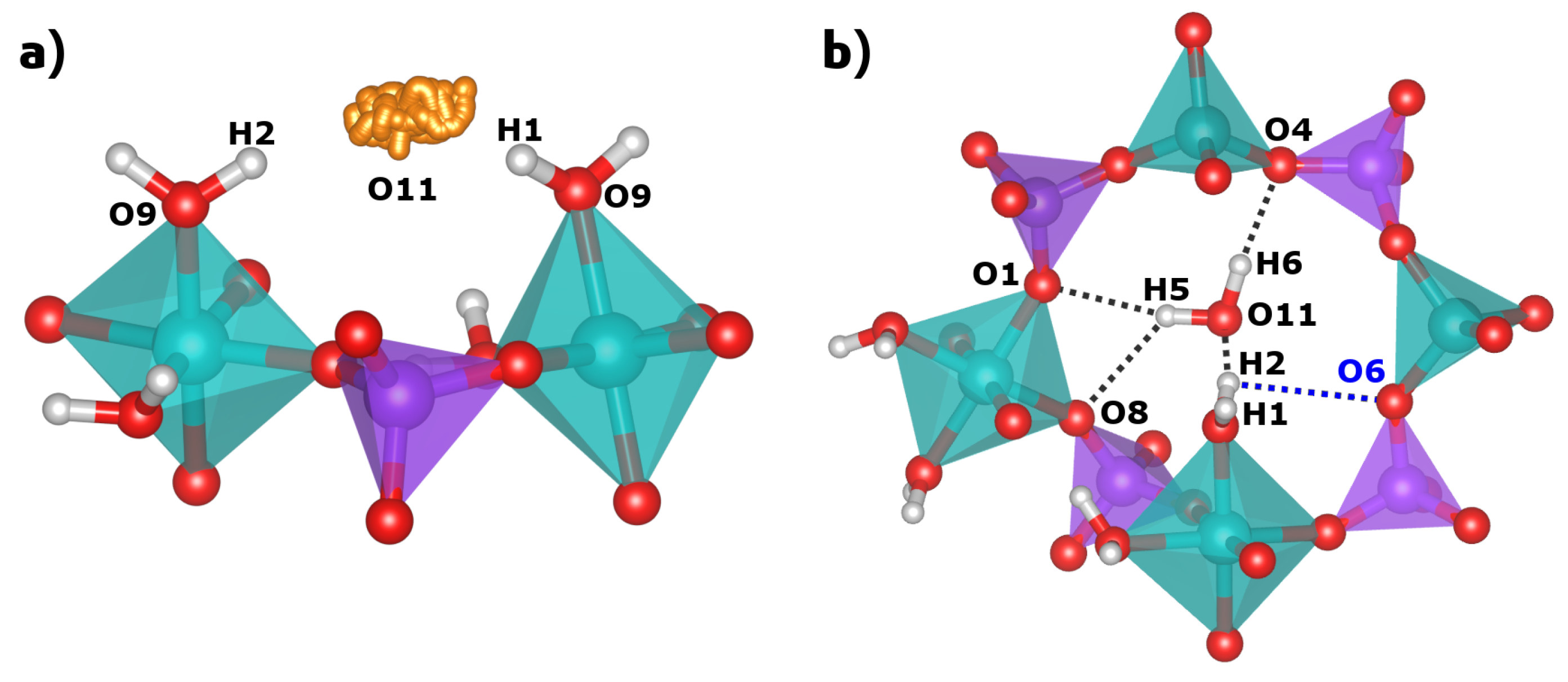
2.2. Hydrogen Bonds
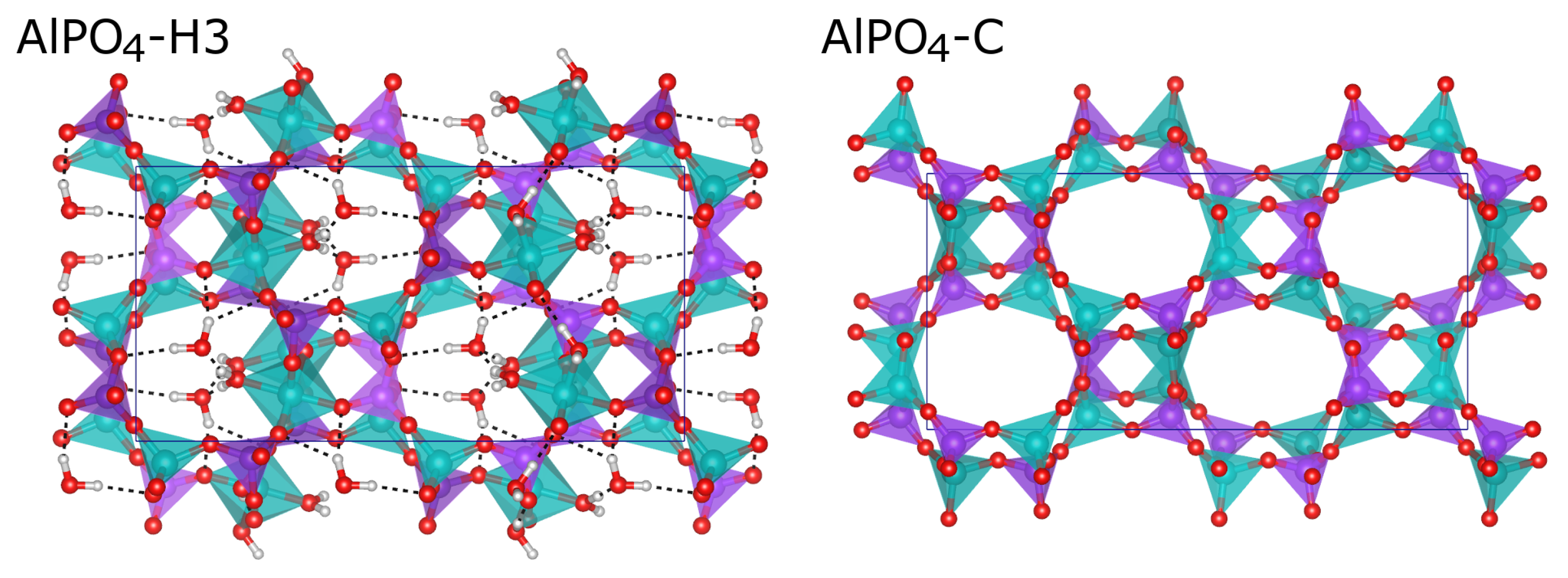
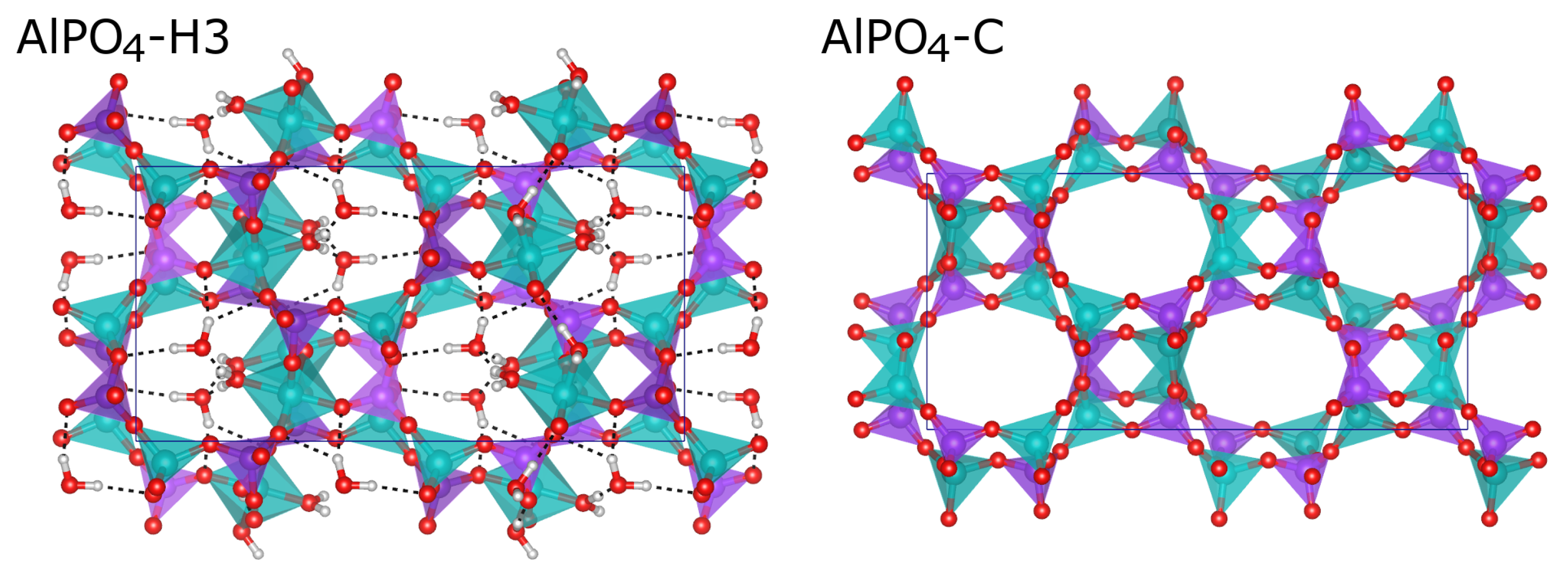
2.3. Comparison of Fully and Partially Hydrated APC Phases
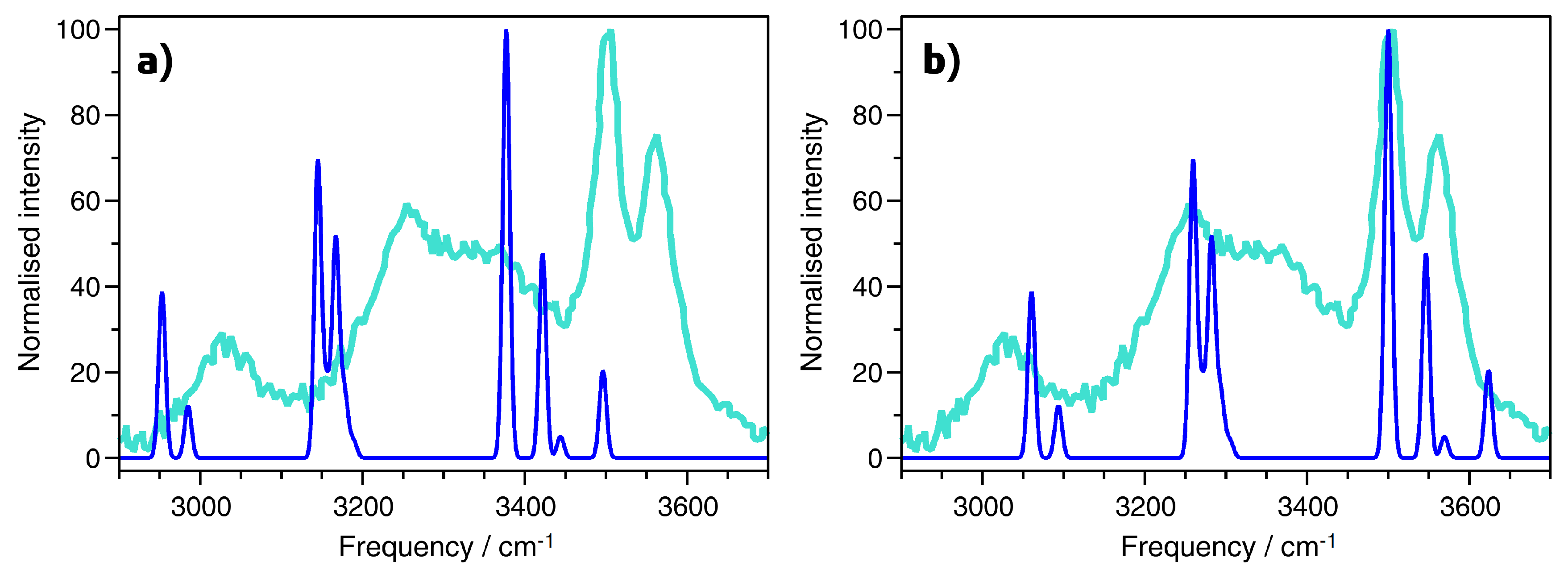
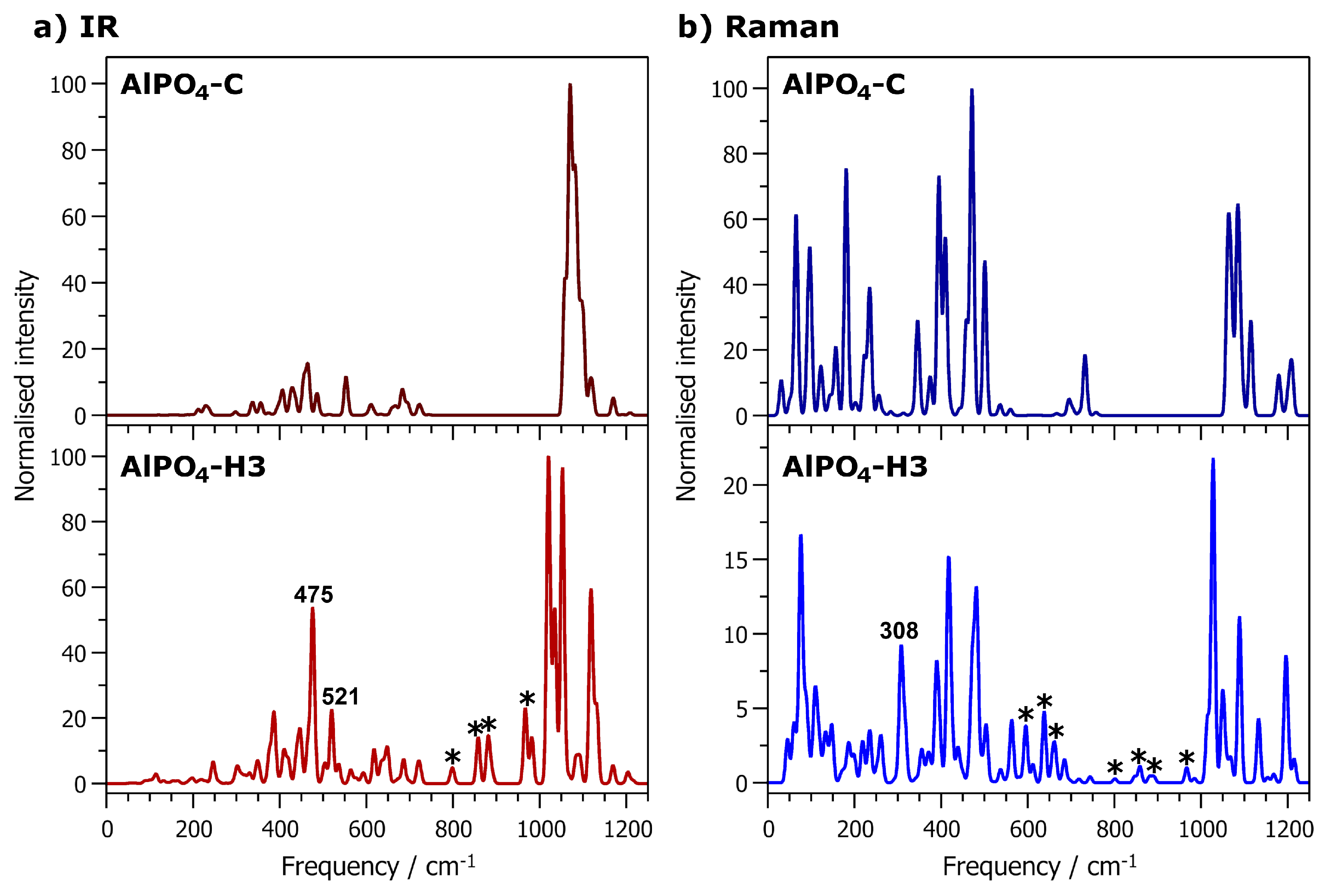
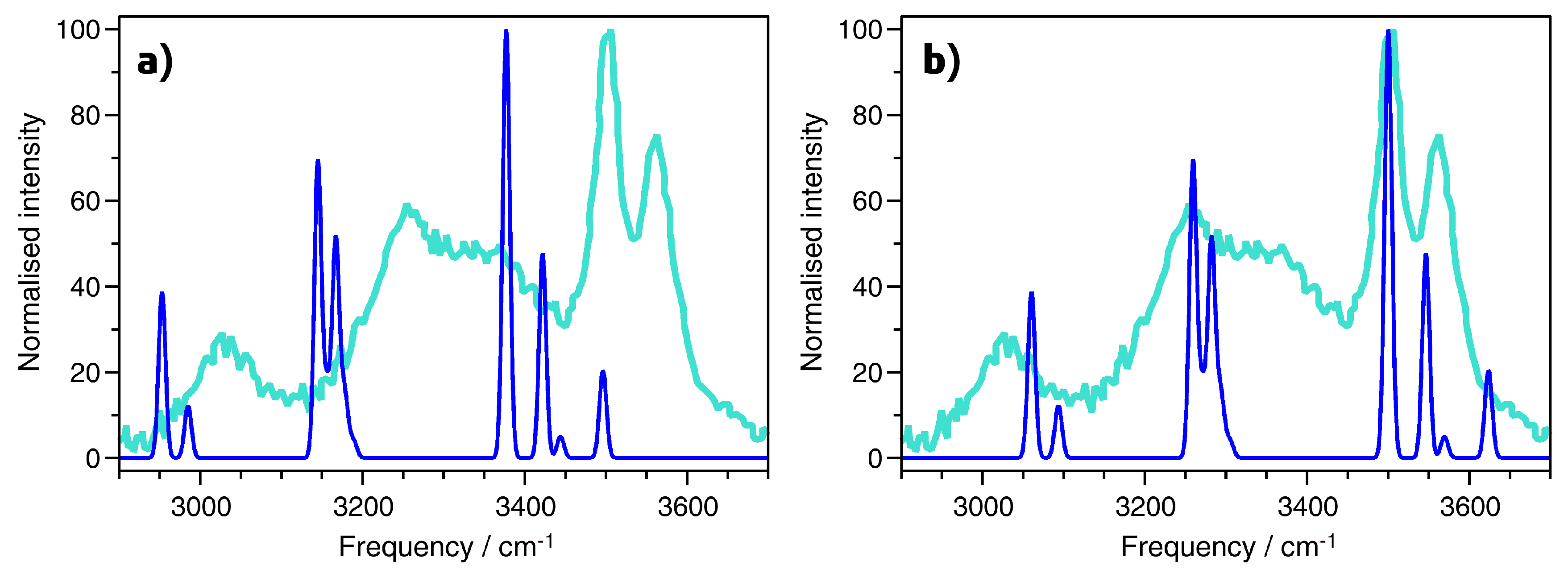
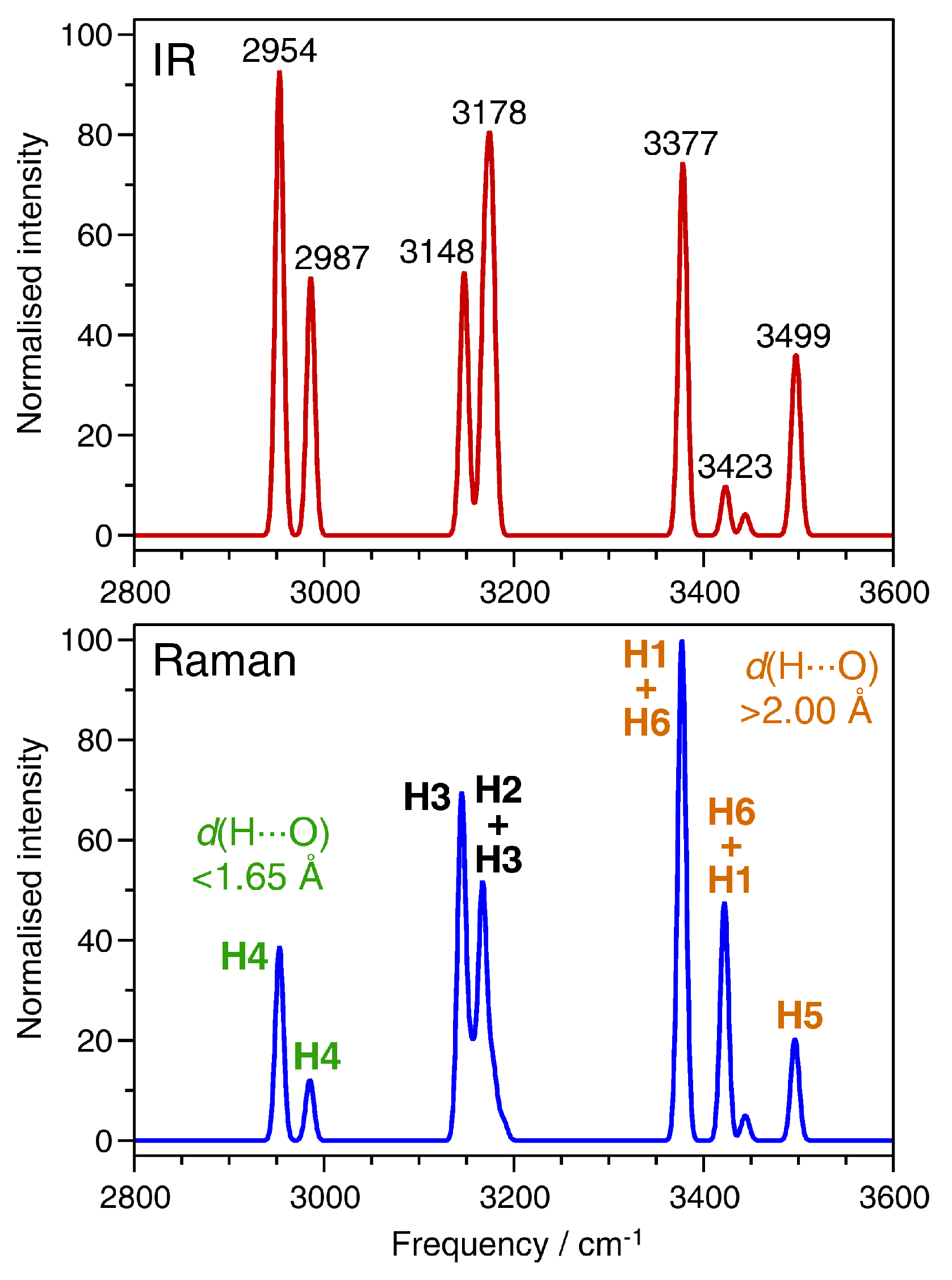
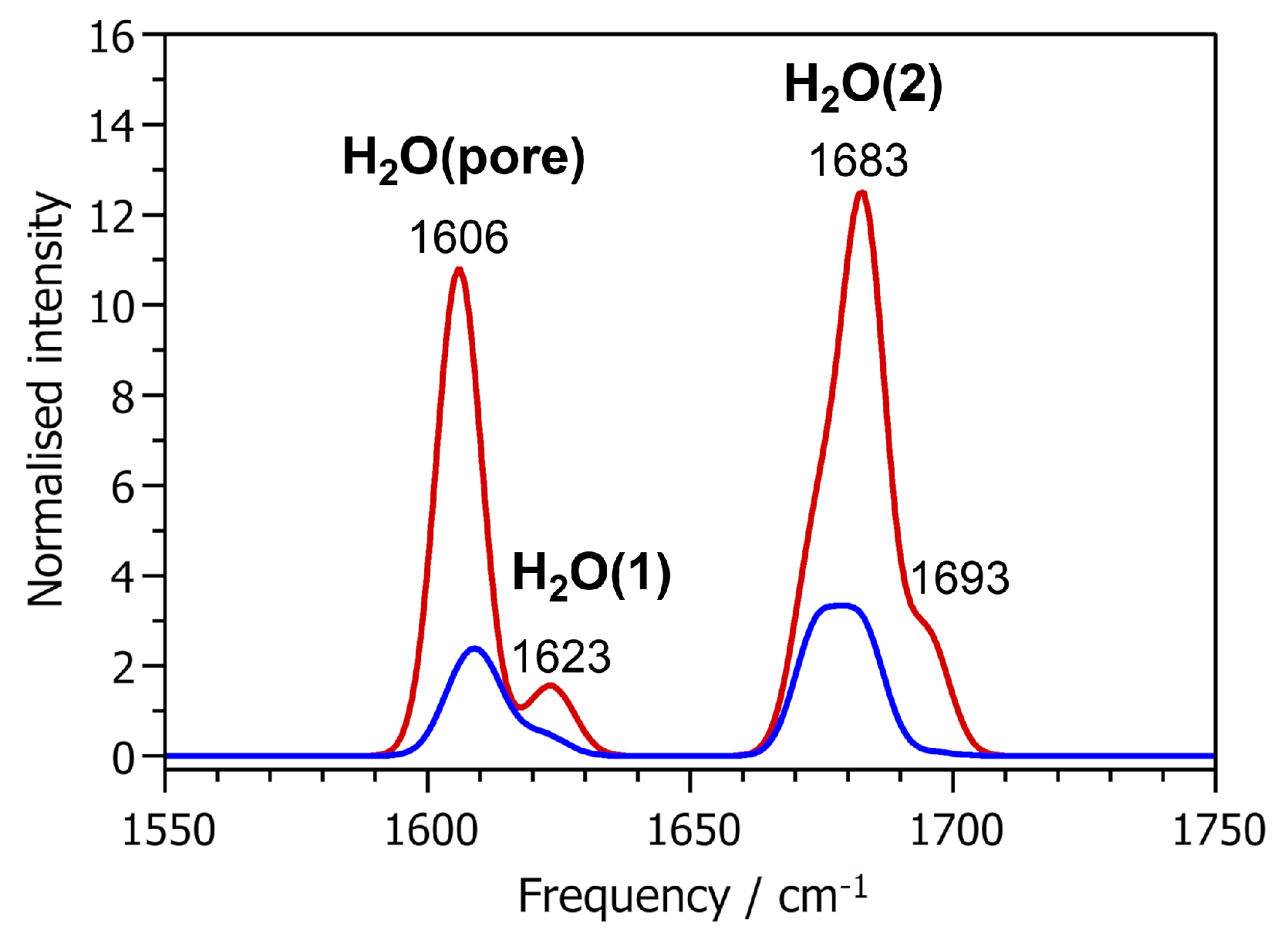
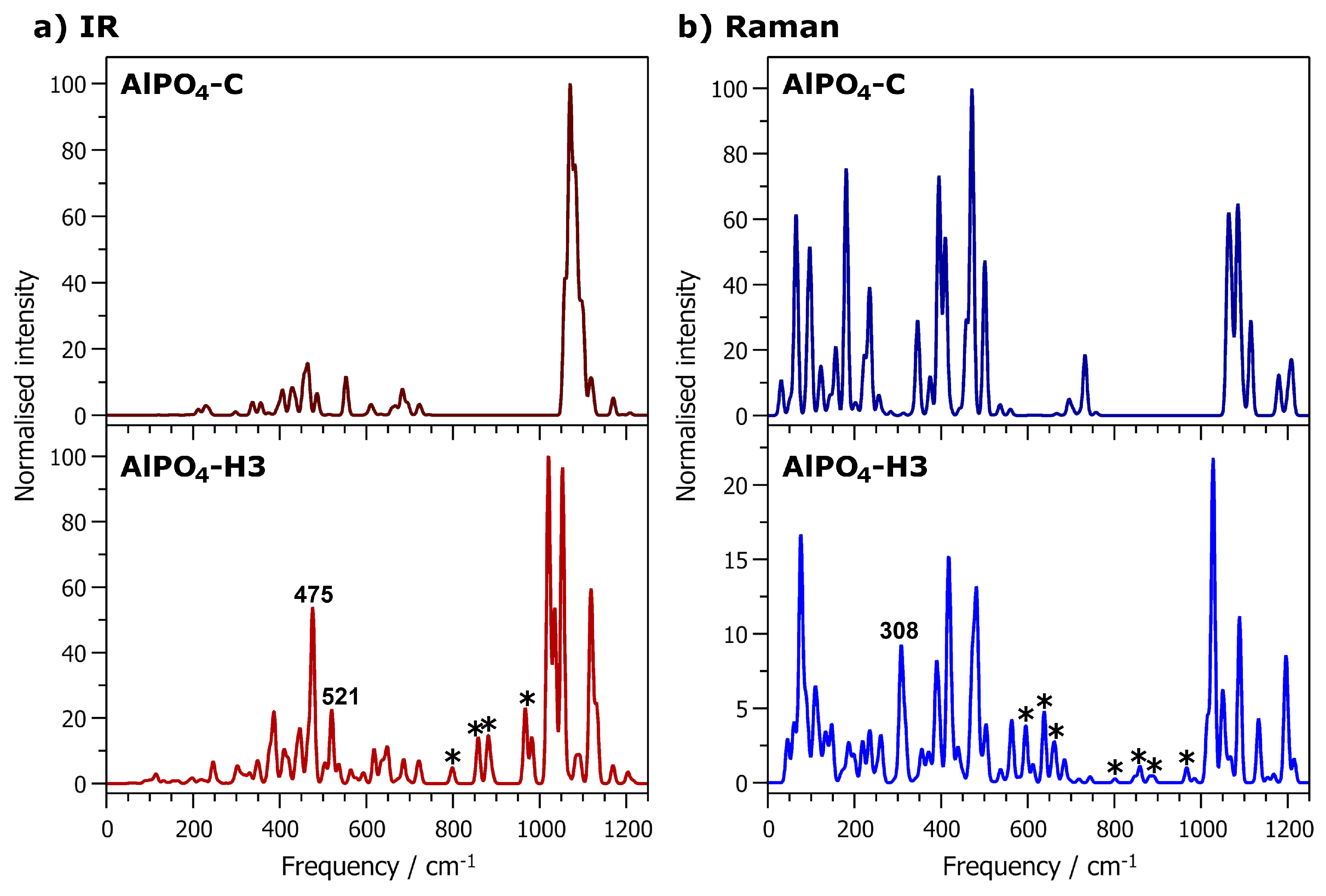
2.4. Vibrational Spectra
2.5. Displacement Parameters and Dynamic Behaviour
3. Discussion
4. Materials and Methods
4.1. Computational Details
4.2. Structure Models
5. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Ng, E.P.; Mintova, S. Nanoporous materials with enhanced hydrophilicity and high water sorption capacity. Microporous Mesoporous Mater. 2008, 114, 1–26. [Google Scholar] [CrossRef]
- Ristić, A.; Zabukovec Logar, N.; Henninger, S.K.; Kaučič, V. The Performance of Small-Pore Microporous Aluminophosphates in Low-Temperature Solar Energy Storage: The Structure-Property Relationship. Adv. Funct. Mater. 2012, 22, 1952–1957. [Google Scholar] [CrossRef]
- Henninger, S.K.; Ernst, S.J.; Gordeeva, L.; Bendix, P.; Fröhlich, D.; Grekova, A.D.; Bonaccorsi, L.; Aristov, Y.; Jaenchen, J. New materials for adsorption heat transformation and storage. Renew. Energy 2017, 110, 59–68. [Google Scholar] [CrossRef]
- Blackwell, C.S.; Patton, R.L. Aluminum-27 and Phosphorus-31 Nuclear Magnetic Resonance Studies of Aluminophosphate Molecular Sieves. J. Phys. Chem. 1984, 88, 6135–6139. [Google Scholar] [CrossRef]
- Meinhold, R.H.; Tapp, N.J. An NMR study of the reaction of water with AlPO4-5. J. Chem. Soc. Chem. Commun. 1990, 3, 219–220. [Google Scholar] [CrossRef]
- Peeters, M.P.J.; van de Ven, L.J.M.; de Haan, J.W.; van Hooff, J.H.C. A combined NMR and XRD study of AFI and AEL type molecular sieves. J. Phys. Chem. 1993, 97, 8254–8260. [Google Scholar] [CrossRef]
- Knops-Gerrits, P.P.; Toufar, H.; Li, X.Y.; Grobet, P.; Schoonheydt, R.A.; Jacobs, P.A.; Goddard, W.A. The Structure of Water in Crystalline Aluminophosphates: Isolated Water and Intermolecular Clusters Probed by Raman Spectroscopy, NMR and Structural Modeling. J. Phys. Chem. A 2000, 104, 2410–2422. [Google Scholar] [CrossRef]
- Poulet, G.; Tuel, A.; Sautet, P. A combined experimental and theoretical evaluation of the structure of hydrated microporous aluminophosphate AlPO4-18. J. Phys. Chem. B 2005, 109, 22939–22946. [Google Scholar] [CrossRef] [PubMed]
- Varlec, J.; Krajnc, A.; Mazaj, M.; Ristić, A.; Vanatalu, K.; Oss, A.; Samoson, A.; Kaučič, V.; Mali, G. Dehydration of AlPO4-34 studied by variable-temperature NMR, XRD and first-principles calculations. New J. Chem. 2016, 40, 4178–4186. [Google Scholar] [CrossRef]
- Pluth, J.J.; Smith, J.V. Hydrated aluminophosphate (AlPO4 · 1.5H2O) with PO4, AlO4 and AlO4(H2O)2 groups and encapsulated water. Acta Cryst. Sect. C Cryst. Struct. Commun. 1986, 42, 1118–1120. [Google Scholar] [CrossRef]
- McCusker, L.; Baerlocher, C.; Jahn, E.; Bülow, M. The triple helix inside the large-pore aluminophosphate molecular sieve VPI-5. Zeolites 1991, 11, 308–313. [Google Scholar] [CrossRef]
- Poulet, G.; Sautet, P.; Tuel, A. Structure of Hydrated Microporous Aluminophosphates: Static and Molecular Dynamics Approaches of AlPO4-34 from First Principles Calculations. J. Phys. Chem. B 2002, 106, 8599–8608. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Krajnc, A.; Varlec, J.; Mazaj, M.; Ristić, A.; Logar, N.Z.; Mali, G. Superior Performance of Microporous Aluminophosphate with LTA Topology in Solar-Energy Storage and Heat Reallocation. Adv. Energy Mater. 2017, 7, 1601815. [Google Scholar] [CrossRef]
- Kniep, R.; Mootz, D. Metavariscite—A redetermination of its crystal structure. Acta Cryst. Sect. B Struct. Crystallogr. Cryst. Chem. 1973, 29, 2292–2294. [Google Scholar] [CrossRef]
- Kniep, R.; Mootz, D.; Vegas, A. Variscite. Acta Cryst. Sect. B Struct. Crystallogr. Cryst. Chem. 1977, 33, 263–265. [Google Scholar] [CrossRef]
- D’Yvoire, F. Étude des phosphates d’aluminium et de fer trivalent. I. L’orthophosphate neutre d’aluminium. Bull. Soc. Chim. Fr. 1961, 1762–1776. [Google Scholar]
- Baerlocher, C.; McCusker, L. Database of Zeolite Structures. 2018. Available online: http://www.iza-structure.org/databases/ (accessed on 28 October 2018).
- Anurova, N.A.; Blatov, V.A.; Ilyushin, G.D.; Proserpio, D.M. Natural Tilings for Zeolite-Type Frameworks. J. Phys. Chem. C 2010, 114, 10160–10170. [Google Scholar] [CrossRef]
- Lagno, F.; Demopoulos, G.P. Synthesis of Hydrated Aluminum Phosphate AlPO4 · 1.5H2O (AlPO4-H3) by Controlled Reactive Crystallization in Sulfate Media. Ind. Eng. Chem. Res. 2005, 44, 8033–8038. [Google Scholar] [CrossRef]
- Keller, E.; Meier, W.; Kirchner, R.M. Synthesis, structures of AlPO4-C and AlPO4-D, and their topotactic transformation. Solid State Ionics 1990, 43, 93–102. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Evers, F.O.; Formalik, F.; Olejniczak, A. Benchmarking DFT-GGA calculations for the structure optimisation of neutral-framework zeotypes. Theor. Chem. Acc. 2016, 135, 257. [Google Scholar] [CrossRef]
- Fischer, M.; Angel, R.J. Accurate structures and energetics of neutral-framework zeotypes from dispersion-corrected DFT calculations. J. Chem. Phys. 2017, 146, 174111. [Google Scholar] [CrossRef] [PubMed]
- Downs, R.T.; Gibbs, G.V.; Bartelmehs, K.L.; Boisen, M.B. Variations of bond lengths and volumes of silicate tetrahedra with temperature. Am. Miner. 1992, 77, 751–757. [Google Scholar]
- Alecu, I.M.; Zheng, J.; Zhao, Y.; Truhlar, D.G. Computational thermochemistry: Scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries. J. Chem. Theor. Comput. 2010, 6, 2872–2887. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, M.K.; Brauer, B.; Martin, J.M.L. Frequency and Zero-Point Vibrational Energy Scale Factors for Double-Hybrid Density Functionals (and Other Selected Methods): Can Anharmonic Force Fields Be Avoided? J. Phys. Chem. A 2015, 119, 1701–1714. [Google Scholar] [CrossRef] [PubMed]
- Csonka, G.I.; Ruzsinszky, A.; Perdew, J.P.; Grimme, S. Improved description of stereoelectronic effects in hydrocarbons using semilocal density functional theory. J. Chem. Theory Comput. 2008, 4, 888–891. [Google Scholar] [CrossRef]
- Ireta, J.; Neugebauer, J.; Scheffler, M. On the Accuracy of DFT for Describing Hydrogen Bonds: Dependence on the Bond Directionality. J. Phys. Chem. A 2004, 108, 5692–5698. [Google Scholar] [CrossRef]
- Fischer, M. Structure and bonding of water molecules in zeolite hosts: Benchmarking plane-wave DFT against crystal structure data. Z. Kristallogr. Cryst. Mater. 2015, 230, 325–336. [Google Scholar] [CrossRef]
- Chen, M.; Ko, H.Y.; Remsing, R.C.; Calegari Andrade, M.F.; Santra, B.; Sun, Z.; Selloni, A.; Car, R.; Klein, M.L.; Perdew, J.P.; et al. Ab initio theory and modeling of water. Proc. Natl. Acad. Sci. USA 2017, 114, 10846–10851. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Interaction of water with (silico)aluminophosphate zeotypes: A comparative investigation using dispersion-corrected DFT. Phys. Chem. Chem. Phys. 2016, 18, 15738–15750. [Google Scholar] [CrossRef] [PubMed]
- Henninger, S.K.; Schmidt, F.P.; Henning, H.M. Characterisation and improvement of sorption materials with molecular modeling for the use in heat transformation applications. Adsorption 2011, 17, 833–843. [Google Scholar] [CrossRef]
- Román-Román, E.I.; Zicovich-Wilson, C.M. The role of long-range van der Waals forces in the relative stability of SiO2-zeolites. Chem. Phys. Lett. 2015, 619, 109–114. [Google Scholar] [CrossRef]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D, Version 14. 2014. Available online: http://www.jmol.org/ (accessed on 28 October 2018).
- Desiraju, G.R. A Bond by Any Other Name. Angew. Chem. Int. Ed. 2011, 50, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Frost, R.L.; Weier, M.L.; Erickson, K.L.; Carmody, O.; Mills, S.J. Raman spectroscopy of phosphates of the variscite mineral group. J. Raman Spectr. 2004, 35, 1047–1055. [Google Scholar] [CrossRef]
- Yang, H.; Walton, R.I.; Biedasek, S.; Antonijevic, S.; Wimperis, S.; Ramirez-Cuesta, A.J.; Li, J.; Kolesnikov, A.I. Experimental Observations of Water-Framework Interactions in a Hydrated Microporous Aluminum Phosphate. J. Phys. Chem. B 2005, 109, 4464–4469. [Google Scholar] [CrossRef]
- Tabacchi, G. Supramolecular Organization in Confined Nanospaces. ChemPhysChem 2018, 19, 1249–1297. [Google Scholar] [CrossRef] [PubMed]
- Fois, E.; Gamba, A.; Tilocca, A. Structure and Dynamics of the Flexible Triple Helix of Water inside VPI-5 Molecular Sieves. J. Phys. Chem. B 2002, 106, 4806–4812. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Bennett, J.W. Discovery and Design of Functional Materials: Integration of Database Searching and First Principles Calculations. Phys. Procedia 2012, 34, 14–23. [Google Scholar] [CrossRef]
- Fischer, M. Water adsorption in SAPO-34: Elucidating the role of local heterogeneities and defects using dispersion-corrected DFT calculations. Phys. Chem. Chem. Phys. 2015, 17, 25260–25271. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Computational evaluation of aluminophosphate zeotypes for CO2/N2 separation. Phys. Chem. Chem. Phys. 2017, 19, 22801–22812. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Template effects on the pressure-dependent behavior of chabazite-type fluoroaluminophosphates: A computational approach. Phys. Chem. Miner. 2019. [Google Scholar] [CrossRef]
- Porezag, D.; Pederson, M.R. Infrared intensities and Raman-scattering activities within density-functional theory. Phys. Rev. B 1996, 54, 7830–7836. [Google Scholar] [CrossRef]
- Gonze, X. First-principles responses of solids to atomic displacements and homogeneous electric fields: Implementation of a conjugate-gradient algorithm. Phys. Rev. B 1997, 55, 10337–10354. [Google Scholar] [CrossRef]
- Gonze, X.; Lee, C. Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory. Phys. Rev. B 1997, 55, 10355–10367. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; Dal Corso, A. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Refson, K.; Tulip, P.R.; Clark, S.J. Variational density-functional perturbation theory for dielectrics and lattice dynamics. Phys. Rev. B 2006, 73, 155114. [Google Scholar] [CrossRef]
- Hofstetter, A.; Emsley, L. Positional Variance in NMR Crystallography. J. Am. Chem. Soc. 2017, 139, 2573–2576. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. CP2K: Atomistic simulations of condensed matter systems. WIREs Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Krack, M. Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals. Theor. Chem. Acc. 2005, 114, 145–152. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Herrera-Perez, G.; Zicovich-Wilson, C.; Ramirez-Solis, A. Periodic DFT Studies of AlPO-11: The Role of Hydration on Structural Properties. J. Phys. Chem. C 2007, 111, 9664–9670. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AlPO4-H3 | AlPO4-C | |||
|---|---|---|---|---|
| sc-XRD [10] | DFT | PXRD [21] | DFT | |
| Unit cell parameters | ||||
| a/Å | 19.353 | 19.498 | 19.816 | 20.392 |
| b/Å | 9.727 | 9.751 | 10.047 | 9.648 |
| c/Å | 9.762 | 9.795 | 8.935 | 8.825 |
| V/Å3 | 1837.7 | 1862.3 | 1778.9 | 1736.3 |
| T-O bond lengths | ||||
| daver(P-Ofw)/Å | 1.524 (1.530) | 1.540 | 1.523 | 1.539 |
| daver(Al1-Ofw)/Å | 1.733 (1.739) | 1.758 | 1.730 | 1.757 |
| daver(Al2-Ofw)/Å | 1.847 (1.851) | 1.873 | - | - |
| d(Al2-O9)/Å | 1.967 (1.977) | 1.981 | - | - |
| d(Al2-O10)/Å | 1.951 (1.956) | 1.965 | - | - |
| sc-XRD | DFT | |
|---|---|---|
| H2O(1) | ||
| d(O9O11(A)) | 2.893 Å | 3.020 Å |
| d(H1O11(A)) | 2.070 Å | 2.055 Å |
| (O9-H1O11(A)) | 162.8 | 167.7 |
| d(O9O11(B)) | 3.011 Å | 2.791 Å |
| d(H2O11(B)) | 2.183 Å | 1.821 Å |
| (O9-H2O11(B)) | 164.5 | 165.1 |
| H2O(2) | ||
| d(O10O5) | 2.752 Å | 2.722 Å |
| d(H3O5) | 1.990 Å | 1.752 Å |
| (O10-H3O5) | 148.7 | 163.9 |
| d(O10O7) | 2.603 Å | 2.578 Å |
| d(H4O7) | 1.790 Å | 1.603 Å |
| (O10-H4O7) | 159.4 | 162.2 |
| H2O(pore) | ||
| d(O11O8) | 3.047 Å | 3.061 Å |
| d(H5O8) | - | 2.349 Å |
| (O11-H5O8) | - | 129.1 |
| d(O11O1) | 3.213 Å | 3.267 Å |
| d(H5O1) | - | 2.369 Å |
| (O11-H5O1) | - | 152.4 |
| d(O11O4) | 3.013 Å | 2.995 Å |
| d(H6O4) | - | 2.027 Å |
| (O11-H6O4) | - | 169.6 |
| H2O(1) | H2O(2) | H2O(pore) | /kJ mol−1 | /kJ mol−1 | |
|---|---|---|---|---|---|
| Fully hydrated | 8 | 8 | 8 | −81.8 | −67.3 |
| 2/3 hydrated | 8 | 8 | 0 | −76.7 | −62.5 |
| 8 | 0 | 8 | −70.6 | −56.0 | |
| 0 | 8 | 8 | −76.6 | −62.8 | |
| 1/3 hydrated | 8 | 0 | 0 | −44.8 | −30.6 |
| 0 | 8 | 0 | −71.5 | −57.7 | |
| 0 | 0 | 8 | −67.8 | −53.3 |
| d(O-H)/Å | d(HO)/Å | Mode Frequencies/cm−1 | |
|---|---|---|---|
| H2O molecule | 0.9668 | - | 3584 (symmetric stretching) |
| 3699 (asymmetric stretching) | |||
| AlPO4-H3 | |||
| H1 | 0.9799 | 2.055 | 3377 to 3379 (secondary: ∼3445) |
| H2 | 0.9921 | 1.821 | 3168 to 3189 |
| H3 | 0.9949 | 1.752 | 3145 to 3148 and 3178 to 3189 |
| H4 | 1.0047 | 1.603 | 2954 to 2987 |
| H5 | 0.9778 | 2.349/2.369 | 3496 to 3499 |
| H6 | 0.9790 | 2.027 | 3423 to 3445 (secondary: ∼3375) |
| (H-O-H)/ | Mode Frequencies/cm−1 | |
|---|---|---|
| H2O molecule | 104.21 | 1607 |
| AlPO4-H3 | ||
| H2O(1) | 112.39 | 1619 to 1625 |
| H2O(2) | 102.90 | 1674 to 1696 |
| H2O(pore) | 105.04 | 1606 to 1611 |
| RMSD(AIMD)/Å | (AIMD)/Å2 | (phonon)/Å2 | (sc-XRD)/Å2 | |
|---|---|---|---|---|
| Framework | ||||
| Al1 | 0.143 ± 0.015 | 0.0068 | 0.0079 | 0.0089 |
| Al2 | 0.135 ± 0.015 | 0.0061 | 0.0069 | 0.0079 |
| P1 | 0.142 ± 0.014 | 0.0067 | 0.0072 | 0.0084 |
| P2 | 0.133 ± 0.015 | 0.0059 | 0.0070 | 0.0078 |
| O1 | 0.211 ± 0.017 | 0.0148 | 0.0154 | 0.0146 |
| O2 | 0.227 ± 0.020 | 0.0172 | 0.0193 | 0.0174 |
| O3 | 0.228 ± 0.021 | 0.0173 | 0.0230 | 0.0163 |
| O4 | 0.198 ± 0.017 | 0.0131 | 0.0140 | 0.0134 |
| O5 | 0.183 ± 0.019 | 0.0112 | 0.0152 | 0.0114 |
| O6 | 0.222 ± 0.016 | 0.0164 | 0.0190 | 0.0160 |
| O7 | 0.203 ± 0.018 | 0.0137 | 0.0126 | 0.0133 |
| O8 | 0.178 ± 0.018 | 0.0106 | 0.0148 | 0.0112 |
| H2O(1) | ||||
| O9 | 0.244 ± 0.020 | 0.0198 | 0.0226 | 0.022 |
| H1 | 0.357 ± 0.030 | 0.0425 | 0.0397 | 0.20 |
| H2 | 0.506 ± 0.051 | 0.0853 | 0.0365 | 0.15 |
| H2O(2) | ||||
| O10 | 0.188 ± 0.016 | 0.0118 | 0.0121 | 0.0155 |
| H3 | 0.242 ± 0.022 | 0.0195 | 0.0278 | 0.066 |
| H4 | 0.231 ± 0.022 | 0.0178 | 0.0242 | 0.054 |
| H2O(pore) | ||||
| O11 | 0.552 ± 0.065 | 0.1016 | 0.0311 | 0.0568 |
| H5 | 0.590 ± 0.075 | 0.1160 | 0.0602 | - |
| H6 | 0.556 ± 0.063 | 0.1030 | 0.0465 | - |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, M. First-Principles Study of AlPO4-H3, a Hydrated Aluminophosphate Zeotype Containing Two Different Types of Adsorbed Water Molecules. Molecules 2019, 24, 922. https://doi.org/10.3390/molecules24050922
Fischer M. First-Principles Study of AlPO4-H3, a Hydrated Aluminophosphate Zeotype Containing Two Different Types of Adsorbed Water Molecules. Molecules. 2019; 24(5):922. https://doi.org/10.3390/molecules24050922
Chicago/Turabian StyleFischer, Michael. 2019. "First-Principles Study of AlPO4-H3, a Hydrated Aluminophosphate Zeotype Containing Two Different Types of Adsorbed Water Molecules" Molecules 24, no. 5: 922. https://doi.org/10.3390/molecules24050922
APA StyleFischer, M. (2019). First-Principles Study of AlPO4-H3, a Hydrated Aluminophosphate Zeotype Containing Two Different Types of Adsorbed Water Molecules. Molecules, 24(5), 922. https://doi.org/10.3390/molecules24050922