Multimerization Increases Tumor Enrichment of Peptide–Photosensitizer Conjugates


Abstract
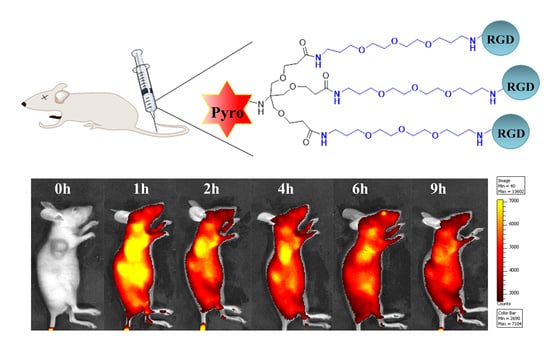
1. Introduction
2. Results and Discussion
2.1. Molecular Design
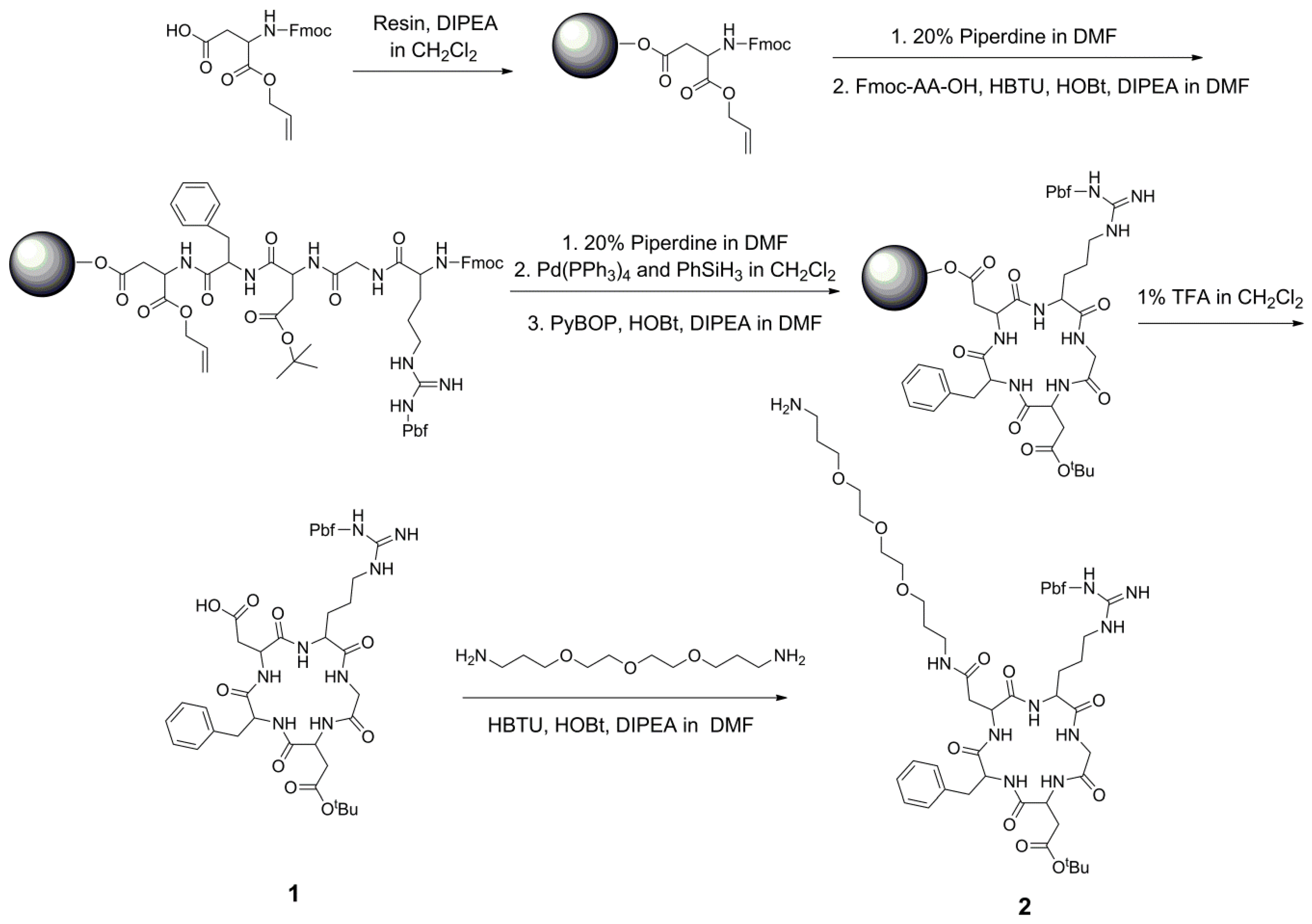
2.2. Solid-Phase Synthesis of Side Chain-Protected Cyclic RGD Pentapeptide 1 and 2
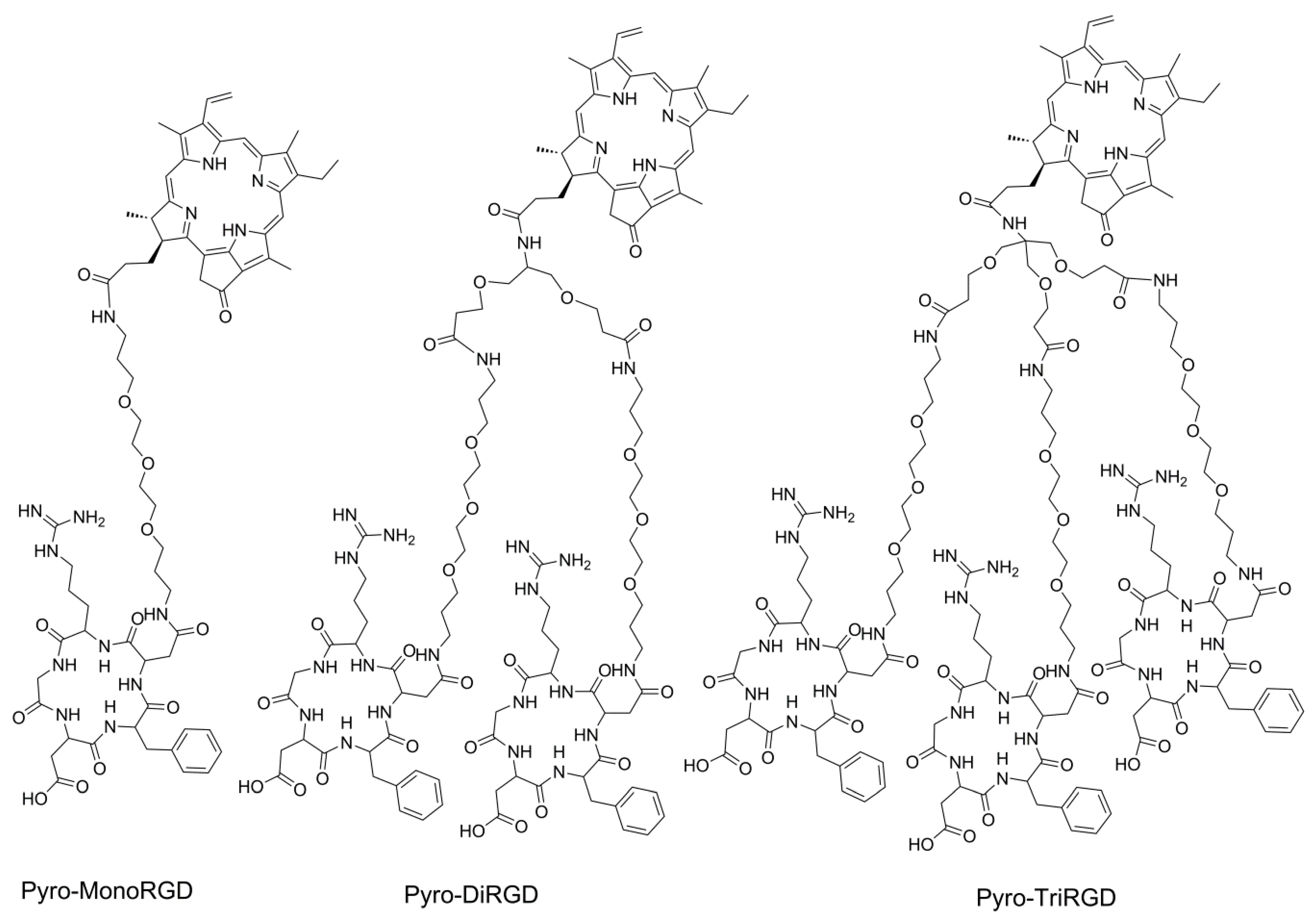
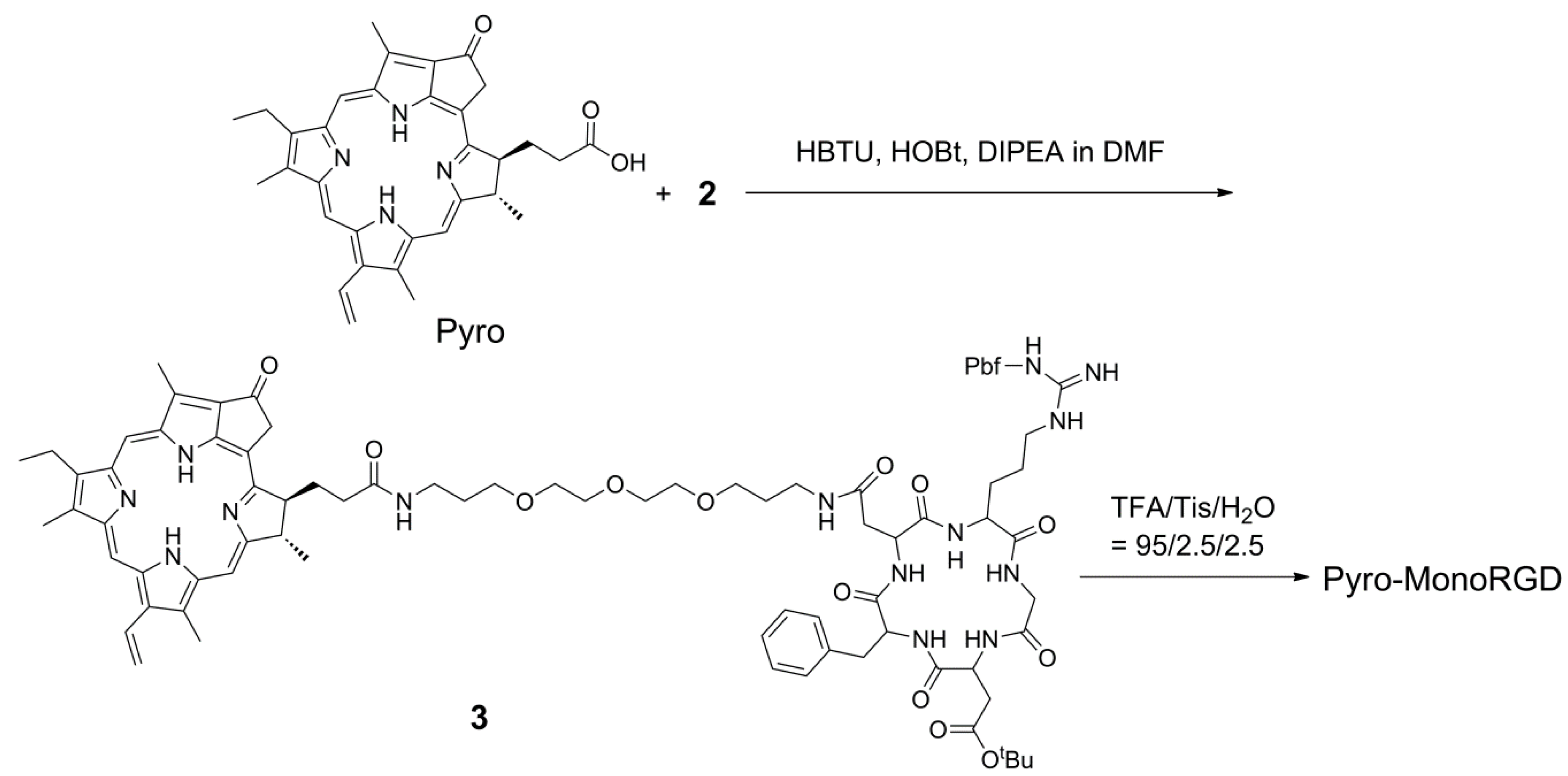
2.3. Synthesis of Mono-RGD Conjugate Pyro-MonoRGD
2.4. Synthesis of the Dimeric-RGD Conjugate, Pyro-DiRGD
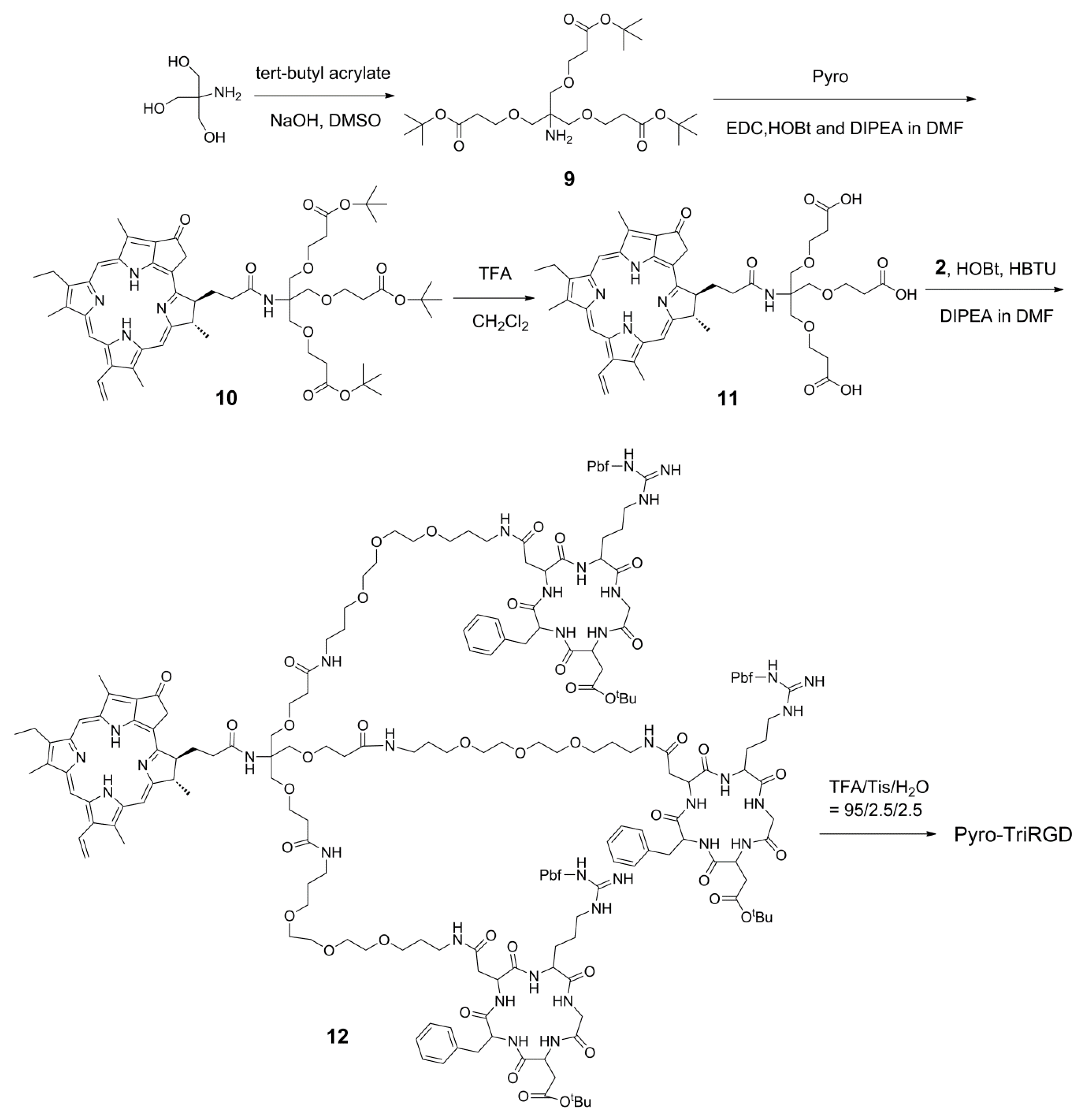
2.5. Synthesis of the Trimeric-RGD Conjugate, Pyro-TriRGD
2.6. Photophysical and Photochemical Properties
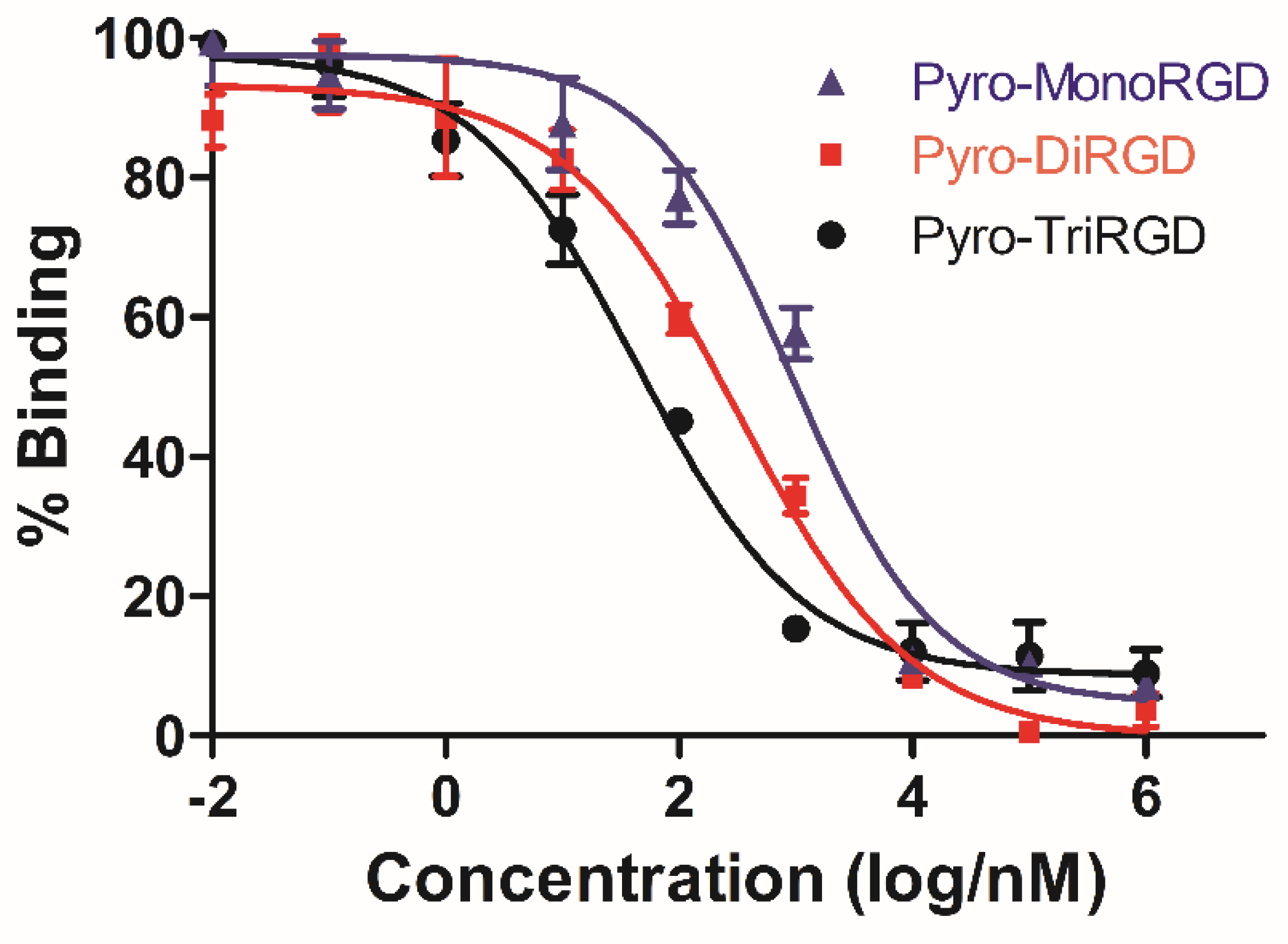
2.7. Receptor Binding Assay
2.8. In Vivo Distribution Analysis
3. Experimental
3.1. Instruments and Materials
3.2. Solid-Phase Synthesis of Side Chain-Protected Cyclic RGD Pentapeptide Cyclo(-Arg[Pbf]-Gly-Asp[tBu]-D-Phe-Asp-)
3.3. Synthesis of Amino-Modified Cyclic Pentapeptide Cyclo(-Arg[Pbf]-Gly-Asp[tBu]-D-Phe-Asp[PEG-amine]-)
3.4. Synthesis of Monomeric RGD Conjugate Pyro-MonoRGD
3.5. Synthesis of Tert-butyl (1,3-dihydroxypropan-2-yl)carbamate
3.6. Synthesis of Di-tert-butyl-3,3′-((2-((tert-butoxycarbonyl)amino)propane-1,3-diyl)bis(oxy))dipropionate
3.7. Synthesis of Dimeric Linker 3,3′-((2-Aminopropane-1,3-diyl)bis(oxy))dipropionic acid hydrochloride
3.8. Synthesis of Pyro-Conjugated Dimeric Linker 7
3.9. Synthesis of Conjugate Pyro-DiRGD
3.10. Synthesis of Tris((2-(tert-butoxycarbonyl)ethoxyl)methyl)methylamine
3.11. Synthesis of Pyro-Conjugated Compound
3.12. Synthesis of Deprotected Pyro-Conjugated Trimeric Linker
3.13. Synthesis of the Trimeric Conjugate Pyro-TriRGD
3.14. Determination of the Photophysical and Photochemical Properties
3.15. Receptor Binding Assay
3.16. In Vivo Tumor Enrichment Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Allison, P.R. Photodynamic therapy: Oncologic horizons. Future Oncol. 2014, 10, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Celli, J.P.; Spring, B.Q.; Rizvi, I.; Evans, C.L.; Samkoe, K.S.; Verma, S.; Pogue, B.W.; Hasan, T. Imaging and photodynamic therapy: Mechanisms, monitoring, and optimization. Chem. Rev. 2010, 110, 2795–2838. [Google Scholar] [CrossRef] [PubMed]
- Juarranz, A.; Jaen, P.; Sanz-Rodriguez, F.; Cuevas, J.; Gonzalez, S. Photodynamic therapy of cancer: Basic principles and applications. Clin. Transl. Oncol. 2008, 10, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Josefsen, L.B.; Boyle, R.W. Unique diagnostic and therapeutic roles of porphyrins and phthalocyanines in photodynamic therapy, imaging and theranostics. Theranostics 2012, 2, 916–966. [Google Scholar] [CrossRef] [PubMed]
- van Dam, G.M.; Themelis, G.; Crane, L.M.; Harlaar, N.J.; Pleijhuis, R.G.; Kelder, W.; Sarantopoulos, A.; de Jong, J.S.; Arts, H.J.; van der Zee, A.G.; et al. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-alpha targeting: First in-human results. Nat. Med. 2011, 17, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 2006, 6, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Mroz, P.; Hamblin, M.R. Photodynamic therapy stimulates anti-tumor immunity in a murine mastocytoma model. Proc. Spie 2008, 6857, 685706. [Google Scholar]
- Bugaj, A.M. Targeted photodynamic therapy-a promising strategy of tumor treatment. Photochem. Photobiol. Sci. 2011, 10, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Yoon, I.; Li, J.Z.; Shim, Y.K. Advance in photosensitizers and light delivery for photodynamic therapy. Clin. Endosc. 2013, 46, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, P.; Nomula, R.; Zhao, J.Z. Activatable triplet photosensitizers: Magic bullets for targeted photodynamic therapy. J. Mater. Chem. C 2014, 2, 5982–5997. [Google Scholar] [CrossRef]
- Srinivasarao, M.; Low, P.S. Ligand-targeted drug delivery. Chem. Rev. 2017, 117, 12133–12164. [Google Scholar] [CrossRef] [PubMed]
- Sharman, W.M.; van Lier, J.E.; Allen, C.M. Targeted photodynamic therapy via receptor mediated delivery systems. Adv. Drug Deliv. Rev. 2004, 56, 53–76. [Google Scholar] [CrossRef] [PubMed]
- Mitsunaga, M.; Ogawa, M.; Kosaka, N.; Rosenblum, L.T.; Choyke, P.L.; Kobayashi, H. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat. Med. 2011, 17, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Srinivasarao, M.; Galliford, C.V.; Low, P.S. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat. Rev. Drug Discov. 2015, 14, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Casi, G.; Neri, D. Antibody−drug conjugates and small molecule−drug conjugates: Opportunities and challenges for the development of selective anticancer cytotoxic agents. J. Med. Chem. 2015, 58, 8751–8761. [Google Scholar] [CrossRef] [PubMed]
- Ranyuk, E.; Cauchon, N.; Klarskov, K.; Guerin, B.; van Lier, J.E. Phthalocyanine-peptide conjugates: Receptor-targeting bifunctional agents for imaging and photodynamic therapy. J. Med. Chem. 2013, 56, 1520–1534. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, Q.; Liang, Z.; Wang, J.; Pang, M.; Huang, W.; Wu, W.; Hong, Z. Synthesis and biological evaluation of peptideconjugated phthalocyanine photosensitizers with highly hydrophilic modifications. Org. Biomol. Chem. 2016, 14, 3409–3422. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, J.; Zhang, X.; Guo, W.; Li, F.; Yu, M.; Kong, X.; Wu, W.; Hong, Z. Highly water-soluble and tumor-targeted photosensitizers for photodynamic therapy. Org. Biomol. Chem. 2015, 13, 7681–7694. [Google Scholar] [CrossRef] [PubMed]
- Ongarora, B.G.; Fontenot, K.R.; Hu, X.; Sehgal, I.; Satyanarayana-Jois, S.D.; Vicente, M.G. Phthalocyanine-peptide conjugates for epidermal growth factor receptor targeting. J. Med. Chem. 2012, 55, 3725–3738. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tan, S.; Xing, Y.; Liu, Q.; Li, S.; Chen, Q.; Yu, M.; Wang, F.; Hong, Z. cRGD peptide conjugated pyropheophorbide-a photosensitizers for tumor targeting photodynamic therapy. Mol. Pharm. 2018, 15, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Schottelius, M.; Laufer, B.; Kessler, H.; Wester, H.J. Ligands for mapping αvβ3-integrin expression in vivo. Acc. Chem. Res. 2009, 42, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.R.M.; Pina, A.; Dean, A.; Lerchen, H.; Caruso, M.; Gasparri, F.; Fraietta, I.; Troiani, S.; Arosio, D.; Belvisi, L.; et al. Neutrophil elastase promotes linker cleavage and paclitaxel release from an integrin-targeted conjugate. Chem. Eur. J. 2019, 25, 1696–1700. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Nomoto, T.; Takemoto, H.; Matsui, M.; Tomoda, K.; Nishiyama, N. Effect of multiple cyclic RGD peptides on tumor accumulation and intratumoral distribution of IRDye 700DX-conjugated polymers. Sci. Rep. 2018, 8, 8126. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Svirskis, D.; Sarojini, V.; McGregor, A.L.; Bevitt, J.; Wu, Z. Cyclic-RGDyC functionalized liposomes for dual-targeting of tumor vasculature and cancer cells in glioblastoma: An in vitro boron neutron capture therapy study. Oncotarget 2017, 8, 36614–36627. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, Y.; Shen, Y.; Wang, A.; Wang, S.; Xie, T. The functions and applications of RGD in tumor therapy and tissue engineering. Int. J. Mol. Sci. 2013, 14, 13447–13462. [Google Scholar] [CrossRef] [PubMed]
- Luan, L.; Fang, W.; Liu, W.; Tian, M.; Ni, Y.; Chen, X.; Yua, X. Phthalocyanine-cRGD conjugate: Synthesis, photophysical properties and in vitro biological activity for targeting photodynamic therapy. Org. Biomol. Chem. 2016, 14, 2985–2992. [Google Scholar] [CrossRef] [PubMed]
- Ke, M.R.; Ng, D.K.P.; Lo, P.C. Synthesis and in vitro photodynamic activities of an integrin-targeting cRGD-conjugated zinc(II) phthalocyanine. Chem-Asian J. 2014, 9, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, A.; Ethirajan, M.; Pandey, S.K.; Dubey, S.; Zheng, X.; Liu, T.H.; Shibata, M.; Missert, J.; Morgan, J.; Pandey, R.K. Conjugation of cRGD peptide to chlorophyll a based photosensitizer (HPPH) alters its pharmacokinetics with enhanced tumor-imaging and photosensitizing (PDT) efficacy. Mol. Pharm. 2011, 8, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Mahon, E.; Barboiu, M. Synthetic multivalency for biological applications. Org. Biomol. Chem. 2015, 13, 10590–10599. [Google Scholar] [CrossRef] [PubMed]
- Mammen, M.; Choi, S.-K.; Whitesides, G.M. Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2754. [Google Scholar] [CrossRef]
- Chen, X.; Plasencia, C.; Hou, Y.; Neamati, N. Synthesis and biological evaluation of dimeric RGD peptide-paclitaxel conjugate as a model for integrin-targeted drug delivery. J. Med. Chem. 2005, 48, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Ryppa, C.; Mann-Steinberg, H.; Biniossek, M.L.; Satchi-Fainarob, R.; Kratz, F. In vitro and in vivo evaluation of a paclitaxel conjugate with the divalent peptide E-[c(RGDfK)2] that targets integrin αvβ3. Int. J. Pharm. 2009, 368, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Jasinski, S. Semi-natural and Synthetic Chiral Cycloketo-Porphyrin Systems-Approaching Novel Photosensitizers. Available online: https://opus4.kobv.de/opus4-fau/files/864/StefanJasinskiDissertation.pdf (accessed on 20 January 2019).
- Thumshirn, G.; Hersel, U.; Goodman, S.L.; Kessler, H. Multimeric cyclic RGD peptides as potential tools for tumor targeting: Solid-phase peptide synthesis and chemoselective oxime ligation. Chem. Eur. J. 2003, 9, 2717–2725. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, J.; Kim, Y.; Zhai, S.; Jia, B.; Zhao, H.; Liu, Z.; Wang, F.; Chen, X.; Liu, S. Improving tumor-targeting capability and pharmacokinetics of 99mTc-labeled cyclic RGD dimers with PEG4 linkers. Mol. Pharm. 2009, 6, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Cong, X.; Hu, F.; Liu, K.-G.; Liao, Q.-J.; Yao, Z.-J. Chemoselective deprotection of cyclic N,O-aminals using catalytic bismuth(III) bromide in acetonitrile. J. Org. Chem. 2005, 70, 4514–4516. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, D.; Rodrigues, L.L.; Straβburger, D.; Mezger, M.; Besenius, P. Tuneable transient thermogels mediated by a pH- and redox-regulated supramolecular polymerization. Angew. Chem. Int. Ed. 2017, 56, 15461–15465. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds Pyro, Pyro-MonoRGD, Pyro-DiRGD, and Pyro-TriRGD are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λmax (nm) | λem (nm) | Stokes Shift (nm) | ΦΔ |
|---|---|---|---|---|
| Pyro | 668 | 673 | 5 | 0.52 |
| Pyro-MonoRGD | 668 | 672 | 4 | 0.49 |
| Pyro-DiRGD | 668 | 673 | 5 | 0.47 |
| Pyro-TriRGD | 668 | 673 | 5 | 0.48 |
| Compounds | Pyro-TriRGD | Pyro-DiRGD | Pyro-MonoRGD |
|---|---|---|---|
| IC50 (nM) | 43.8 ± 8.2 | 313.2 ± 23.1 | 950.7 ± 39.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Li, S.; Jin, Y.; Wang, J.Y.; Li, W.; Wu, W.; Hong, Z. Multimerization Increases Tumor Enrichment of Peptide–Photosensitizer Conjugates. Molecules 2019, 24, 817. https://doi.org/10.3390/molecules24040817
Zhao J, Li S, Jin Y, Wang JY, Li W, Wu W, Hong Z. Multimerization Increases Tumor Enrichment of Peptide–Photosensitizer Conjugates. Molecules. 2019; 24(4):817. https://doi.org/10.3390/molecules24040817
Chicago/Turabian StyleZhao, Jisi, Shuang Li, Yingying Jin, Jessica Yijia Wang, Wenjing Li, Wenjie Wu, and Zhangyong Hong. 2019. "Multimerization Increases Tumor Enrichment of Peptide–Photosensitizer Conjugates" Molecules 24, no. 4: 817. https://doi.org/10.3390/molecules24040817
APA StyleZhao, J., Li, S., Jin, Y., Wang, J. Y., Li, W., Wu, W., & Hong, Z. (2019). Multimerization Increases Tumor Enrichment of Peptide–Photosensitizer Conjugates. Molecules, 24(4), 817. https://doi.org/10.3390/molecules24040817