Electronic Peculiarities of a Self-Assembled M12L24 Nanoball (M = Pd+2, Cr, or Mo)

 ,
,
Abstract
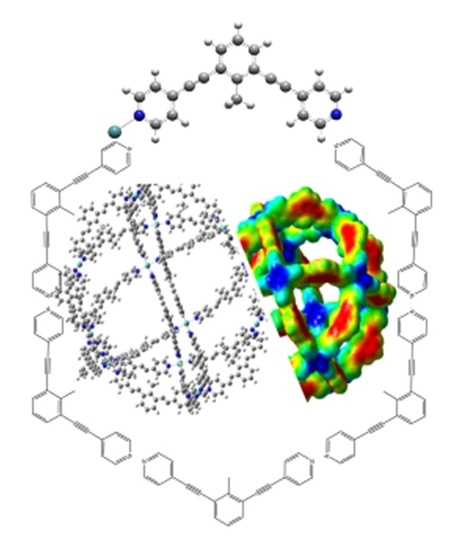
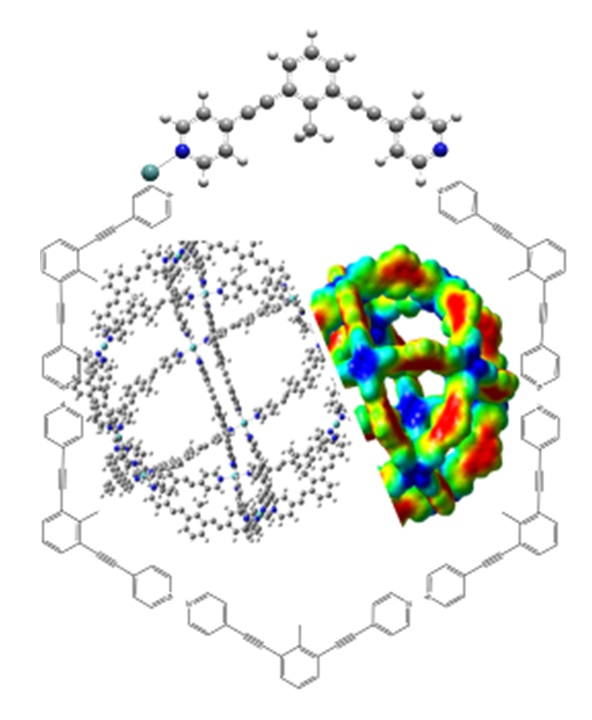
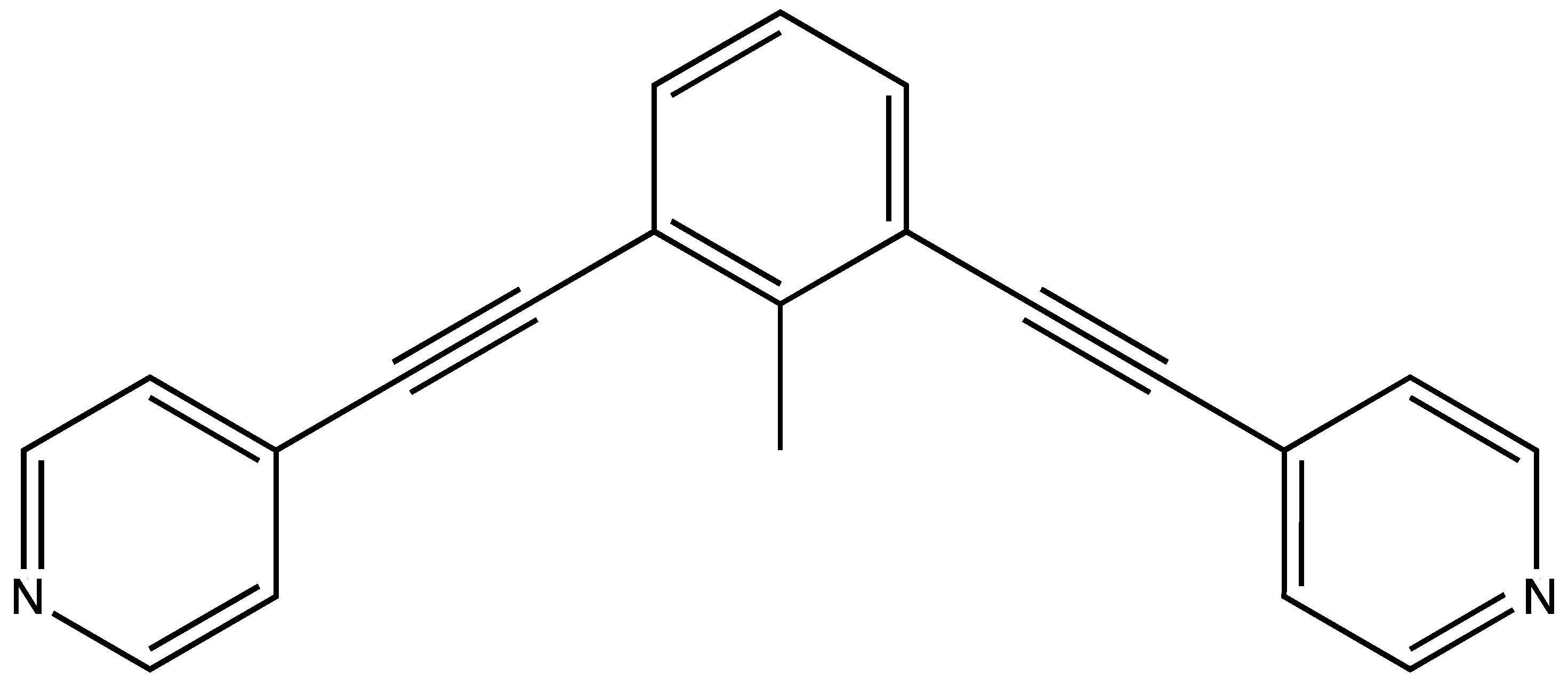
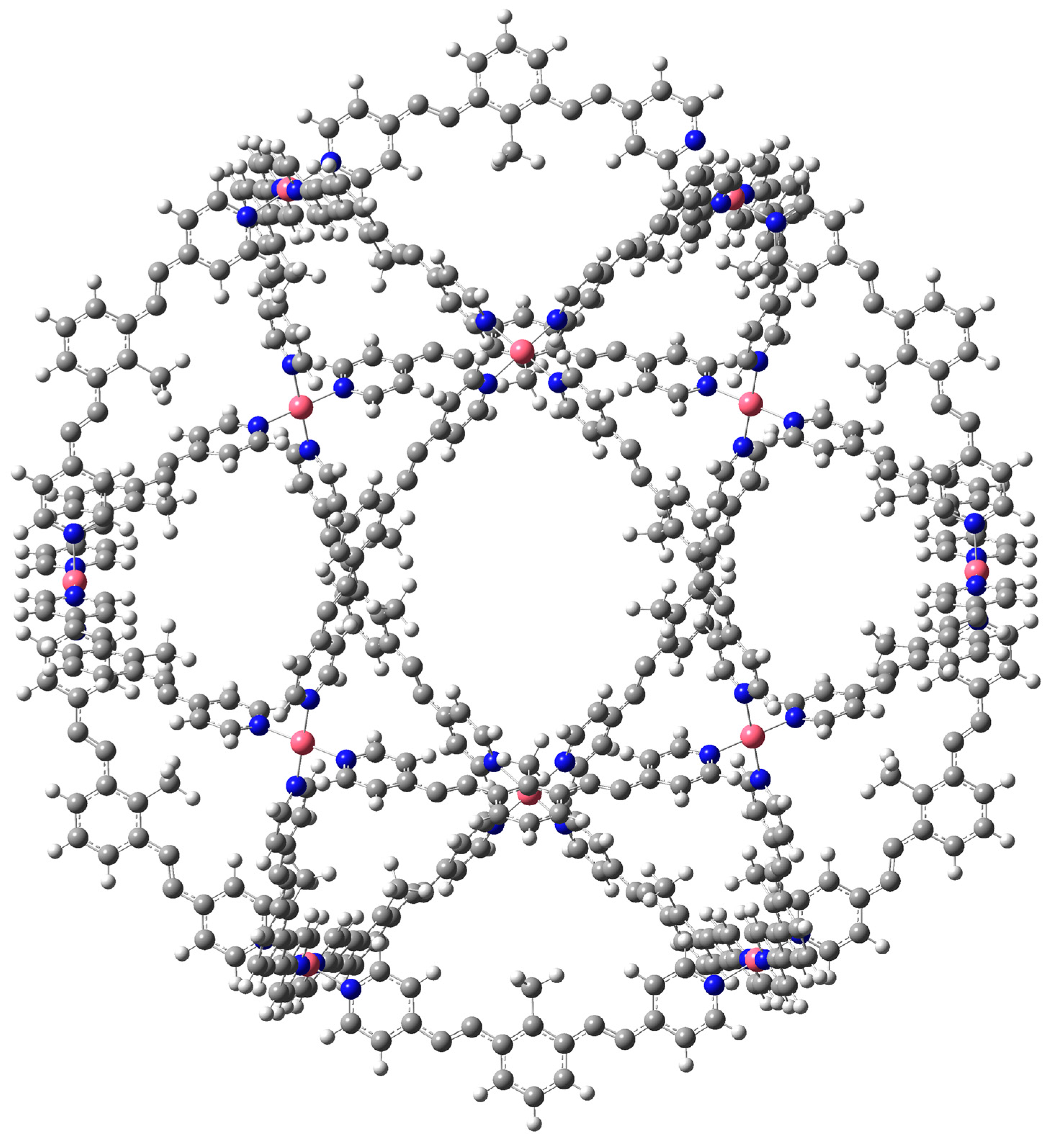
1. Introduction
2. Computational Details
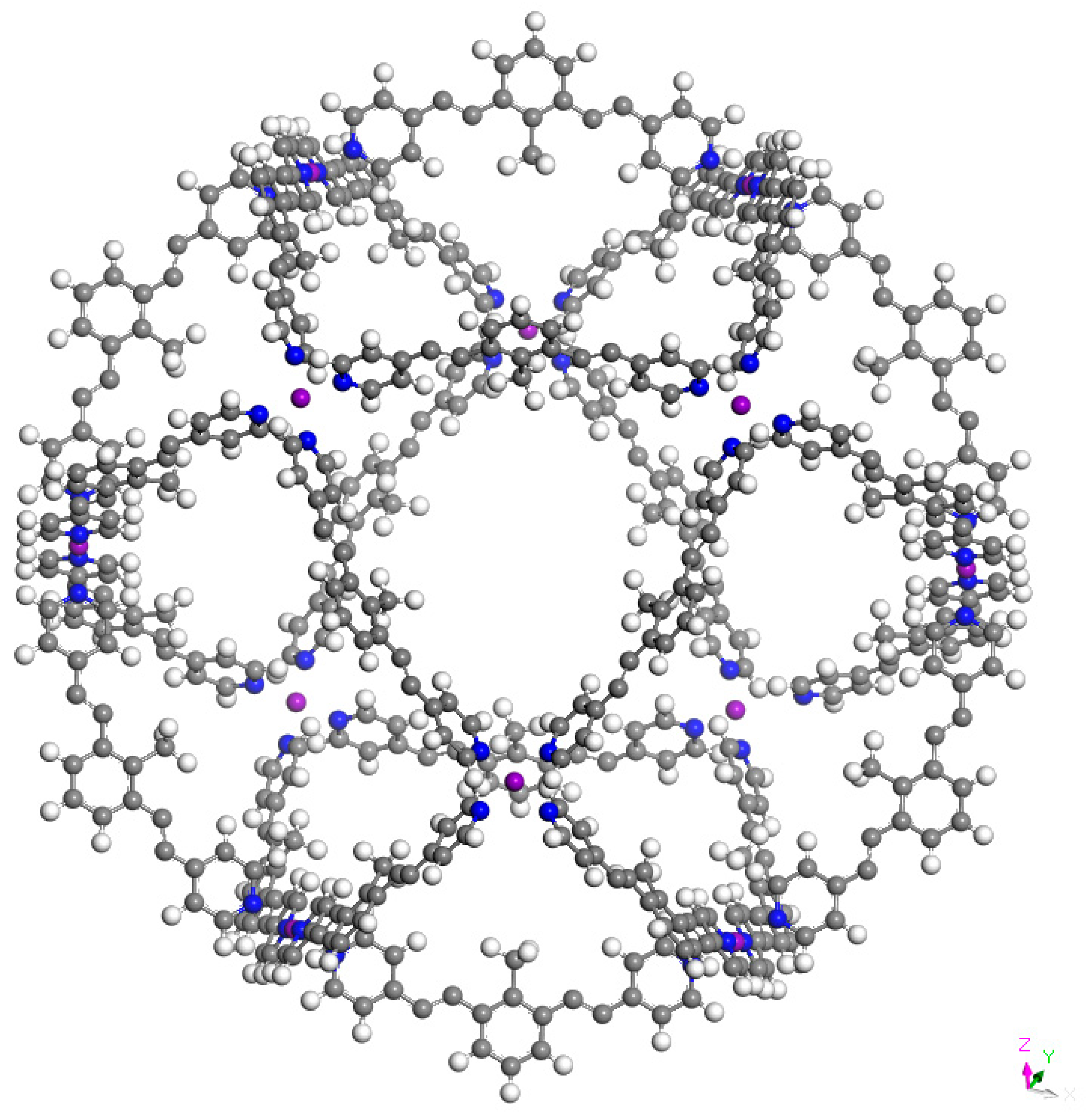
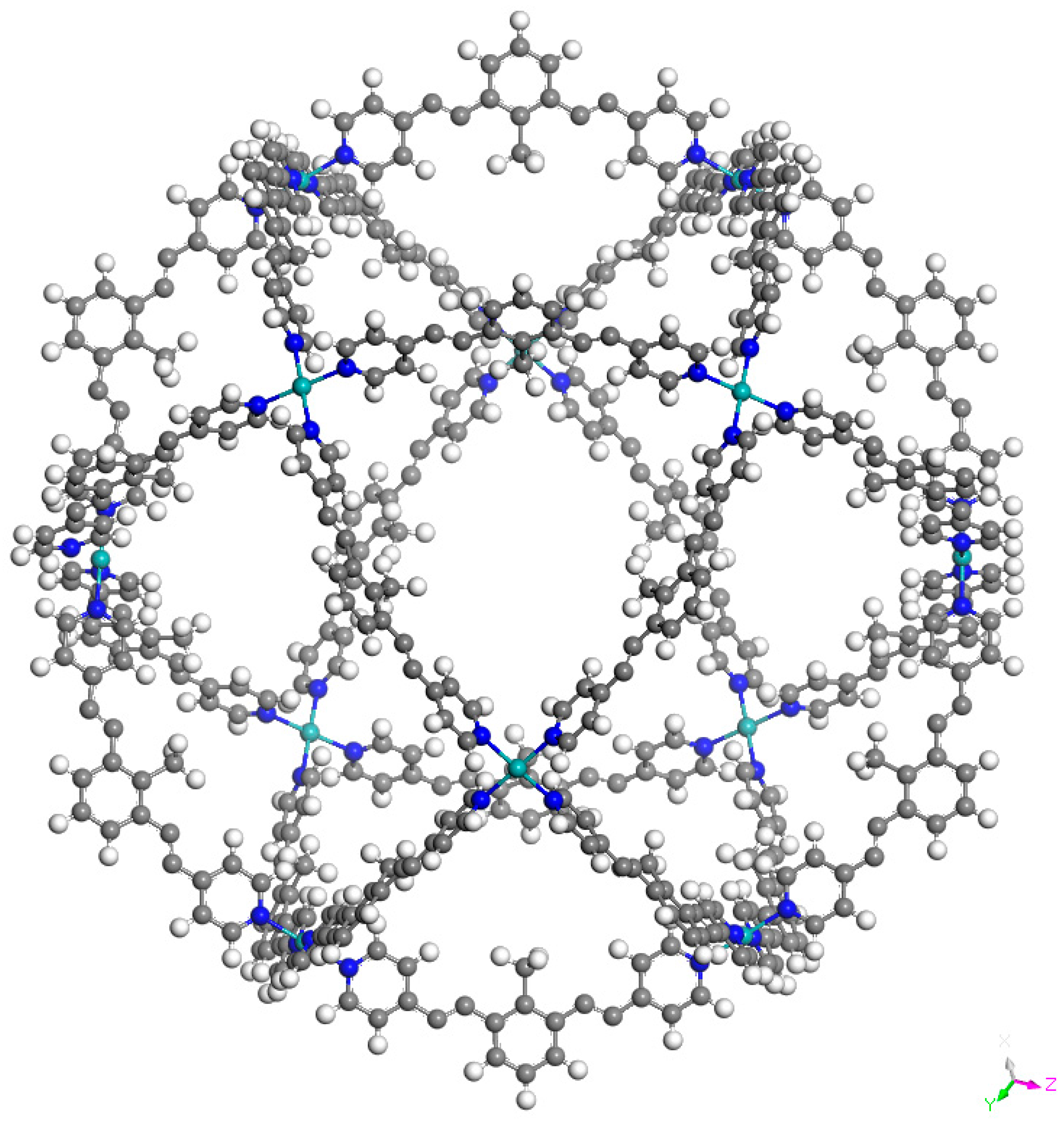
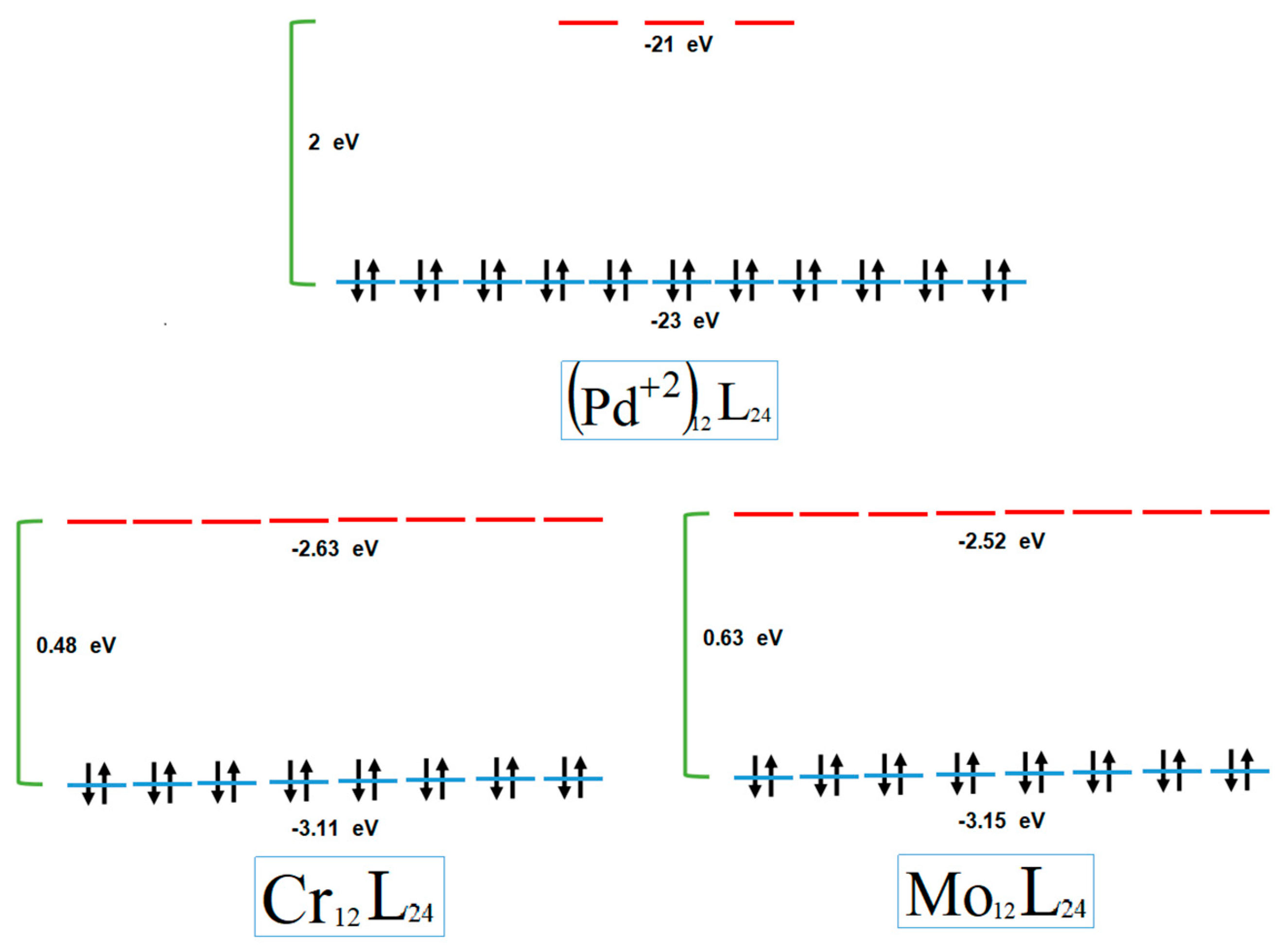
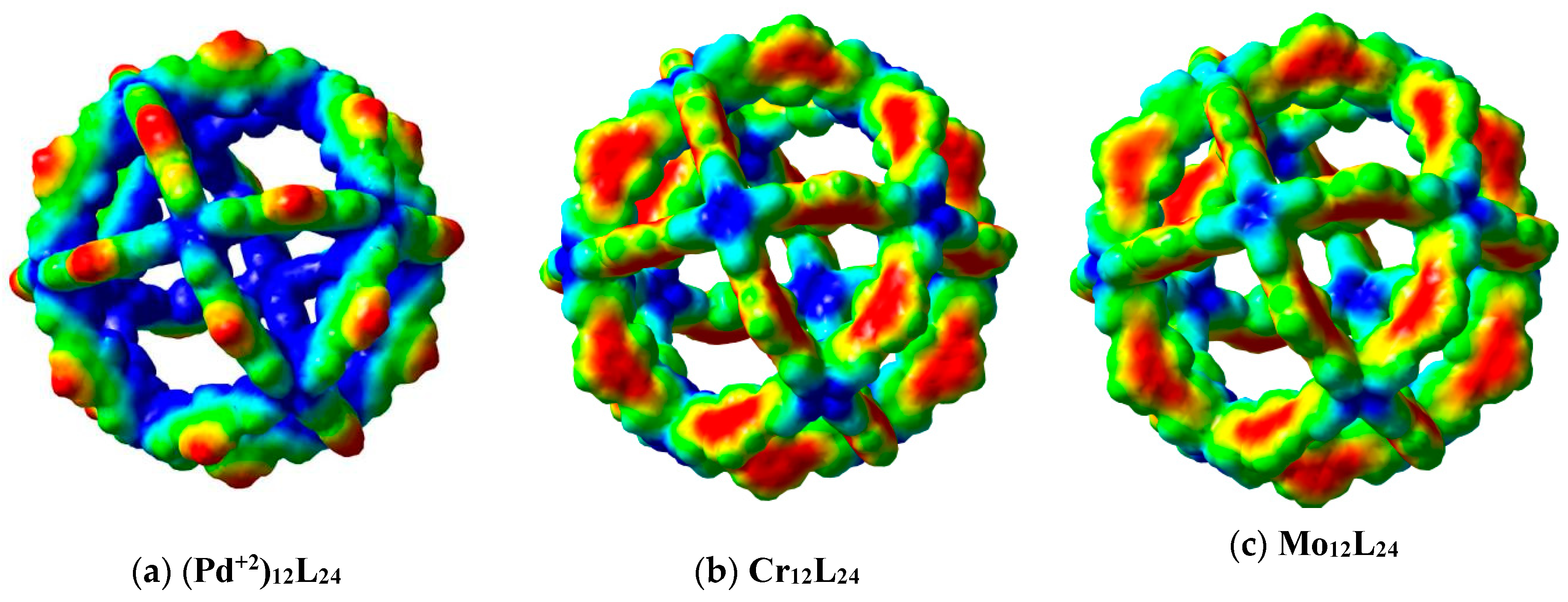
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, H.; Wang, J.; Xie, S.; Liua, W.; Niua, C. Template synthesis of graphitic hollow carbon nanoballs as supports for SnOx nanoparticles towards enhanced lithium storage performance. Nanoscale 2018, 10, 6159–6167. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.D.; Dirtu, M.M.; Léonard, A.; Tinant, B.; Marchand-Brynaert, J.; Su, B-L.; Garcia, Y. Engineering Three-Dimensional Chains of Porous Nanoballs from a 1,2,4-Triazole-carboxylate Supramolecular Synthon. Cryst. Growth Des. 2010, 10, 1798–1807. [Google Scholar] [CrossRef]
- Lai, Q.; Paskevicius, M.; Sheppard, D.A.; Buckley, C.E.; Thornton, A.W.; Hill, M.R.; Gu, Q.; Mao, J.; Huang, Z.; Liu, H.K.; et al. Hydrogen storage materials for mobile and stationary applications: Current state of the art. ChemSusChem 2015, 8, 2789–2825. [Google Scholar] [CrossRef] [PubMed]
- Butova, V.V.; Soldatov, M.A.; Guda, A.A.; Lomachenko, K.A.; Lamberti, C. Metal-organic frameworks: Structure, properties, methods of synthesis and characterization. Russ. Chem. Rev. 2016, 85, 280. [Google Scholar] [CrossRef]
- Yaghi, O.M. Reticular chemistry—Construction, properties, and precision reactions of frameworks. J. Am. Chem. Soc. 2016, 138, 15507–15509. [Google Scholar] [CrossRef] [PubMed]
- Byrne, K.; Zubair, M.; Zhu, N.; Zhou, X.P.; Fox, D.S.; Zhang, H.; Twamley, B.; Lennox, M.J.M.; Düren, T.; Shmitt, W. Ultra-large supramolecular coordination cages composed of endohedral Archimedean and Platonic bodies. Nat. Commun. 2017, 8, 15268. [Google Scholar] [CrossRef] [PubMed]
- Senkovska, I.; Kaskel, S. Ultrahigh porosity in mesoporous MOFs: Promises and limitations. Chem. Commun. 2014, 50, 7089–7098. [Google Scholar] [CrossRef] [PubMed]
- Grünker, R.; Bon, V.; Müller, P.; Stoeck, U.; Krause, S.; Mueller, U.; Senkovska, I.; Kaskel, S. A new metal-organic framework with ultra-high surface area. Chem. Commun. 2014, 50, 3450–3452. [Google Scholar] [CrossRef]
- Sun, L.; Campbell, M.G.; Dinca, M. Electrically conductive porous metal-organic frameworks. Angew. Chem. Int. Ed. 2016, 55, 3566–3579. [Google Scholar] [CrossRef]
- Lehaire, M.L.; Scopelliti, R.; Piotrowski, H.; Severin, K. Selective recognition of fluoride anion using a Li+-metallacrown complex. Angew. Chem. Int. Ed. 2002, 41, 1419–1422. [Google Scholar] [CrossRef]
- Yabushita, M.; Li, P.; Bernales, V.; Kobayashi, H.; Fukuoka, A.; Gagliardi, L.; Farha, O.K.; Katz, A. Unprecedented selectivity in molecular recognition of carbohydrates by a metal-organic framework. Chem. Commun. 2016, 52, 7094–7097. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.B.; Xia, B.Y.; Yu, L.; Yu, X.Y.; Lou, X.W. Porous molybdenum carbide nano-octahedrons synthesized via confined carburization in metal-organic frameworks for efficient hydrogen production. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.W.; Wang, R. A new green system of HPW@MOFs catalyzed desulfurization using O2 as oxidant. Chin. Chem. Lett. 2016, 27, 655–658. [Google Scholar] [CrossRef]
- Aghaji, M.Z.; Fernandez, M.; Boyd, P.G.; Daff, T.D.; Woo, T.K. Quantitative structure-property relationship models for recognizing metal organic frameworks (MOFs) with High CO2 working capacity and CO2/CH4 selectivity for methane purification. Eur. J. Inorg. Chem. 2016, 27, 4505–4511. [Google Scholar]
- Sumida, K.; Rogow, D.L.; Mason, J.A.; McDonald, T.M.; Bloch, E.D.; Herm, Z.R.; Bae, T.H.; Long, J.F. Carbon dioxide capture in metal-organic frameworks. Chem. Rev. 2012, 112, 724–781. [Google Scholar]
- Salles, F.; Ghoufi, A.; Maurin, G.; Bell, R.G.; Mellot-Draznieks, C.; Férey, G. Molecular dynamics simulations of breathing MOFs: Structural transformation of MIL-53(Cr) upon thermal activation and CO2 adsorption. Angew. Chem. Int. Ed. 2008, 47, 8487–8491. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.K.; Hong, D.Y.; Chang, J.S.; Jhung, S.H.; Seo, Y.K.; Kim, J.; Vimont, A.; Daturi, M.; Serre, C.; Férey, G. Amine grafting on coordinativelly unsaturated metal centers of MOF’s: Consequences for catalysis and metal encapsulation. Angew. Chem. Int. Ed. 2008, 47, 4144–4148. [Google Scholar] [CrossRef]
- Maurin, G.; Serre, C.; Cooper, A.; Férey, G. The new age of MOFs and of their porous-related solids. Chem. Soc. Rev. 2017, 46, 3104–3107. [Google Scholar] [CrossRef]
- Carrano, F.; Chapman, K.; Chen, Z.; Dinca, M.; Easun, T.; Eddaoudi, M.; Farha, O.; Forgan, R.; Gagliardi, L.; Haase, F.; et al. Catalysis in MOFs: General discussion. Faraday Discuss. 2017, 201, 369–394. [Google Scholar] [CrossRef]
- Levchenko, I.; Bazaka, K.; Keidar, M.; Xu, S.; Fang, J. Hierarchical multicomponent inorganic metamaterials: Intrinsically driven self-assembly at the nanoscale. Adv. Mater. 2017, 30–32. [Google Scholar] [CrossRef]
- Northrop, B.H.; Chercka, D.; Stang, P.J. Carbon rich supramolecular metallacycles and metallacages. Tetrahedron 2008, 64, 11495–11502. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M.; Suzuki, K.; Murase, T.; Fujita, M. 24-fold endohedral functionalization of a self-assembled M12L24 coordination nanoball. J. Am. Chem. Soc. 2005, 127, 11950–11951. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.E.M.; Elliot, A.B.S.; McAdam, C.J.; Gordon, K.C.; Crowley, J.D. ‘Click’ to functionalise: Synthesis, characterization and enhancement of the physical properties of a series of exo- and endo-functionalised Pd2L4 nanocages. Chem. Sci. 2014, 5, 1833–1843. [Google Scholar] [CrossRef]
- Otte, M. Size selective molecular-flasks. ACS Catal. 2016, 6, 6491–6510. [Google Scholar] [CrossRef]
- Drev, M.; Grošelj, U.; Ledinek, B.; Perdih, F.; Svete, J.; Štefane, B.; Požgan, F. Ruthenium(II)-catalyzed microwave-promoted multiple C–H activation in synthesis of Hexa(heteroaryl)benzenes in water. Org. Lett. 2018, 20, 5268–5273. [Google Scholar] [CrossRef] [PubMed]
- Holade, Y.; Lehoux, A.; Remita, H.; Kokoh, K.B.; Napporn, T.W. Au@Pt core–shell mesoporous nanoballs and nanoparticles as efficient electrocatalysts toward formic acid and glucose oxidation. J. Phys. Chem. C 2015, 119, 27529–27539. [Google Scholar] [CrossRef]
- Chen, M.; Wang, J.; Liu, D.; Jiang, Z.; Liu, Q.; Wu, T.; Liu, H.; Yu, W.; Yan, Y.; Wang, P. Highly stable spherical metallo-capsule from a branched hexapodal terpyridine and its self-assembled berry-type nanostructure. J. Am. Chem. Soc. 2018, 140, 2555–2561. [Google Scholar] [CrossRef] [PubMed]
- Matsuuraab, K. Synthetic approaches to construct viral capsid-like spherical nanomaterials. J. Chem. Commun. 2018. [Google Scholar] [CrossRef]
- Brynda, M.; Gagliardi, L.; Roos, B.O. Analysing the chromium-chromium multiple bonds using multiconfigurational quantum chemistry. Chem. Phys. Lett. 2009, 471, 1–10. [Google Scholar] [CrossRef]
- Yi, M.; Zhang, C. The synthesis of MoS2 particles with different morphologies for tribological applications. Tribol. Int. 2017, 116, 285–294. [Google Scholar] [CrossRef]
- Zhang, X.; Luo, X.; Duan, Y.; Huang, Y.; Zhang, N.; Zhao, L.; Wu, J. Two new hybrid molybdenum arsenate derivative constructed from [As2Mo6O26]6- building: Synthesis, structural characterization and photocatalysis property. J. Mol. Struct. 2017, 1141, 245–251. [Google Scholar] [CrossRef]
- Park, J.S.; Kang, Y.C. Multicomponent (Mo, Ni) metal sulfide and selenide microspheres with empty nanovoids as anode materials for Na-ion batteries. J. Mater. Chem. 2017. [Google Scholar] [CrossRef]
- Zhang, L.; Stephens, A.J.; Nussbaumer, A.L.; Lemonnier, J.F.; Jurček, P.; Vitorica-Yrezabal, I.J.; Leigh, D.A. Stereoselective synthesis of a composite knot with nine crossings. Nat. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Szymanska-Buzar, T. Carbonyl complexes of molybdenum and their catalytic activity. Curr. Org. Chem. 2012, 16, 3–15. [Google Scholar] [CrossRef]
- Beamson, G.; Papworth, A.J.; Philipps, C.; Smith, A.M.; Whyman, R. Selective hydrogenation of amides using ruthenium/molybdenum catalysts. Adv. Synth. Catal. 2010, 352, 869–883. [Google Scholar] [CrossRef]
- Fenlon, E.E. What tangled webs we weave. Nat. Chem. 2018, 10, 1078–1079. [Google Scholar] [CrossRef] [PubMed]
- Fenlon, E.E. Open Problems in Chemical Topology. Eur. J. Org. Chem. 2008, 2008. [Google Scholar] [CrossRef]
- Sauvage, J.P. From chemical topology to molecular machines (Nobel lecture). Angew. Chem. Int. Ed. 2017, 56, 11080–11093. [Google Scholar] [CrossRef]
- Kassem, S.; Lee, A.T.L.; Leigh, D.A.; Marcos, V.; Palmer, L.I.; Pisano, S. Stereodivergent synthesis with a programmable molecular machine. Nature 2017, 549, 374–378. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 (Revision A.03); Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Rappé, A.K.; Goddard, W.A., III. Charge equilibration for molecular dynamics simulations. J. Phys. Chem. 1991, 95, 3358–3363. [Google Scholar] [CrossRef]
- Rappé, A.K.; Casewit, C.J. Molecular Mechanics across Chemistry; University Science Books: California, CA, USA, 1996; ISBN 0-935702-77-6. [Google Scholar]
- Rappé, A.K.; Colwell, K.S.; Casewit, C.J. Application of a universal force field to metal complexes. Inor. Chem. 1993, 32, 3438–3450. [Google Scholar] [CrossRef]
- TURBOMOLE V6.2 2010, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007. Available online: http://www.turbomole.com (accessed on 20 December 2018).
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Design of density functionals that are broadly accurate for thermochemistry, thermochemical kinetics, and nonbonded interactions. J. Phys. Chem. A 2005, 109, 5656–5667. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 18, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Schröder, H.; Hühnert, J.; Schwabe, T. Evaluation of DFT-D3 dispersion corrections for various structural benchmark sets. J. Chem. Phys. 2017, 146, 044115. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Truhlar, D.G. Performance of effective core potentials for density functional calculations on 3d transition metals. J. Chem. Theory Comput. 2012, 8, 80–90. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef]
- Parr, R.J.; Gázquez, J.L. Hardness functional. J. Phys. Chem. 1993, 97, 3939–3940. [Google Scholar] [CrossRef]
Sample Availability: This work is fully theoretical and there are no physical samples. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Binding Energy (Hartrees) | Total Energy (eV) |
|---|---|---|
| Pd+2 | −3.06 | −636,353.1585 |
| Cr | −2.05 | −623,010.6125 |
| Mo | −1.39 | −617,264.9439 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
del Castillo, R.M.; Salcedo, R.; Martínez, A.; Ramos, E.; Sansores, L.E. Electronic Peculiarities of a Self-Assembled M12L24 Nanoball (M = Pd+2, Cr, or Mo). Molecules 2019, 24, 771. https://doi.org/10.3390/molecules24040771
del Castillo RM, Salcedo R, Martínez A, Ramos E, Sansores LE. Electronic Peculiarities of a Self-Assembled M12L24 Nanoball (M = Pd+2, Cr, or Mo). Molecules. 2019; 24(4):771. https://doi.org/10.3390/molecules24040771
Chicago/Turabian Styledel Castillo, Roxana Mitzayé, Roberto Salcedo, Ana Martínez, Estrella Ramos, and Luis Enrique Sansores. 2019. "Electronic Peculiarities of a Self-Assembled M12L24 Nanoball (M = Pd+2, Cr, or Mo)" Molecules 24, no. 4: 771. https://doi.org/10.3390/molecules24040771
APA Styledel Castillo, R. M., Salcedo, R., Martínez, A., Ramos, E., & Sansores, L. E. (2019). Electronic Peculiarities of a Self-Assembled M12L24 Nanoball (M = Pd+2, Cr, or Mo). Molecules, 24(4), 771. https://doi.org/10.3390/molecules24040771