
Stratification of Breast Cancer by Integrating Gene Expression Data and Clinical Variables



Abstract
1. Introduction
2. Results
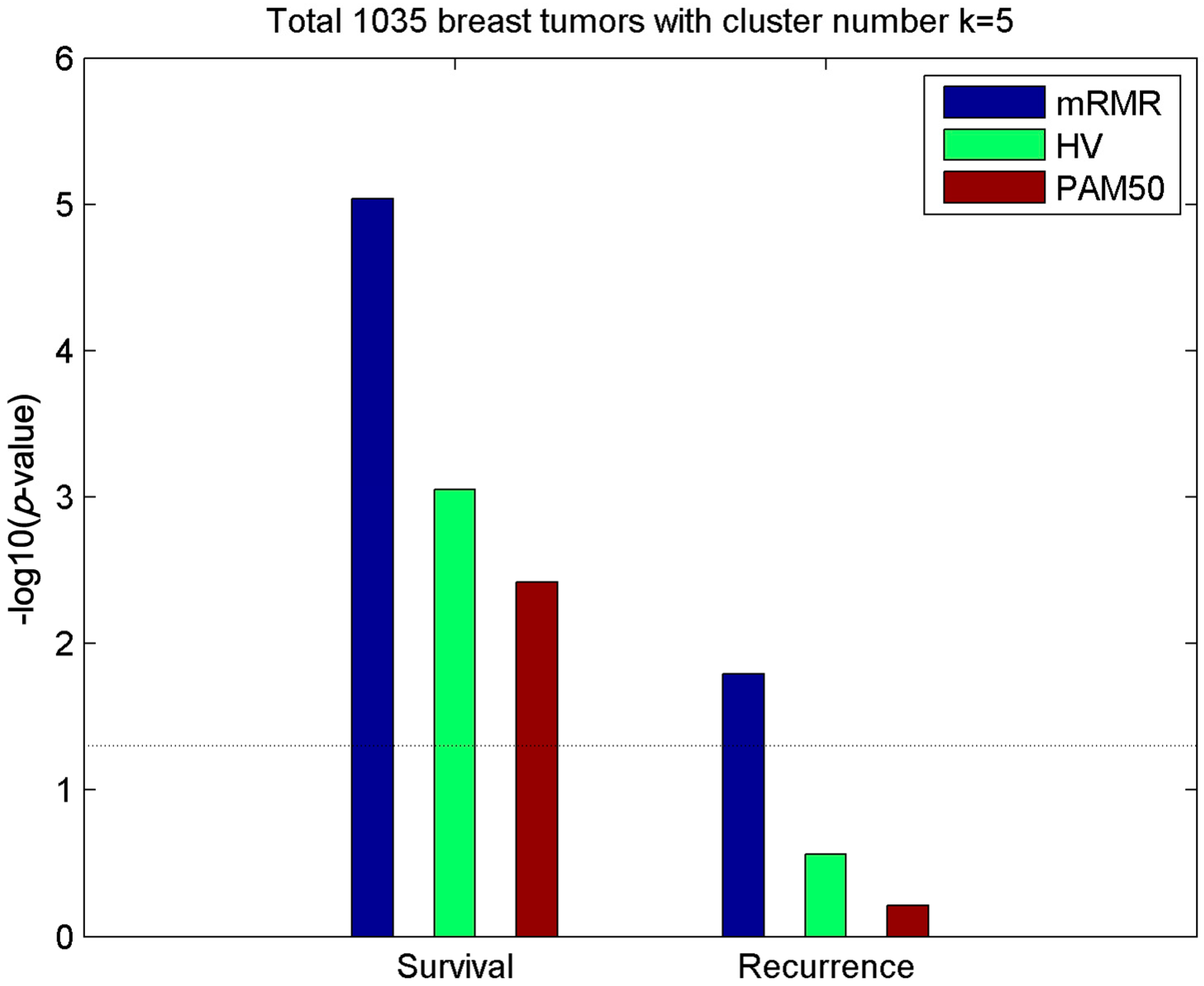
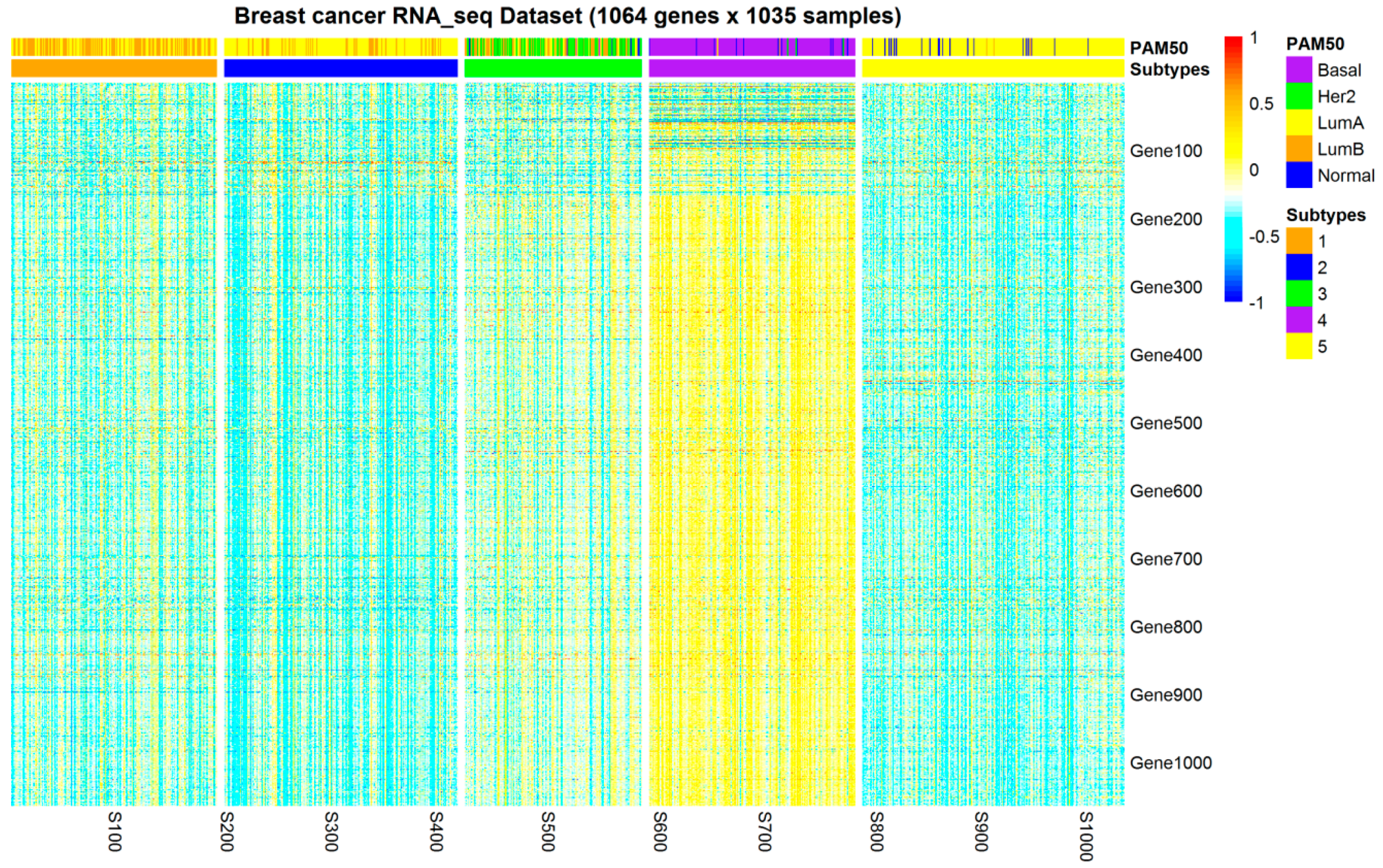
2.1. Subtypes of Breast Cancer
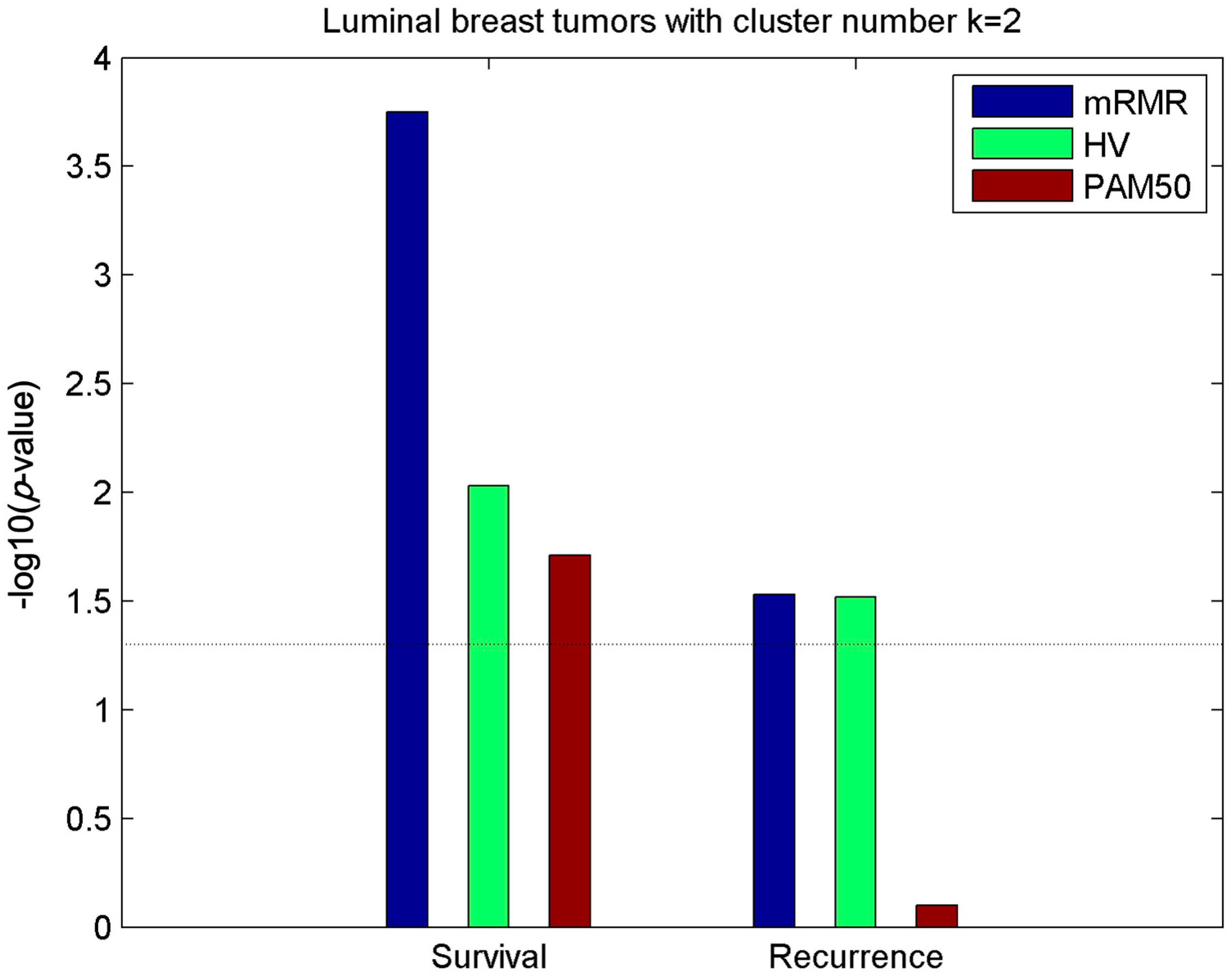
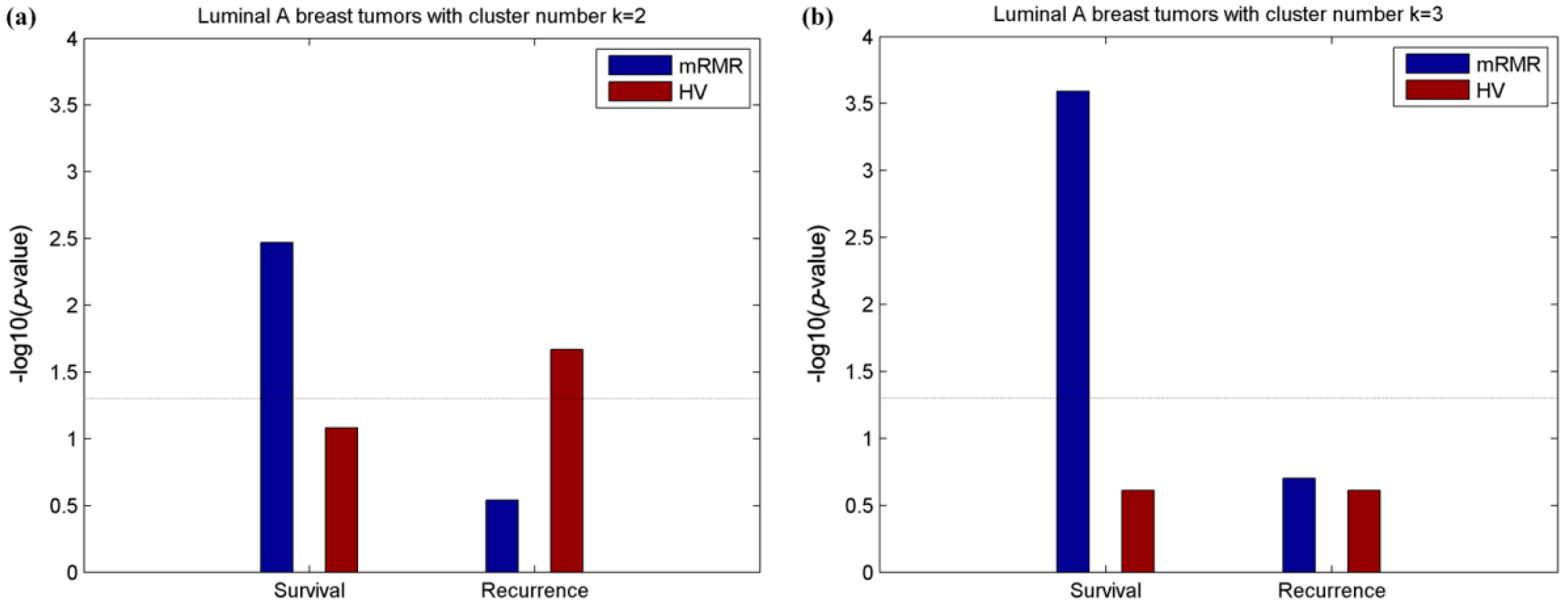
2.2. Clusters of Luminal Samples Predict Survival and Recurrence Better than PAM50
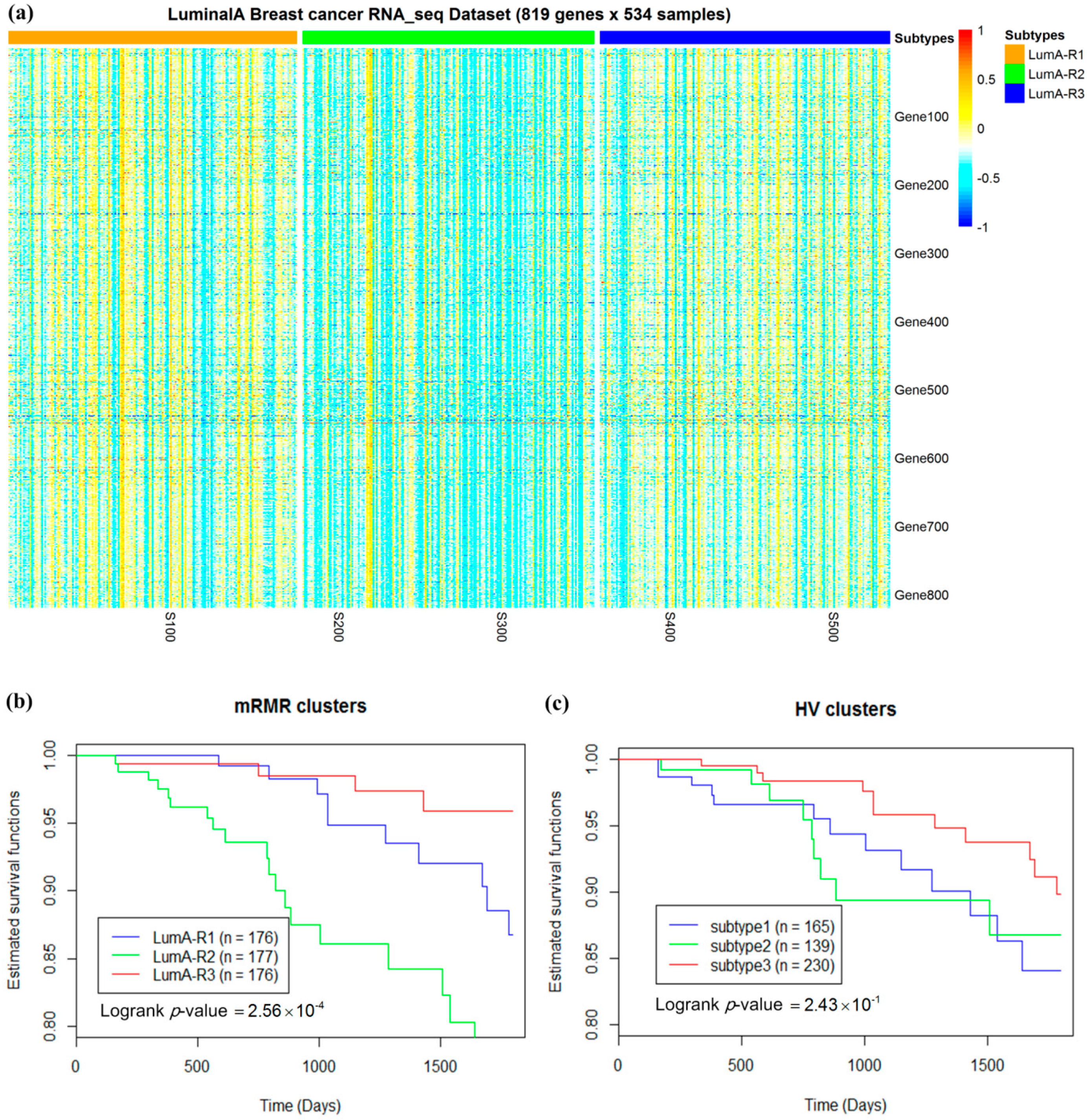
2.3. Luminal-A Samples Have Three Distinct Classes Predictive of Survival
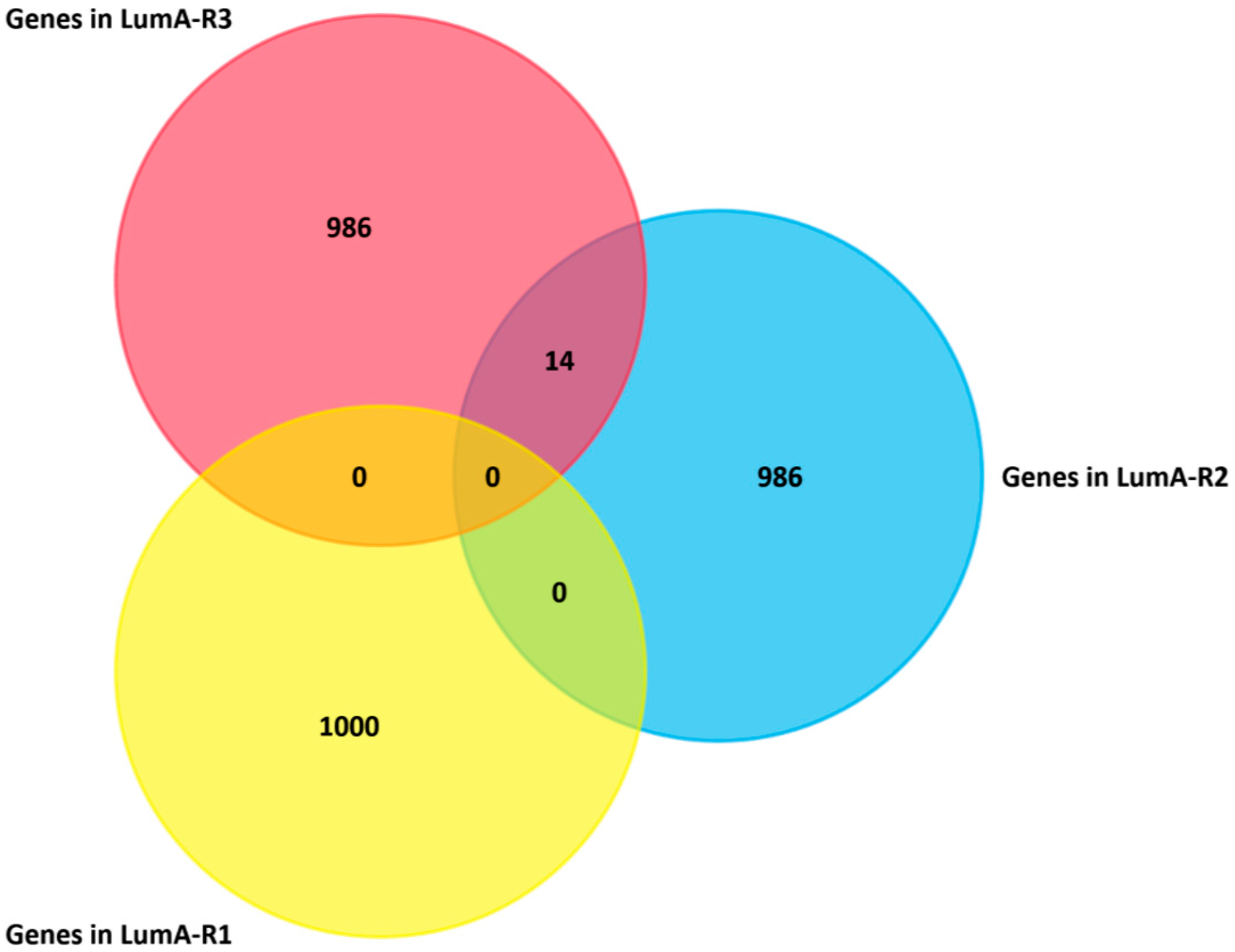
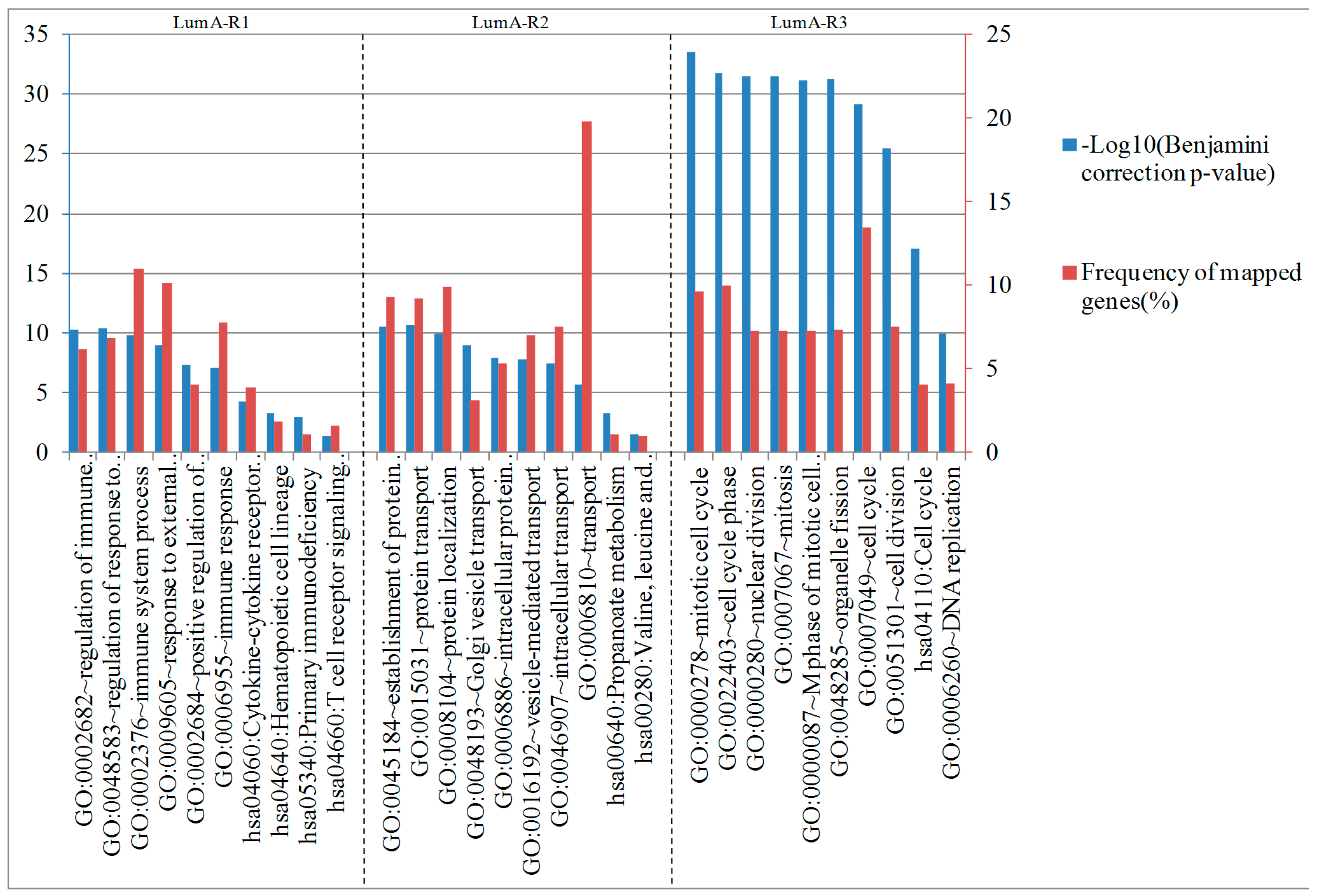
2.4. Analysis of Differentially Expressed Genes in Three Luminal-A Subgroups
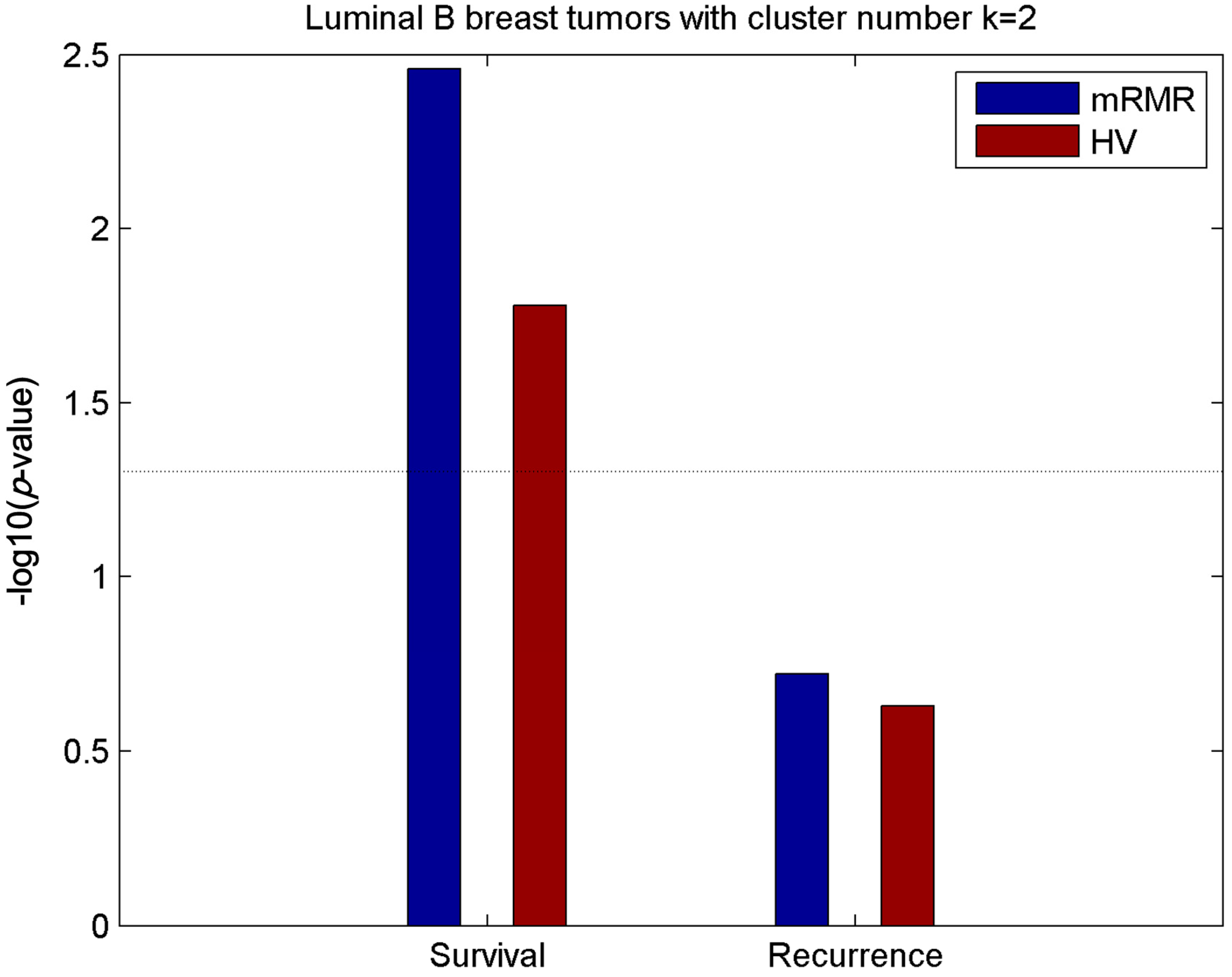
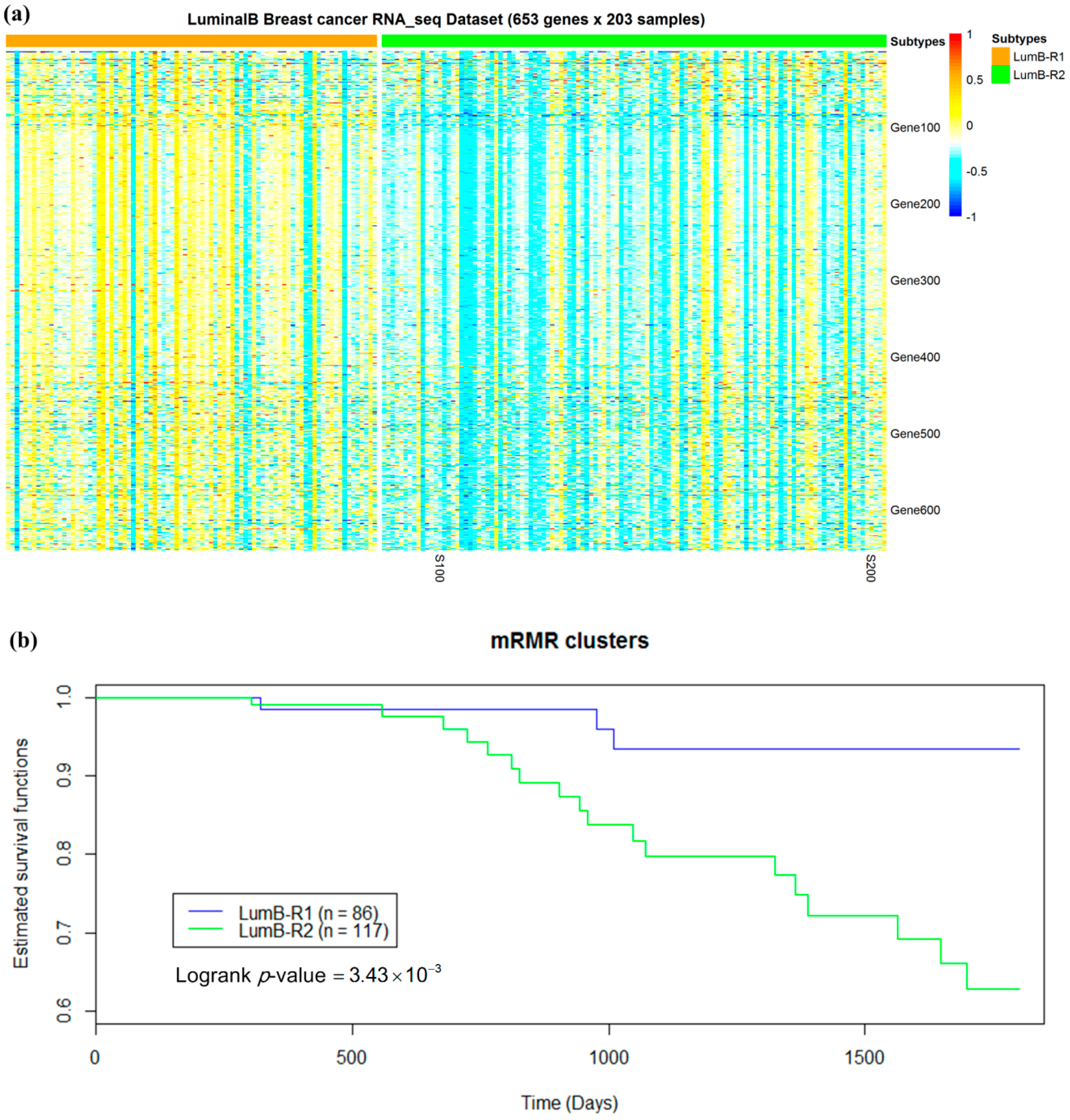
2.5. Luminal-B Samples have Two Distinct Subtypes Predictive of Survival
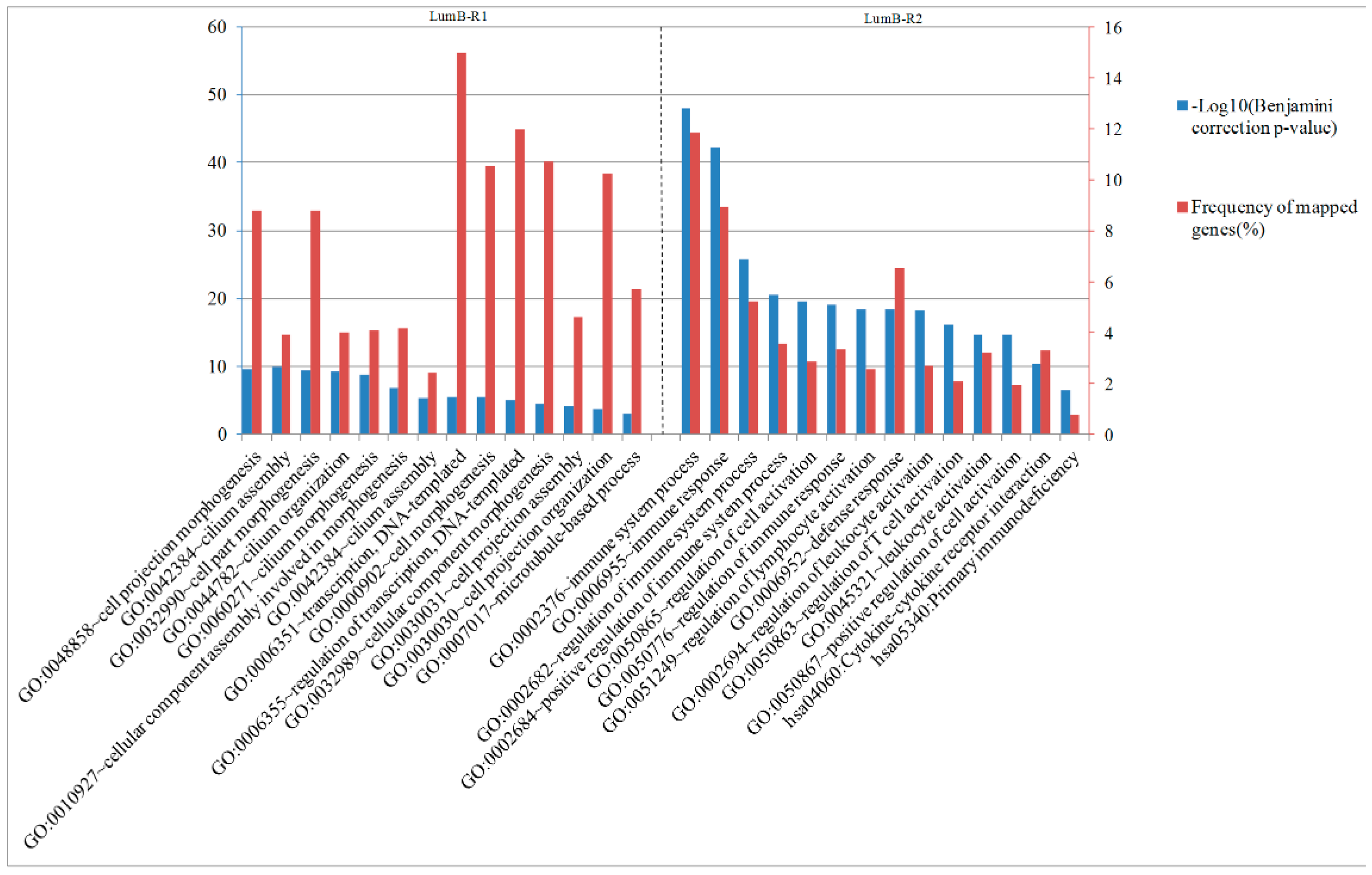
2.6. Analysis of Differentially Expressed Genes between Two Luminal-B Subgroups
3. Discussion
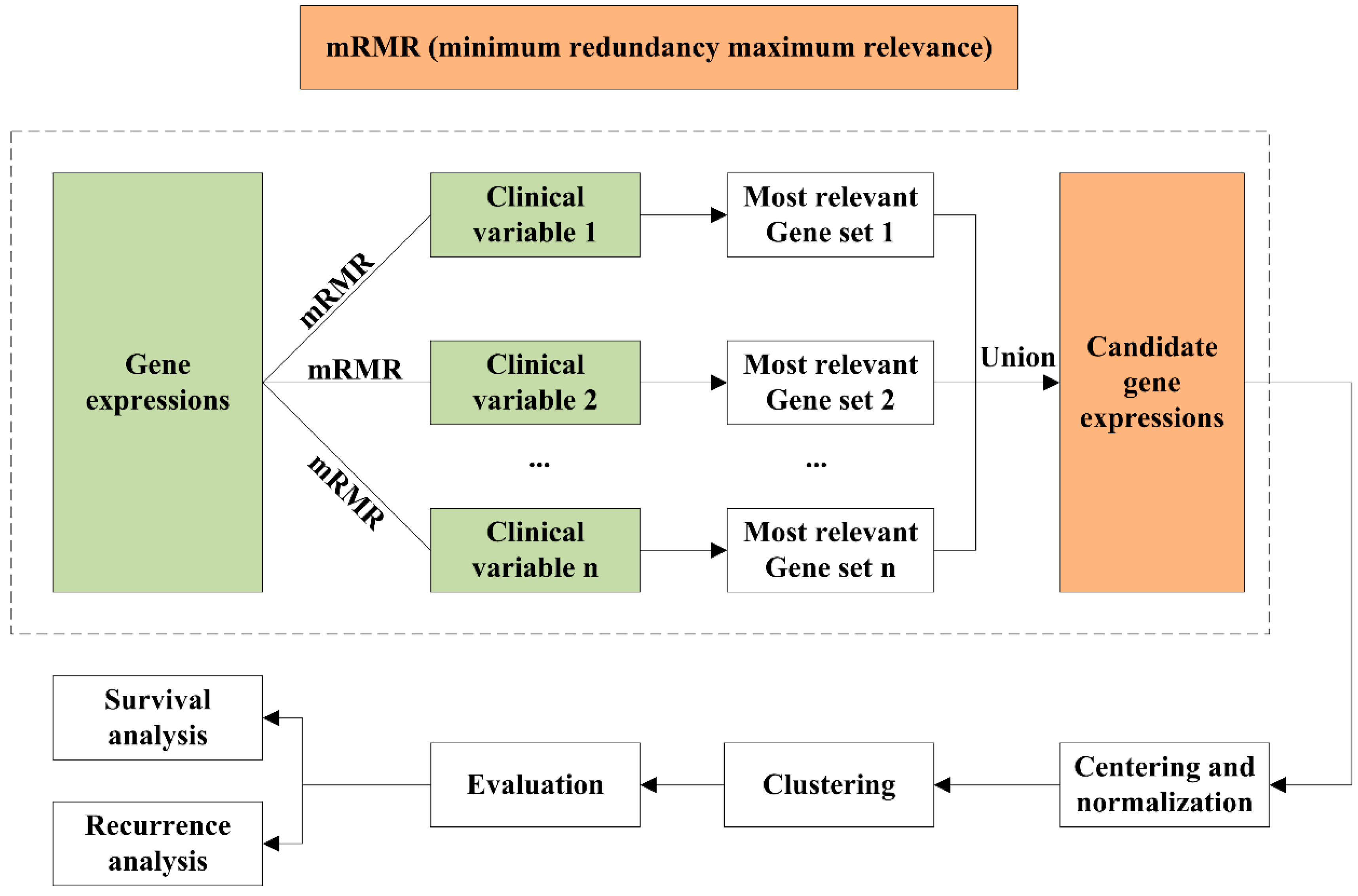
4. Materials and Methods
4.1. Data Preprocessing
4.2. mRMR Feature Selection
4.3. Unsupervised Clustering
4.4. Clustering Assignment and Clinical Analysis
4.5. Identification of Differentially Expressed Genes and Gene Enrichment
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bertos, N.R.; Park, M. Breast cancer—One term, many entities? J. Clin. Invest. 2011, 121, 3789–3796. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, X.S.; Zhang, S. Breast tumor subgroups reveal diverse clinical prognostic power. Sci. Rep. 2014, 4, 4002. [Google Scholar] [CrossRef] [PubMed]
- Hofree, M.; Shen, J.P.; Carter, H.; Gross, A.; Ideker, T. Network-based stratification of tumor mutations. Nat. Methods 2013, 10, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Gupta, R.; Steinbach, M.; Kumar, V. Integration of Clinical and Genomic Data: A Methodological Survey; Technical Report; Department of Computer Science and Engineering, University of Minnesota: Minneapolis, MA, USA, 20 February 2013. [Google Scholar]
- Xi, J.; Li, A.; Wang, M. A novel unsupervised learning model for detecting driver genes from pan-cancer data through matrix tri-factorization framework with pairwise similarities constraints. Neurocomputing 2018, 296, 64–73. [Google Scholar] [CrossRef]
- Hastie, T. L1-regularization path algorithm for generalized linear models. J. R. Statist. Soc. Series B 2007, 69, 659–677. [Google Scholar] [CrossRef]
- Lee, D.; Seung, H. Learning the parts of objects by non-negative matrix factorization. Nature 1999, 401, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T.; Perou, C.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.; van de Rijn, M.; Jeffrey, S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA. 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Therese, S.; Robert, T.; Joel, P.; Trevor, H.; Marron, J.S.; Andrew, N.; Shibing, D.; Hilde, J.; Robert, P.; Stephanie, G. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA. 2003, 100, 8418–8423. [Google Scholar] [CrossRef]
- Therese, S.R. Molecular portraits of breast cancer: Tumour subtypes as distinct disease entities. Eur. J. Cancer 2004, 40, 2667–2675. [Google Scholar] [CrossRef]
- Weigelt, B.; Mackay, A.; A’Hern, R.; Natrajan, R.; Tan, D.S.; Dowsett, M.; Ashworth, A.; Reisfilho, J.S. Breast cancer molecular profiling with single sample predictors: A retrospective analysis. Lancet Oncol. 2010, 11, 339–349. [Google Scholar] [CrossRef]
- Holm, K.; Hegardt, C.; Staaf, J.; Vallonchristersson, J.; Jönsson, G.; Olsson, H.; Borg, Å.; Ringnér, M. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010, 12, R36. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Wang, W.; Jinming, L.I. Identifying molecular subtypes in human colon cancer using gene expression and DNA methylation microarray data. Int. J. Oncol. 2016, 48, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Netanely, D.; Avraham, A.; Ben-Baruch, A.; Evron, E.; Shamir, R. Expression and methylation patterns partition luminal-A breast tumors into distinct prognostic subgroups. Breast Cancer Res. BCR 2016, 18, 74. [Google Scholar] [CrossRef] [PubMed]
- Bernard, F.; Redmond, C.K.; Fisher, E.R. Evolution of knowledge related to breast cancer heterogeneity: A 25-year retrospective. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 2068. [Google Scholar] [CrossRef]
- Weigel, M.T.; Dowsett, M. Current and emerging biomarkers in breast cancer: Prognosis and prediction. Endocr. Relat. Cancer 2010, 17, R245–R262. [Google Scholar] [CrossRef]
- Russnes, H.G.; Lingjaerde, O.C.; Børresen-Dale, A.L.; Caldas, C. Breast cancer molecular stratification—From intrinsic subtypes to integrative clusters. Am. J. Pathol. 2017, 187, 2152–2162. [Google Scholar] [CrossRef]
- Yuan, Y.; Allen, E.M.; Van Larsson, O.; Nikhil, W.; Ali, A.M.; Artem, S.; Byers, L.A.; Yanxun, X.; Hess, K.R.; Lixia, D. Assessing the clinical utility of cancer genomic and proteomic data across tumor types. Nat. Biotechnol. 2014, 32, 644. [Google Scholar] [CrossRef]
- Xu, X.; Huang, L.; Chan, C.H.; Yu, T.; Miao, R.; Liu, C. Assessing the clinical utility of genomic expression data across human cancers. Oncotarget 2016, 7, 45926–45936. [Google Scholar] [CrossRef]
- Zheng, Y. Methodologies for cross-domain data fusion: An overview. IEEE Trans. Big Data 2015, 1, 16–34. [Google Scholar] [CrossRef]
- Drysdale, A.; Grosenick, L.; Downar, J.; Dunlop, K.; Mansouri, F.; Meng, Y.; Fetcho, R.; Zebley, B.; Oathes, D.; Etkin, A.; et al. Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nat. Med. 2017, 23, 28–38. [Google Scholar] [CrossRef]
- Radovic, M.; Ghalwash, M.; Filipovic, N.; Obradovic, Z. Minimum redundancy maximum relevance feature selection approach for temporal gene expression data. Bmc Bioinform. 2017, 18, 9. [Google Scholar] [CrossRef]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.; Craft, B.; Swatloski, T.; Cline, M.; Morozova, O.; Diekhans, M.; Haussler, D.; Zhu, J. The UCSC cancer genomics browser: Update 2015. Nucleic Acids Res. 2015, 43, D812–D817. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Hanchuan, P.; Fuhui, L.; Chris, D. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Hubert, L.; Arabie, P. Comparing partitions. J. Classif. 1985, 2, 193–218. [Google Scholar] [CrossRef]
- Kim, J.; Stanescu, D.; Won, K. CellBIC: Bimodality-based top-down clustering of single-cell RNA sequencing data reveals hierarchical structure of the cell type. Nucleic Acids Res. 2018, 46, e124. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, E.L.; Meier, P. Nonparametric estimation from incomplete observations. J. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- Mantel, N.; Haenszel, W. Statistical aspects of the analysis of data from retrospective studies of disease. J. Natl. Cancer Inst. 1959, 22, 719–748. [Google Scholar] [CrossRef]
- Bland, J.M.; Altman, D.G. The logrank test. BMJ 2004, 328, 1073. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA. 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Screening Method | Number of Features | Adjusted Rand Index with PAM50 |
|---|---|---|
| mRMR | 1064 | 0.3337 |
| HV | 2000 | 0.3306 |
| Clinical Characteristic | Number of Patients (%) |
|---|---|
| Age | 58 years (range 26–90) |
| ER.status | |
| Positive | 761 (74%) |
| Negative | 227 (22%) |
| Unknown | 47 (4%) |
| PR.status | |
| Positive | 658 (64%) |
| Negative | 326 (31%) |
| Unknown | 51 (5%) |
| HER2.status | |
| Positive | 108 (11%) |
| Negative | 649 (63%) |
| Unknown | 278 (26%) |
| Stage | |
| Stage 1 | 176 (17%) |
| Stage 2 | 589 (55%) |
| Stage 3 | 234 (23%) |
| Stage 4 | 16 (2%) |
| Stage 5 | 13 (2%) |
| Unknown | 7 (1%) |
| Histological.type | |
| Infiltrating Ductal | 753 (73%) |
| Infiltrating Lobular | 182 (17%) |
| Medullary | 5 (1%) |
| Metaplastic | 4 (1%) |
| Mixed Histology | 29 (2%) |
| Mucinous | 16 (2%) |
| Unknown | 46 (4%) |
| Pathologic_N | |
| N0 | 491 (47%) |
| N1 | 339 (33%) |
| N2 | 114 (11%) |
| N3 | 71 (7%) |
| Unknown | 20 (2%) |
| Pathologic_T | |
| T1 | 269 (26%) |
| T2 | 600 (58%) |
| T3 | 126 (12%) |
| T4 | 36 (3%) |
| Unknown | 4 (1%) |
| Cancer_Status | |
| Tumor free | 883 (85%) |
| With tumor | 115 (11%) |
| Unknown | 37 (4%) |
| Lymph_node_count | 10 (range 0–44) |
| Tissue_source_site | 10 (range 1–31) |
| Year of initial diagnose | 2007 (range 1988–2013) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Z.; Zhang, J.; Yuan, X.; Xi, J.; Liu, Z.; Zhang, Y. Stratification of Breast Cancer by Integrating Gene Expression Data and Clinical Variables. Molecules 2019, 24, 631. https://doi.org/10.3390/molecules24030631
He Z, Zhang J, Yuan X, Xi J, Liu Z, Zhang Y. Stratification of Breast Cancer by Integrating Gene Expression Data and Clinical Variables. Molecules. 2019; 24(3):631. https://doi.org/10.3390/molecules24030631
Chicago/Turabian StyleHe, Zongzhen, Junying Zhang, Xiguo Yuan, Jianing Xi, Zhaowen Liu, and Yuanyuan Zhang. 2019. "Stratification of Breast Cancer by Integrating Gene Expression Data and Clinical Variables" Molecules 24, no. 3: 631. https://doi.org/10.3390/molecules24030631
APA StyleHe, Z., Zhang, J., Yuan, X., Xi, J., Liu, Z., & Zhang, Y. (2019). Stratification of Breast Cancer by Integrating Gene Expression Data and Clinical Variables. Molecules, 24(3), 631. https://doi.org/10.3390/molecules24030631