
Drug Sensitivity Screening and Targeted Pathway Analysis Reveal a Multi-Driver Proliferative Mechanism and Suggest a Strategy of Combination Targeted Therapy for Colorectal Cancer Cells

Abstract
1. Introduction
2. Results
2.1. Protein Kinase Inhibitors are Selected to Probe the Oncogenic Signaling of CRC Cell Lines
2.2. Some CRC Cells Are Not Sensitive to Kinase Inhibitors
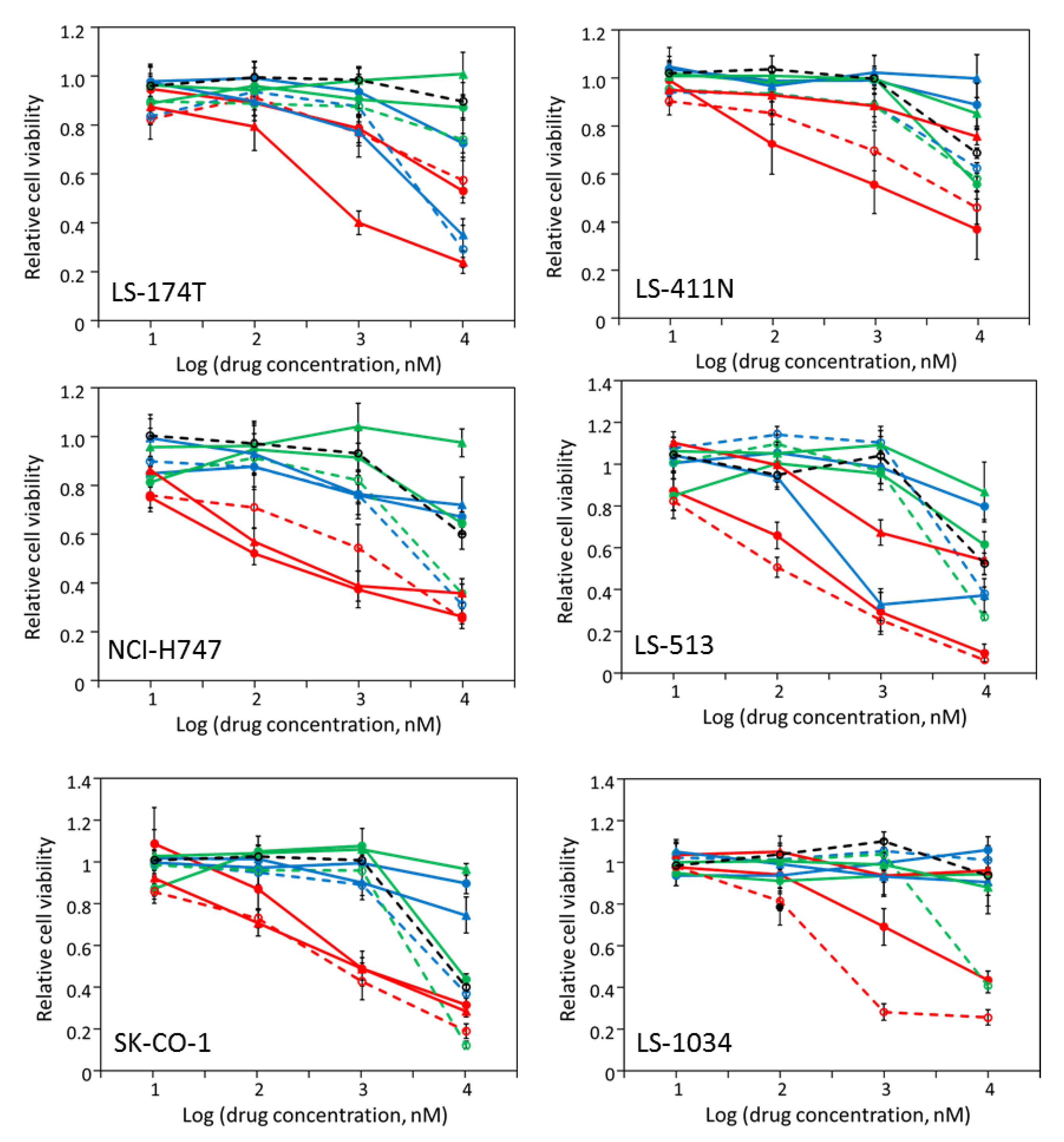
2.3. Most CRC Cells Are Partially Sensitive to Multiple Drugs
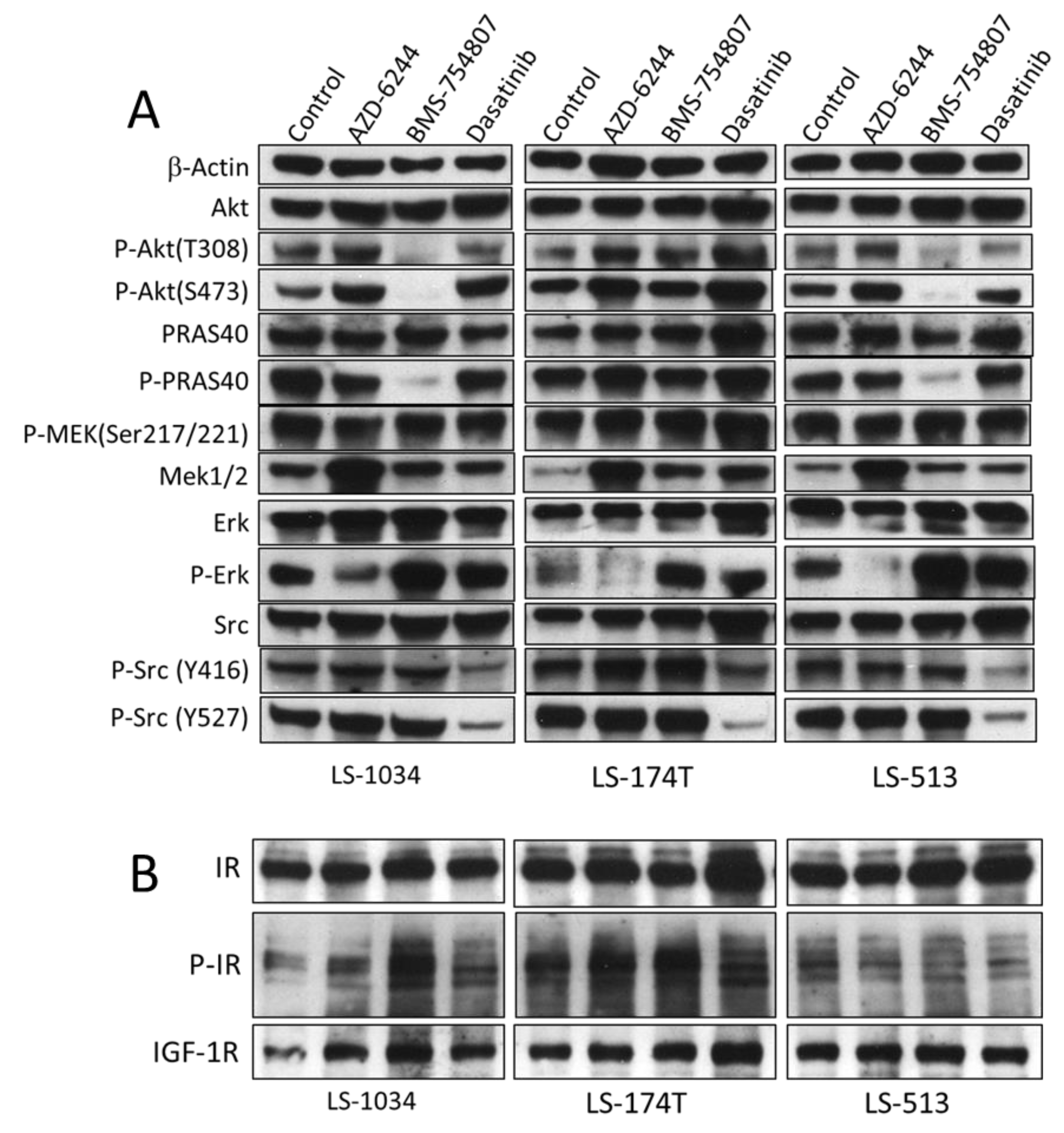
2.4. AZD-6244, BMS-754807 and Dasatinib Block Independent Signaling Pathways in CRC Cell Lines
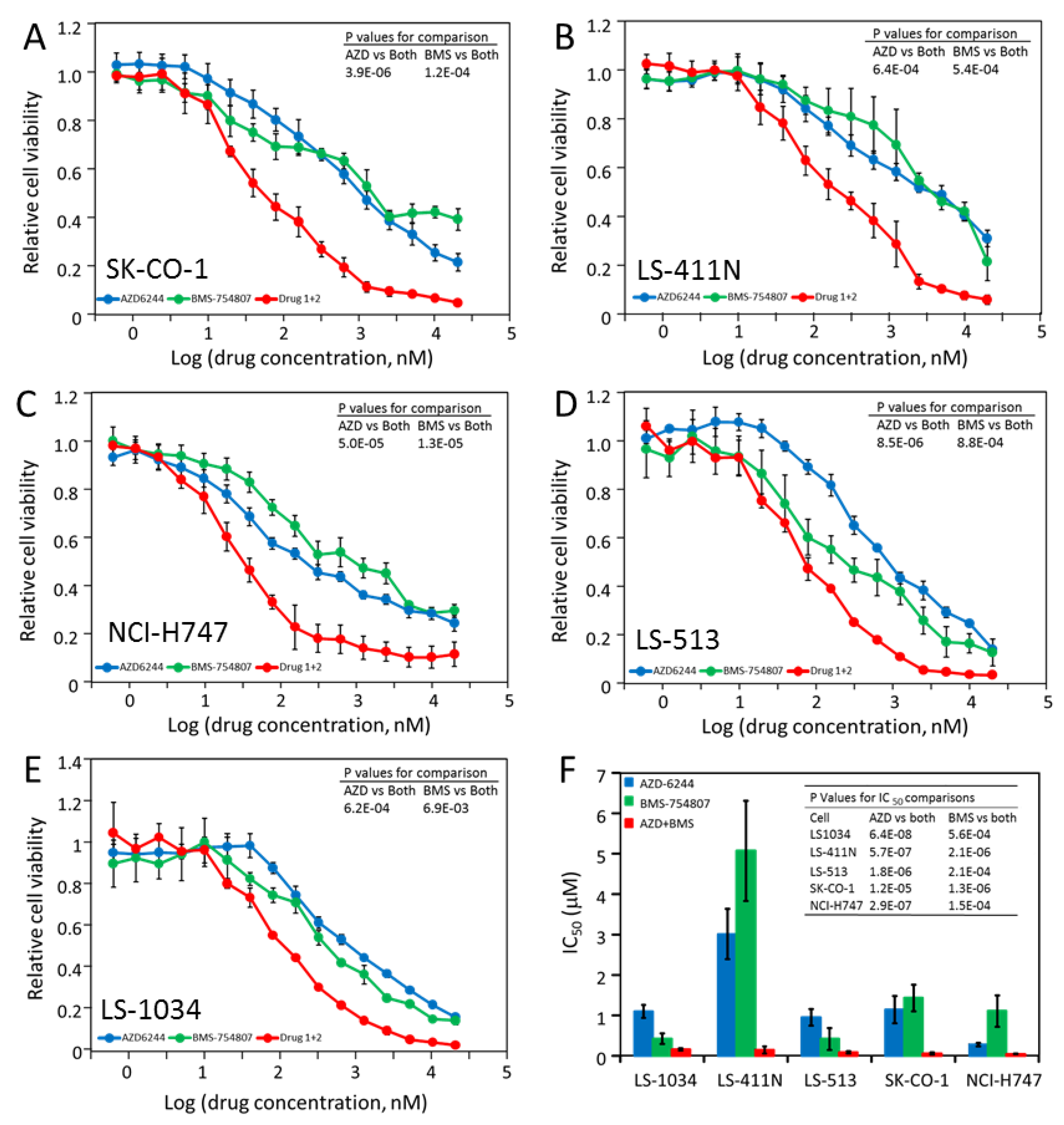
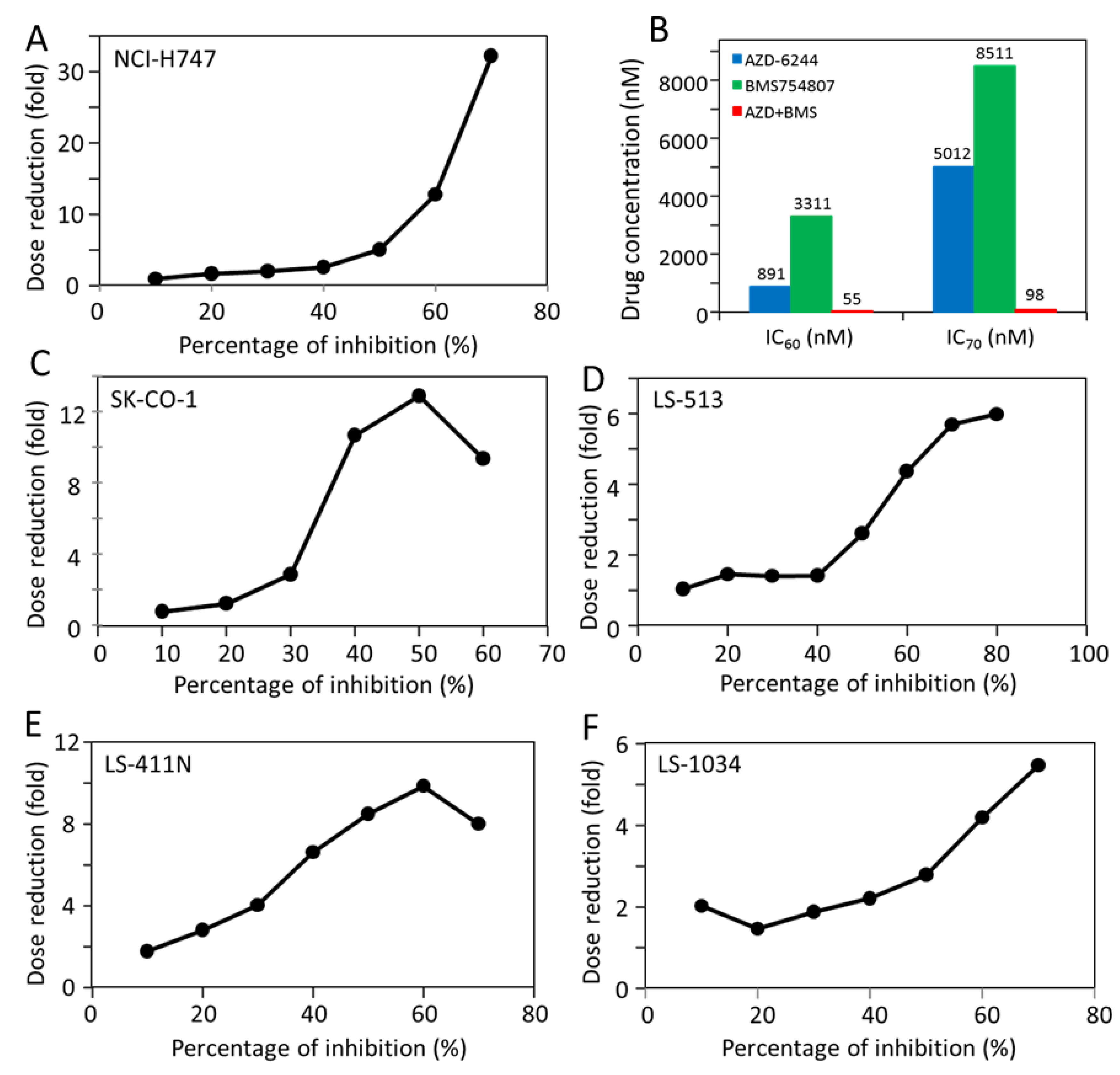
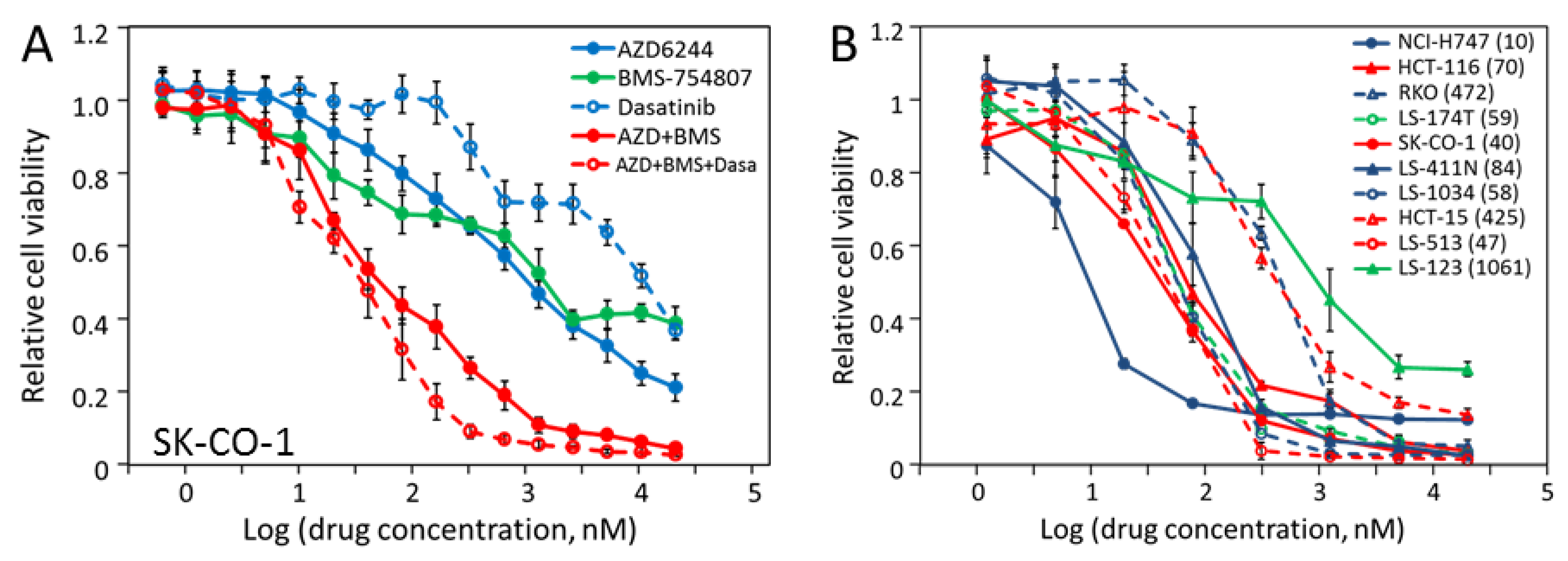
2.5. Most CRC Cells Are Potently and Synergistically Inhibited by the Combination of AZD-6244 and BMS-754807
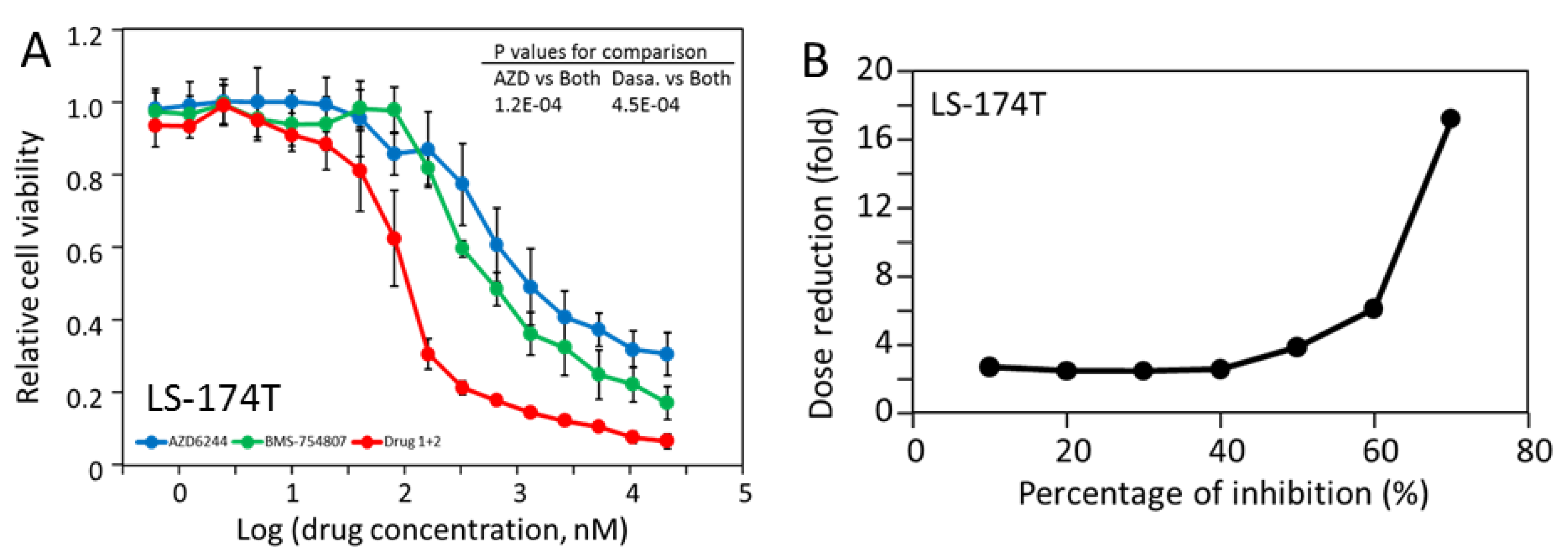
2.6. LS-174T Cells Are Sensitive to Inhibition by the Combination of AZD-6244 and Dasatinib
2.7. The Triple Combination of AZD-6244, BMS-754807 and Dasatinib Is Potently and Broadly Effective in Blocking CRC Cell Proliferation
3. Discussion
3.1. Screening with Kinase Inhibitors Identify Potential Molecular Targets for Blocking CRC Proliferation
3.2. Hypermutation De-Sensitizes Cancer Cell Response to Kinase Inhibitors
3.3. CRC Cells Are Dependent on MAP Kinase Pathway, IR/IGF-1R/AKT, and/or Src Kinases for Proliferation
3.4. A Rationale for Combination Therapy Using Multiple Signaling Inhibitors
3.5. A Triple Combination for CRC Targeted Therapy
4. Materials and Methods
4.1. Cell Lines, Media and Drugs
4.2. Cell Culture and Viability Assays
4.3. Initial Four-Concentration Screening and IC50 Determination
4.4. Western Blot Analysis of Drug Effects on Cell Signaling
4.5. Drug Synergy Analysis and Combination Index Calculation
4.6. Statistical Analysis
4.7. Mutation Information of CRC Cell Lines
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CI | combination index |
| CRC DRI | colorectal cancer dose reduction index |
| IGF-1R | insulin-like growth factor 1 receptor |
| IR | insulin receptor |
| MAP kinase | mitogen activated protein kinase |
| PI 3-kinase | phosphatidylinositol 3-kinase |
| PTK | receptor protein tyrosine kinase |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Mariotto, A.B.; Yabroff, K.R.; Shao, Y.; Feuer, E.J.; Brown, M.L. Projections of the cost of cancer care in the United States: 2010-2020. J. Natl. Cancer Inst. 2011, 103, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Loree, J.M.; Kopetz, S. Recent developments in the treatment of metastatic colorectal cancer. Ther. Adv. Med. Oncol. 2017, 9, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Wu, C.; Bekaii-Saab, T. Targeting BRAF in metastatic colorectal cancer: Maximizing molecular approaches. Cancer Treat. Rev. 2017, 60, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Seeber, A.; Gastl, G. Targeted Therapy of Colorectal Cancer. Oncol. Res. Treatment 2016, 39, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2018, 135, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Linnekamp, J.F.; Wang, X.; Medema, J.P.; Vermeulen, L. Colorectal cancer heterogeneity and targeted therapy: A case for molecular disease subtypes. Cancer Res. 2015, 75, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Tortora, G. A novel approach in the treatment of cancer: Targeting the epidermal growth factor receptor. Clin. Cancer Res. 2001, 7, 2958–2970. [Google Scholar] [PubMed]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, N. Targeting angiogenesis in cancer treatments: Where do we stand? J. Pharm. Pharm. Sci. 2016, 19, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Alberts, S.R.; Sargent, D.J.; Nair, S.; Mahoney, M.R.; Mooney, M.; Thibodeau, S.N.; Smyrk, T.C.; Sinicrope, F.A.; Chan, E.; Gill, S.; et al. Effect of oxaliplatin, fluorouracil, and leukovorin with or without cetuximab on survival among patients with resected stage III colon cancer: A randomized trial. JAMA 2012, 307, 1383–1393. [Google Scholar]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Shirley, M.; Keating, G.M. Regorafenib: A review of its use in patients with advanced gastrointestinal stromal tumours. Drugs 2015, 75, 1009–1017. [Google Scholar] [CrossRef]
- Jacoby, E.; Tresadern, G.; Bembenek, S.; Wroblowski, B.; Buyck, C.; Neefs, J.M.; Rassokhin, D.; Poncelet, A.; Hunt, J.; van Vlijmen, H. Extending kinome coverage by analysis of kinase inhibitor broad profiling data. Drug Discov. Today 2015, 20, 652–658. [Google Scholar] [CrossRef]
- Wittman, M.D.; Carboni, J.M.; Yang, Z.; Lee, F.Y.; Antman, M.; Attar, R.; Balimane, P.; Chang, C.; Chen, C.; Discenza, L.; et al. Discovery of a 2,4-disubstituted pyrrolo[1,2-f][1,2,4]triazine inhibitor (BMS-754807) of insulin-like growth factor receptor (IGF-1R) kinase in clinical development. J. Med. Chem. 2009, 52, 7360–7363. [Google Scholar] [CrossRef] [PubMed]
- Carboni, J.M.; Wittman, M.; Yang, Z.; Lee, F.; Greer, A.; Hurlburt, W.; Hillerman, S.; Cao, C.; Cantor, G.H.; Dell-John, J.; et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther. 2009, 8, 3341–3349. [Google Scholar] [CrossRef] [PubMed]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.C.; Marsh, V.; Bernat, B.A.; Ballard, J.; Colwell, H.; Evans, R.J.; Parry, J.; Smith, D.; Brandhuber, B.J.; Gross, S.; et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin. Cancer Res. 2007, 13, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; McDonnell, S.R.; Yamazaki, S.; Koudriakova, T.B.; Alton, G.; Cui, J.J.; Kung, P.P.; et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007, 67, 4408–4417. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, L.J.; Lee, F.Y.; Chen, P.; Norris, D.; Barrish, J.C.; Behnia, K.; Castaneda, S.; Cornelius, L.A.; Das, J.; Doweyko, A.M.; et al. Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 2004, 47, 6658–6661. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liang, C.; Shirazian, S.; Zhou, Y.; Miller, T.; Cui, J.; Fukuda, J.Y.; Chu, J.Y.; Nematalla, A.; Wang, X.; et al. Discovery of 5-[5-fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenemethyl]-2,4-dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J. Med. Chem. 2003, 46, 1116–1119. [Google Scholar] [PubMed]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res. 2008, 68, 3077–3080. [Google Scholar] [CrossRef]
- Tonon, G. From oncogene to network addiction: The new frontier of cancer genomics and therapeutics. Future Oncol. 2008, 4, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.S.; Kahana, J.A.; Kumar, R. AKT inhibitor, GSK690693, induces growth inhibition and apoptosis in acute lymphoblastic leukemia cell lines. Blood 2009, 113, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Beran, M.; Cao, X.; Estrov, Z.; Jeha, S.; Jin, G.; O’Brien, S.; Talpaz, M.; Arlinghaus, R.B.; Lydon, N.B.; Kantarjian, H. Selective inhibition of cell proliferation and BCR-ABL phosphorylation in acute lymphoblastic leukemia cells expressing Mr 190,000 BCR-ABL protein by a tyrosine kinase inhibitor (CGP-57148). Clin. Cancer Res. 1998, 4, 1661–1672. [Google Scholar] [PubMed]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Hakuno, F.; Takahashi, S.I. IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.C.; Lin, P.C.; Wu, H.Y.; Lin, K.T.; Wu, C.; Bekaii-Saab, T.; Lin, Y.J.; Lee, C.T.; Lee, J.C.; Chen, C.S. Mutant KRAS promotes liver metastasis of colorectal cancer, in part, by upregulating the MEK-Sp1-DNMT1-miR-137-YB-1-IGF-IR signaling pathway. Oncogene 2018, 37, 3440–3455. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Fazenbaker, C.; Chen, C.; Breen, S.; Huang, J.; Yao, X.; Ren, P.; Yao, Y.; Herbst, R.; Hollingsworth, R.E. Overproduction of IGF-2 drives a subset of colorectal cancer cells, which specifically respond to an anti-IGF therapeutic antibody and combination therapies. Oncogene 2017, 36, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Molina-Arcas, M.; Hancock, D.C.; Sheridan, C.; Kumar, M.S.; Downward, J. Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov. 2013, 3, 548–563. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Elfiky, A.; Han, M.; Chen, C.; Saif, M.W. The role of Src in colon cancer and its therapeutic implications. Clin. Colorectal Cancer 2014, 13, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Aligayer, H.; Boyd, D.D.; Heiss, M.M.; Abdalla, E.K.; Curley, S.A.; Gallick, G.E. Activation of Src kinase in primary colorectal carcinoma: An indicator of poor clinical prognosis. Cancer 2002, 94, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Herynk, M.H.; Zhang, J.; Parikh, N.U.; Gallick, G.E. Activation of Src by c-Met overexpression mediates metastatic properties of colorectal carcinoma cells. J. Exp. Ther. Oncol. 2007, 6, 205–217. [Google Scholar] [PubMed]
- Zhang, S.; Yu, D. Targeting Src family kinases in anti-cancer therapies: Turning promise into triumph. Trends Pharmacol. Sci. 2012, 33, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Puls, L.N.; Eadens, M.; Messersmith, W. Current status of SRC inhibitors in solid tumor malignancies. Oncologist 2011, 16, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Peng, W.; Calvo, E. Rational Approaches for Combination Therapy Strategies Targeting the MAP Kinase Pathway in Solid Tumors. Mol. Cancer Ther. 2018, 17, 3–16. [Google Scholar] [CrossRef]
- Hasinoff, B.B.; Patel, D.; Wu, X. The myocyte-damaging effects of the BCR-ABL1-targeted tyrosine kinase inhibitors increase with potency and decrease with specificity. Cardiovas. Toxicol. 2017, 17, 297–306. [Google Scholar] [CrossRef]
- Moslehi, J.J.; Deininger, M. Tyrosine kinase inhibitor-associated cardiovascular toxicity in chronic myeloid leukemia. J. Clin. Oncol. 2015, 33, 4210–4218. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C. Intratumor heterogeneity: Evolution through space and time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds used in this study are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Target Kinases (Kd in nM) |
|---|---|
| BGJ398 | FGFR1 (0.9), FGFR2 (1.4), FGFR3 (1), FGFR4 (60) |
| BMS-754807 | IGF1-R (1.8), insulin receptor (1.7), Met (5.6) |
| Bosutinib | Abl1 (0.12), Abl2 (1.5), EGFR3 (0.77), EphA3 (5.8), Lck (0.59), Src (1), BTK (4.8) |
| Crizotinib | Alk (3.3), Met (2.1) |
| Dasatinib | Abl1 (0.03) Abl2 (0.17), PTK6 (7.8), EGFR3 (18), EphA3 (0.09), CSF1R (0.58), Kit (0.81), PDGFRA (0.47), PDGFRB (0.63), Lck (0.2), Src (0.21), Btk (1.4) |
| Gefitinib Imatinib | EGFR (1) Abl1 (1.1), Abl2 (10), CSF1R (11), Kit (13), PDGFRB (14) |
| Sunitinib | CSF1R (2.5), FLT3 (0.4), Kit (0.4), PDGFRA (0.8), PDGFRB (0.1), Ret (13), ITK (13), VEGFR2 (1.5) |
| AZD-6244 | Mek 1 (14) |
| AZD-6482 | PIK3C (0.69) |
| Cell Line | Pik3ca Mutations | Braf Mutations | KRAS Mutations | Cancer Census Mutations | Coding Mutations |
|---|---|---|---|---|---|
| HCT-15 | E545K, D549N | none | G13D | 321 | 11348 |
| HCT-116 | H1047R | none | G13D | 163 | 4318 |
| LS-1034 | None | none | A146T | 10 | 207 |
| LS-123 | None | none | G12S | 13 | 483 |
| LS-411N | None | V600E | none | 209 | 7141 |
| LS-174T | H1047R | D211G | G12D | 127 | 3517 |
| LS-513 | None | none | G12D | 21 | 444 |
| NCI-H747 | None | none | G13D | 14 | 377 |
| RKO | H1047R | V600E | none | 155 | 4762 |
| SK-CO-1 | None | none | G12V | 16 | 443 |
| Cell | AZD-6244 | BMS-754807 | Dasatinib | Gefitinib | Bi-Combo | ABD Tripo d |
|---|---|---|---|---|---|---|
| LS-1034 | 1.09 ± 0.16 | 0.42 ± 0.13 | >10 | >10 | 0.153 ± 0.027 a | 0.058 ± 0.005 |
| LS-174T | 1.41 ± 0.63 | >10 | 0.61 ± 0.11 | >10 | 0.102 ± 000 b | 0.059 ± 0.006 |
| LS-411N | 3.01 ± 0.62 | 5.07 ± 1.24 | >10 | >10 | 0.146 ± 0.088 a | 0.084 ± 0.028 |
| LS-513 | 0.95 ± 0.21 | 0.42 ± 0.27 | >10 | 0.94 ± 0.44 | 0.086 ± 0.030 a | 0.047 ± 0.040 |
| NCI-H747 | 0.28 ± 0.05 | 1.39 ± 1.02 | 0.26 ± 0.12 | >10 | 0.046 ± 0.005 a | 0.010 ± 0.001 |
| SK-CO-1 | 1.15 ± 0.34 | 1.43 ± 0.33 | 10.4 ± 1.67 | >10 | 0.052 ± 0.021 a | 0.040 ± 0.004 |
| HCT-116 | >10 | >10 | >10 | >10 | ND c | 0.070 ± 0.005 |
| HCT-15 | >10 | >10 | >10 | >10 | ND | 0.425 ± 0.068 |
| LS-123 | >10 | >10 | >10 | >10 | ND | 1.061 ± 0.388 |
| RKO | >10 | >10 | >10 | >10 | ND | 0.472 ± 0.025 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, J.; Li, L.; Yang, T.; Cheng, N.; Sun, G. Drug Sensitivity Screening and Targeted Pathway Analysis Reveal a Multi-Driver Proliferative Mechanism and Suggest a Strategy of Combination Targeted Therapy for Colorectal Cancer Cells. Molecules 2019, 24, 623. https://doi.org/10.3390/molecules24030623
Shen J, Li L, Yang T, Cheng N, Sun G. Drug Sensitivity Screening and Targeted Pathway Analysis Reveal a Multi-Driver Proliferative Mechanism and Suggest a Strategy of Combination Targeted Therapy for Colorectal Cancer Cells. Molecules. 2019; 24(3):623. https://doi.org/10.3390/molecules24030623
Chicago/Turabian StyleShen, Jinyan, Li Li, Tao Yang, Niuliang Cheng, and Gongqin Sun. 2019. "Drug Sensitivity Screening and Targeted Pathway Analysis Reveal a Multi-Driver Proliferative Mechanism and Suggest a Strategy of Combination Targeted Therapy for Colorectal Cancer Cells" Molecules 24, no. 3: 623. https://doi.org/10.3390/molecules24030623
APA StyleShen, J., Li, L., Yang, T., Cheng, N., & Sun, G. (2019). Drug Sensitivity Screening and Targeted Pathway Analysis Reveal a Multi-Driver Proliferative Mechanism and Suggest a Strategy of Combination Targeted Therapy for Colorectal Cancer Cells. Molecules, 24(3), 623. https://doi.org/10.3390/molecules24030623