Simultaneous Determination of Thirteen Q-Markers in Raw and Processed Tussilago farfara L. by UPLC-QQQ-MS/MS Coupled with Chemometrics

 , , ,
, , ,
Abstract
1. Introduction
2. Results
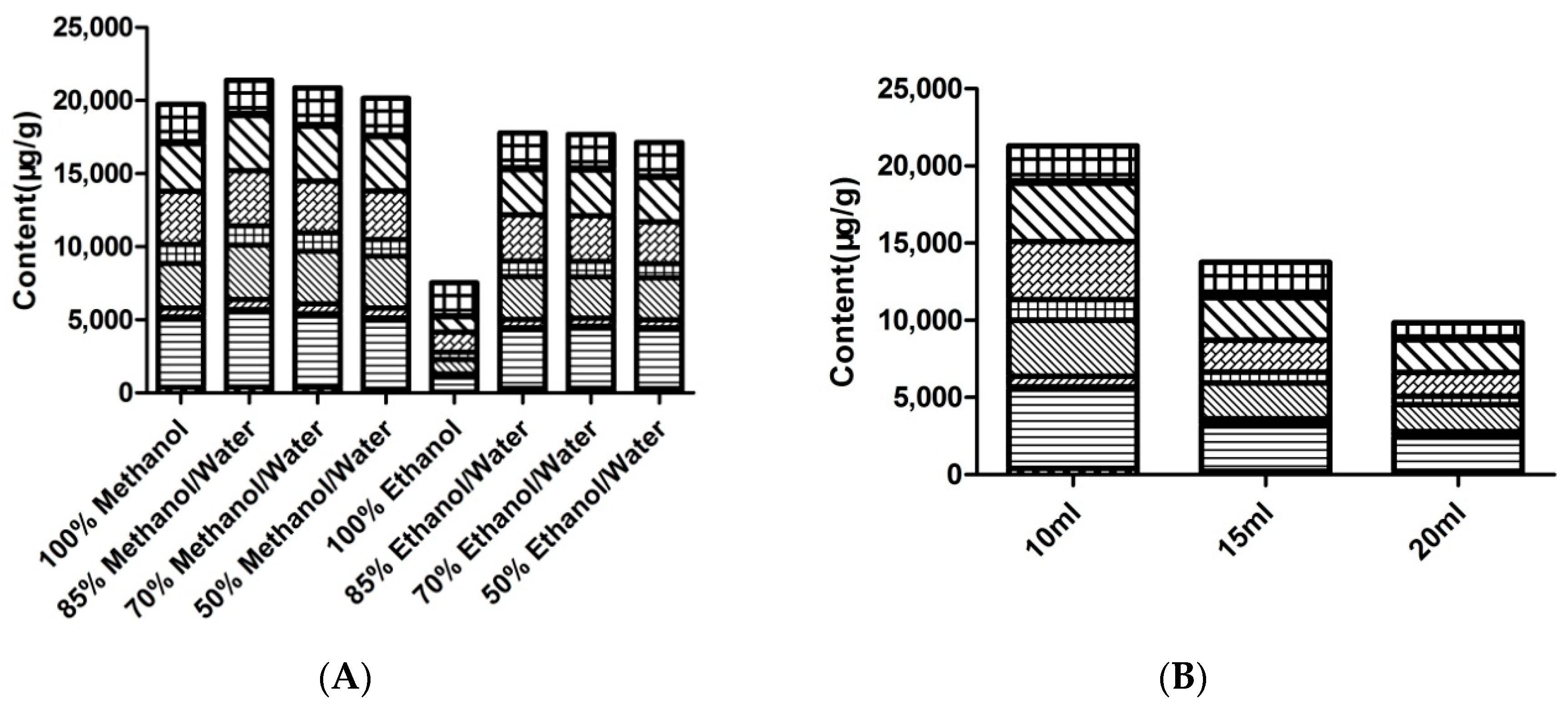
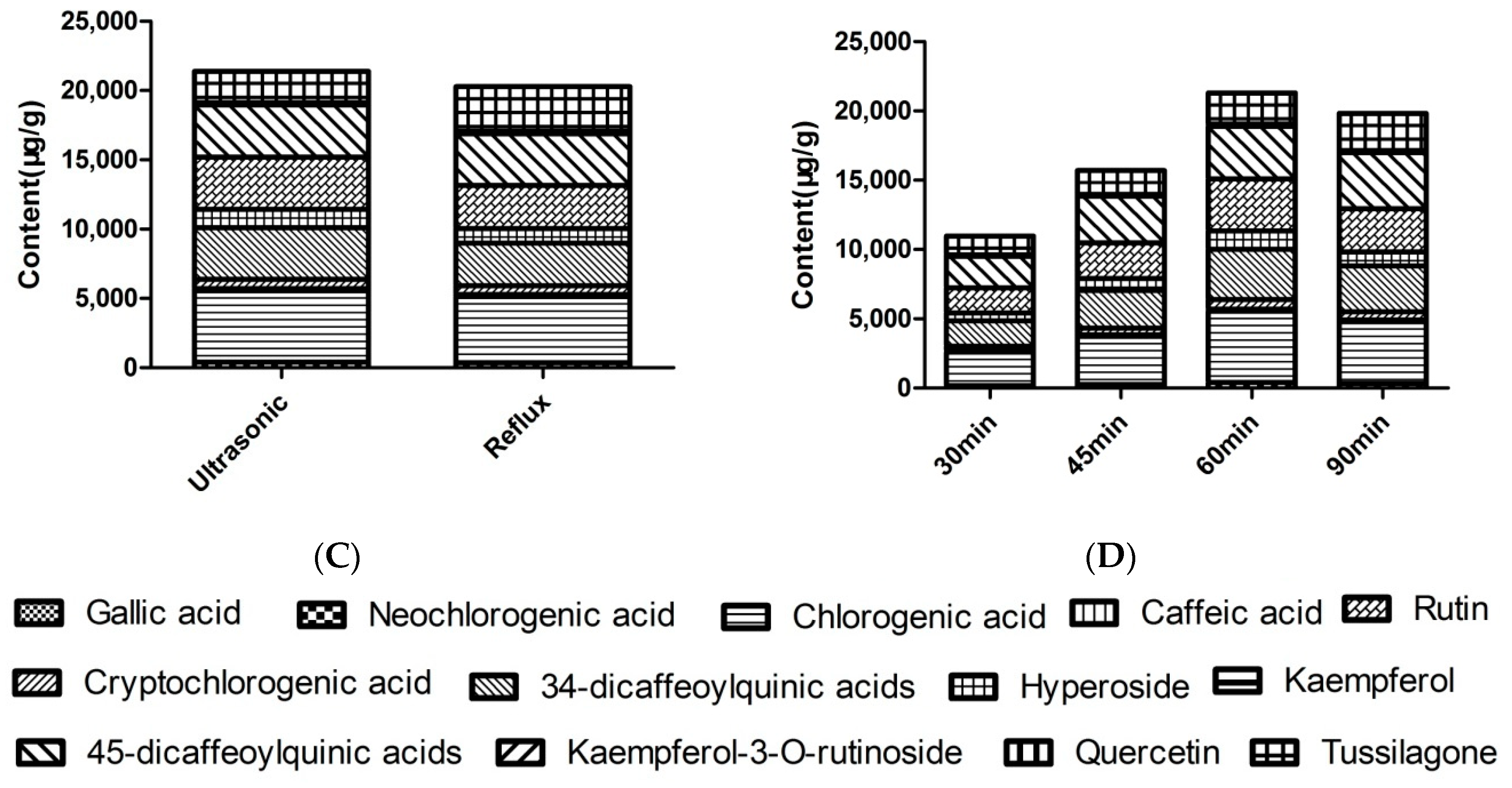
2.1. Optimization of Extraction Conditions
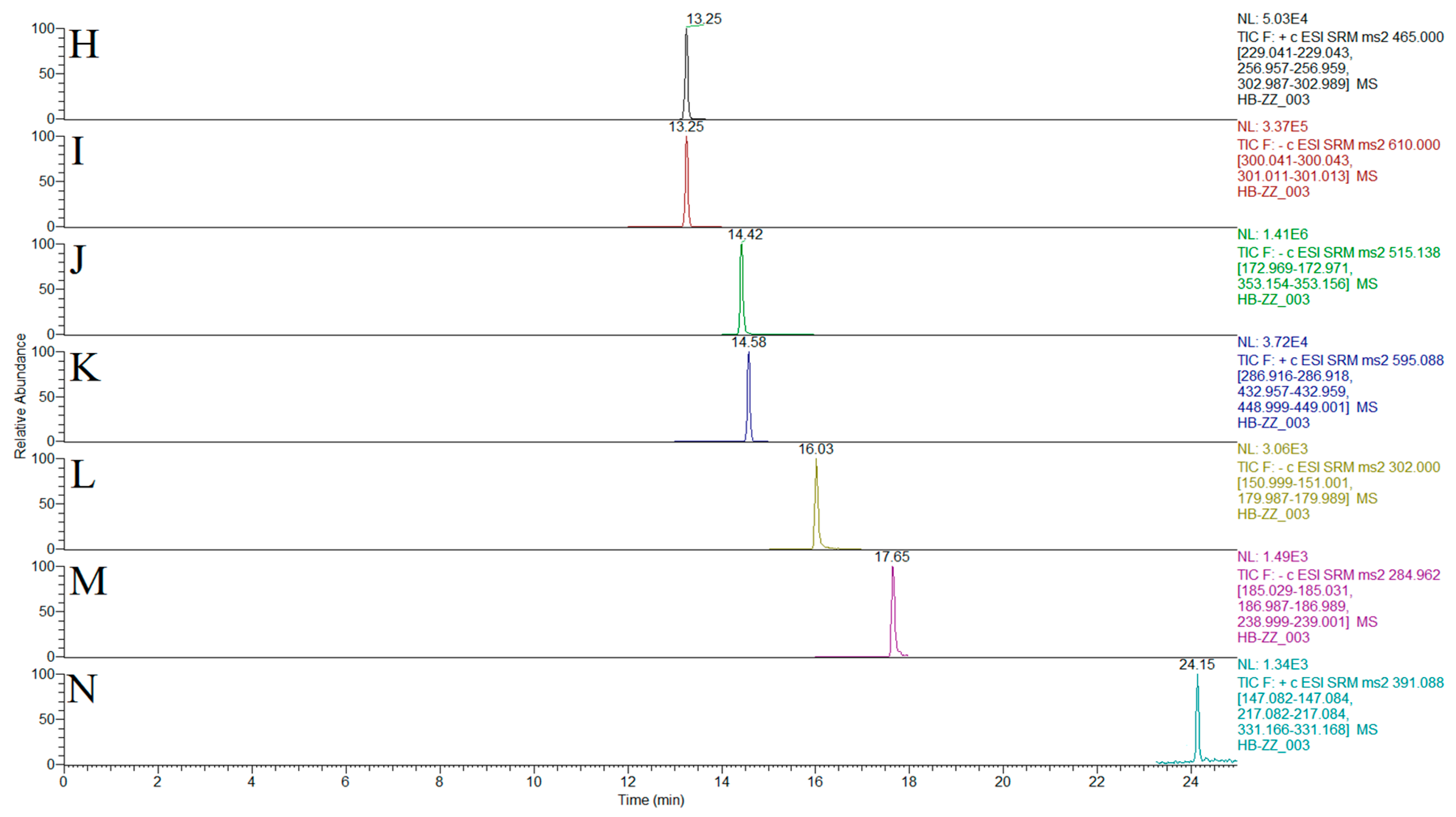
2.2. Optimization of Chromatographic and Mass Spectrometric Conditions
2.3. Method Validation
2.4. Quantitative Analysis
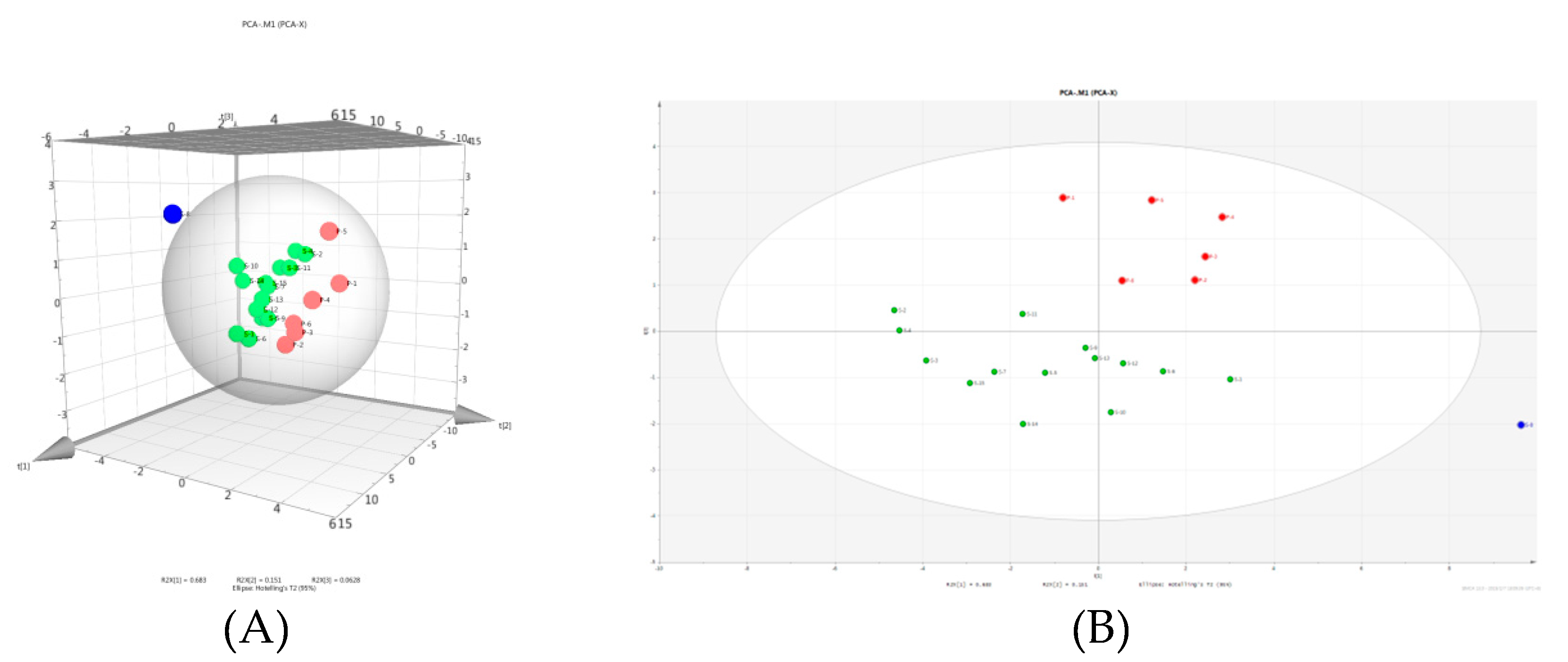
2.5. Chemometrics Analysis
2.5.1. Principal Component Analysis
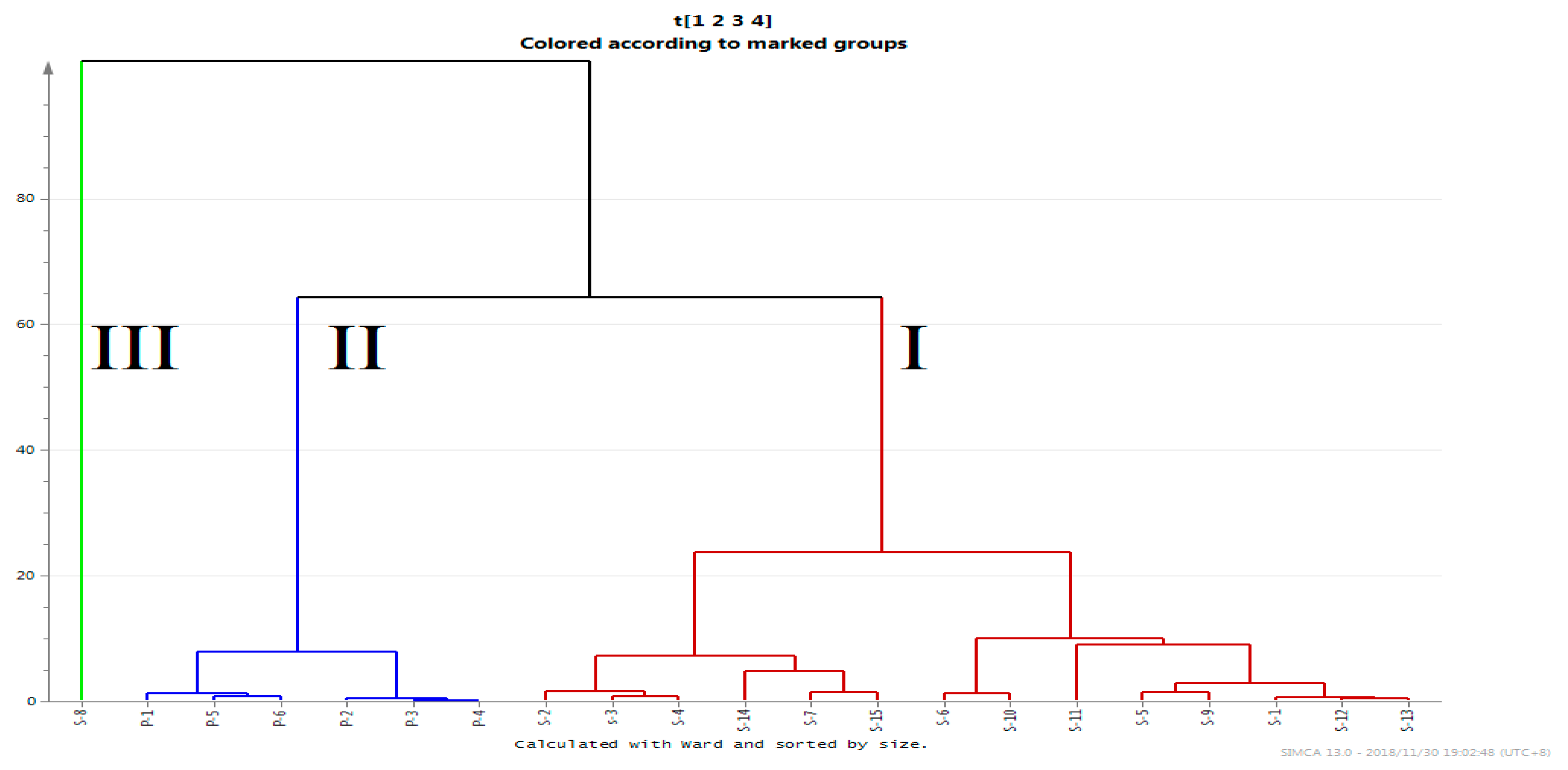
2.5.2. Hierarchical Clustering Analysis
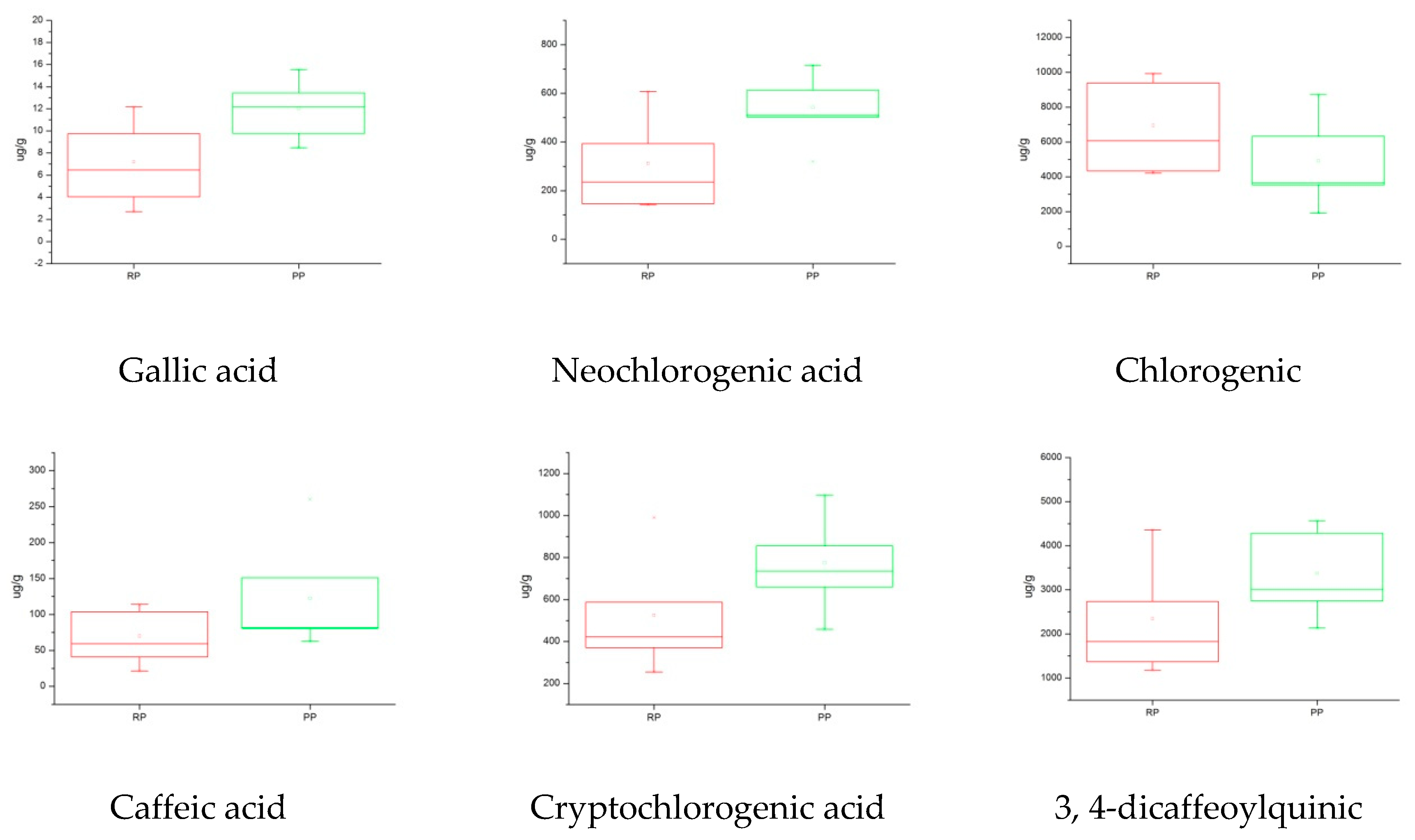
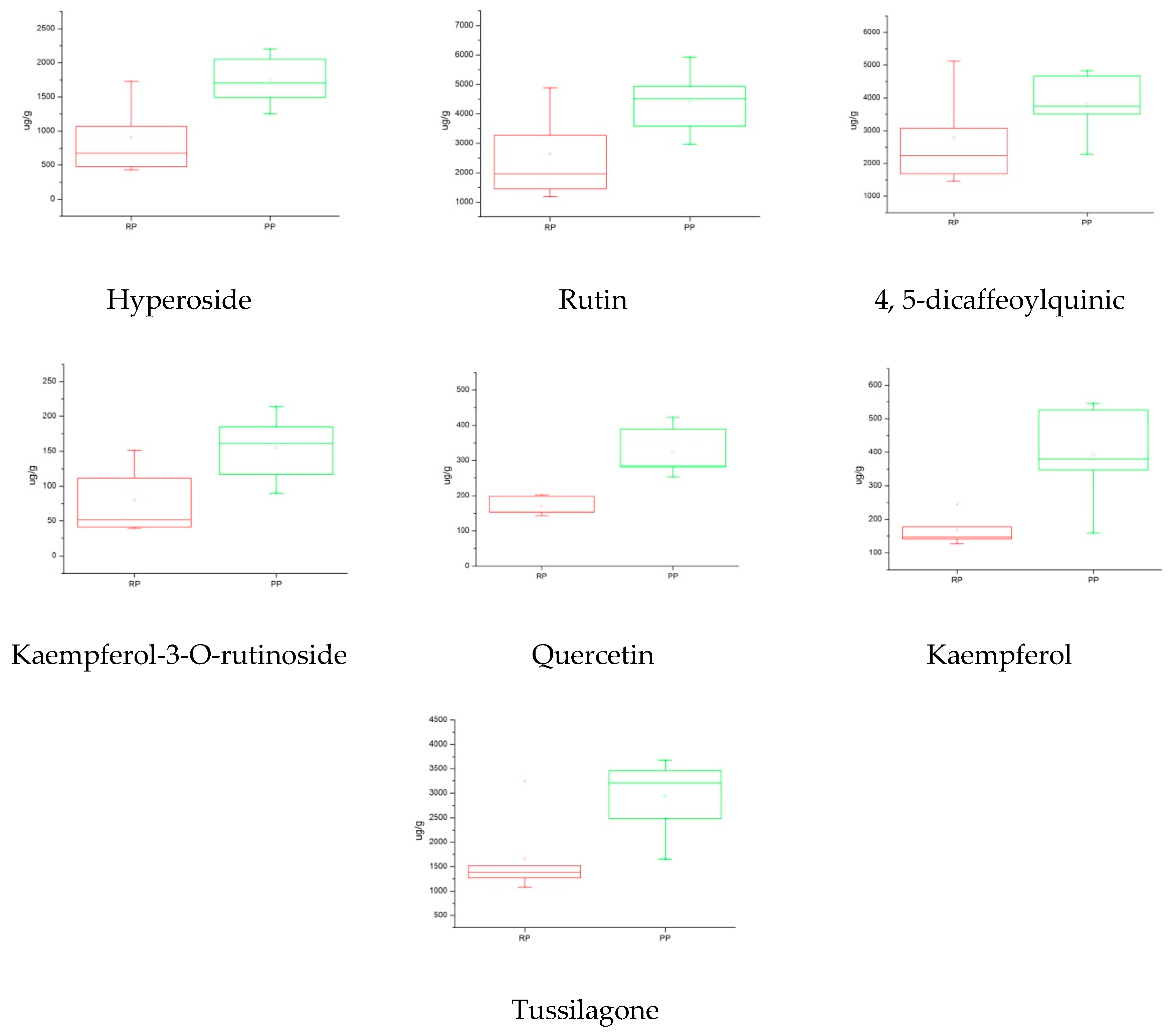
2.5.3. Box Plot Analysis
3. Materials and Methods
3.1. Chemicals, Reagents, and Materials
3.2. Chromatographic and Mass Spectrometric Conditions
3.3. Preparation of Sample Solutions
3.4. Preparation of Standard Solutions
3.5. Method Validation
3.6. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, F.; Wang, B.; Wang, L.; Xiong, Z.Y.; Gao, W.; Li, P.; Li, H.J. Discovery of discriminatory quality control markers for Chinese herbal medicines and related processed products by combination of chromatographic analysis and chemometrics methods: Radix Scutellariae as a case study. J. Pharm. Biomed. Anal. 2017, 138, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Liu, S.; Chen, W. Study of the effect of different processing methods on the content of active ingredients in cortex moutan and its clinical efficacy. Chin. J. Clin. Rat. Drug Use 2017, 10, 15–16. [Google Scholar]
- Cai, B.C.; Qin, K.M.; Wu, H.; Cai, H.; Lu, T.L.; Zhang, X.D. Chemical Mechanism During Chinese Medicine Processing. Prog.Chem. 2012, 77, 637–649. [Google Scholar] [CrossRef]
- Liu, C.X.; Cheng, Y.Y.; Guo, D.A.; Zhang, T.J.; Li, Y.Z.; Hou, W.B.; Huang, L.Q.; Xu, H.Y. A New Concept on Quality Marker for Quality Assessment and Process Control of Chinese Medicines. Chin. Herb. Med. 2017, 9, 3–13. [Google Scholar] [CrossRef]
- Hou, J.J.; Cao, C.M.; Xu, Y.W.; Yao, S.; Cai, L.Y.; Long, H.L.; Bi, Q.R.; Zhen, Y.Y.; Wu, W.Y.; Guo, D.A. Exploring lipid markers of the quality of coix seeds with different geographical origins using supercritical fluid chromatography mass spectrometry and chemometrics. Phytomedicine 2018, 45, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chinese Pharmacopoeia Commission. Pharmacopoeia of People’s Republic of China; Chemical Industry Press: Beijing, China, 2015. [Google Scholar]
- Ding, M.; Li, Z.; Yu, X.A.; Zhang, D.; Li, J.; Wang, H.; He, J.; Gao, X.M.; Chang, Y.X. A network pharmacology-integrated metabolomics strategy for clarifying the difference between effective compounds of raw and processed Farfarae flos by ultra high-performance liquid chromatography-quadrupole-time of flight mass spectrometry. J. Pharm. Bio. Anal. 2018, 156, 349–357. [Google Scholar] [CrossRef]
- Wang, D.; Wang, S.; Chen, X.; Xu, X.; Zhu, J.; Nie, L.; Long, X. Antitussive, expectorant and anti-inflammatory activities of four alkaloids isolated from Bulbus of Fritillaria wabuensis. J. Ethnopharmacol. 2012, 139, 189–193. [Google Scholar] [CrossRef]
- Qu, H.; Yang, W.; Li, J. Structural characterization of a polysaccharide from the flower buds of Tussilago farfara, and its effect on proliferation and apoptosis of A549 human non-small lung cancer cell line. Int. J. Biol. Macromol. 2018, 113, 849–858. [Google Scholar] [CrossRef]
- Lee, J.; Song, K.; HuH, E.; Oh, M.S.; Kim, Y.S. Neuroprotection against 6-OHDA toxicity in PC12 cells and mice through the Nrf2 pathway by a sesquiterpenoid fromTussilago farfara. Redox Biol. 2018, 18, 6–15. [Google Scholar] [CrossRef]
- Liu, X.H.; Gao, C.; Ding, T.L.; Shen, M.H. Experimental study on the effect of three bioactive components in Tussilago farfara L leaf on immune regulation of mice. J. Light Ind. 2018, 33, 35–41. [Google Scholar]
- Liu, K.Y.; Zhang, T.J.; Gao, W.Y.; Chen, H.X.; Zhang, Y.N. Phytochemical and pharmacological research progress in Tussilago farfara. Zhongguo Zhong Yao Za Zhi 2006, 31, 1837–1841. [Google Scholar] [PubMed]
- Hwangbo, C.; Lee, H.S.; Park, J.; Choe, J.; Lee, J.H. The anti-inflammatory effect of tussilagone, from Tussilago farfara, is mediated by the induction of heme oxygenase-1 in murine macrophages. Int. Immunopharmacol. 2009, 9, 1578–1584. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Xing, J.; Lei, Z.H.; Guo, D. Current status in studies on quality evaluation methods of Farfarae Flos. Chin. Tradit. Herbal Drugs 2017, 48, 4578–4583. [Google Scholar]
- Liu, C.H.; Wang, A.L.; Li, Y.Q.; Li, K.; Jia, B.X. Determination of Antioxidation of Polysaccharides in Tussilago Farfara. Chin. J. Mod. Appl. Pharm. 2011, 28, 886–889. [Google Scholar]
- Abdel, M.A.; Ezzat, S.M.; Tadros, M.G.; El-Askary, H.I. In vivo anti-inflammatory activity of caffeoylquinic acid derivatives from Solidago virgaurea in rats. Pharm. Bio. 2016, 54, 2864–2870. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.S.; Spitzer, V.; Schapoval, E.E.; Schenkel, E.P. Antiinflammatory activity of extracts and fractions from the leaves of Gochnatia polymorpha. Phytother. Res. 2015, 14, 638–640. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, S.H.; Jun, C.D.; Suk, K.; Choi, B.J.; Lim, H.; Park, S.; Lee, S.H.; Shin, H.Y.; Kim, D.K.; Shin, T.Y. Gallic acid inhibits histamine release and pro-inflammatory cytokine production in mast cells. Toxicol. Sci. 2006, 91, 123–131. [Google Scholar] [CrossRef]
- Fernandez, M.A.; Saenz, M.T.; Garcia, M.D. Anti-inflammatory activity in rats and mice of phenolic acids isolated from Scrophularia frutescens. J. Pharm. Pharmacol. 2011, 50, 1183–1186. [Google Scholar] [CrossRef]
- Ji, Y.L.; Jang, Y.W.; Kang, H.S.; Moon, H.; Sim, S.S.; Kim, C.J. Anti-inflammatory action of phenolic compounds from Gastrodia elata root. Arch. Pharm. Res. 2006, 29, 849–858. [Google Scholar]
- Xue, S.Y.; Li, Z.Y.; Zhi, H.J.; Sun, H.F.; Zhang, L.Z.; Guo, X.Q.; Qin, X.M. Metabolic fingerprinting investigation of Tussilago farfara L. by GC–MS and multivariate data analysis. Biochem. Systemat. Ecol. 2012, 41, 6–12. [Google Scholar] [CrossRef]
- Li, W.; Yang, X. Simultaneous determination of nine major constituents in Farfarae Flos by HPLC. Chin. J. Pharm. Anal. 2012, 32, 1517–1524. [Google Scholar]
- Li, J.; Li, J.; Jia, J.P.; Zhang, J.J.; Yan, Y.; Gao, X.X.; Qin, X.M.; Li, Z.Y. Comparison of flower buds and leaves of Tussilago farfara L. by UHPLC-Q Extractive Hybrid Quadrupole Orbitrap MS. Acta Pharm. Sin. B 2018. [Google Scholar]
- Zhu, Z.; Tao, W.; Li, J.; Guo, S.; Qian, D.; Shang, E.; Su, S.; Duan, J.A. Rapid determination of flavonoids in licorice and comparison of three licorice species. J. Sep. Sci. 2016, 39, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Jiang, H.; Yan, M.; Xing, X.; Guo, X.; Yang, B.; Wang, Q.; Kuang, H. UHPLC-MS/MS Quantification Combined with Chemometrics for Comparative Analysis of Different Batches of Raw, Wine-Processed, and Salt-Processed Radix Achyranthis Bidentatae. Molecules 2018, 23, 758. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Duan, L.; Liu, K.; Liu, E.H.; Li, P. Chemical comparison of Tripterygium wilfordii and Tripterygium hypoglaucum based on quantitative analysis and chemometrics methods. J. Pharm. Biomed. Anal. 2014, 95, 220–228. [Google Scholar] [CrossRef]
- Zhao, X.; Kong, W.; Zhou, Y.; Wei, J.; Yang, M. Evaluation and quantitative analysis of 11 compounds in Morinda officinalis using ultra high performance liquid chromatography and photodiode array detection coupled with chemometrics. J. Sep. Sci. 2017, 40, 3996–4003. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.S.; Wan, J.Z.; Ding, W.J.; Wu, X.F.; Xie, Z.Y. Multi-responses extraction optimization combined with high-performance liquid chromatography-diode array detection–electrospray ionization-tandem mass spectrometry and chemometrics techniques for the fingerprint analysis of Aloe barbadensis Miller. J. Pharm. Biomed. Anal. 2015, 107, 131–140. [Google Scholar] [CrossRef]
- Williamson, D.F.; Parker, R.A.; Kendrick, J.S. The box plot: a simple visual method to interpret data. Ann. Intern. Med. 1989, 110, 916–921. [Google Scholar] [CrossRef]
- Datar, P.A.; Waghmare, R.U. Development and validation of an analytical method for the stability of duloxetine hydrochloride. J. Taibah Univ. Sci. 2014, 8, 357–363. [Google Scholar] [CrossRef]
Sample Availability: Not available. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
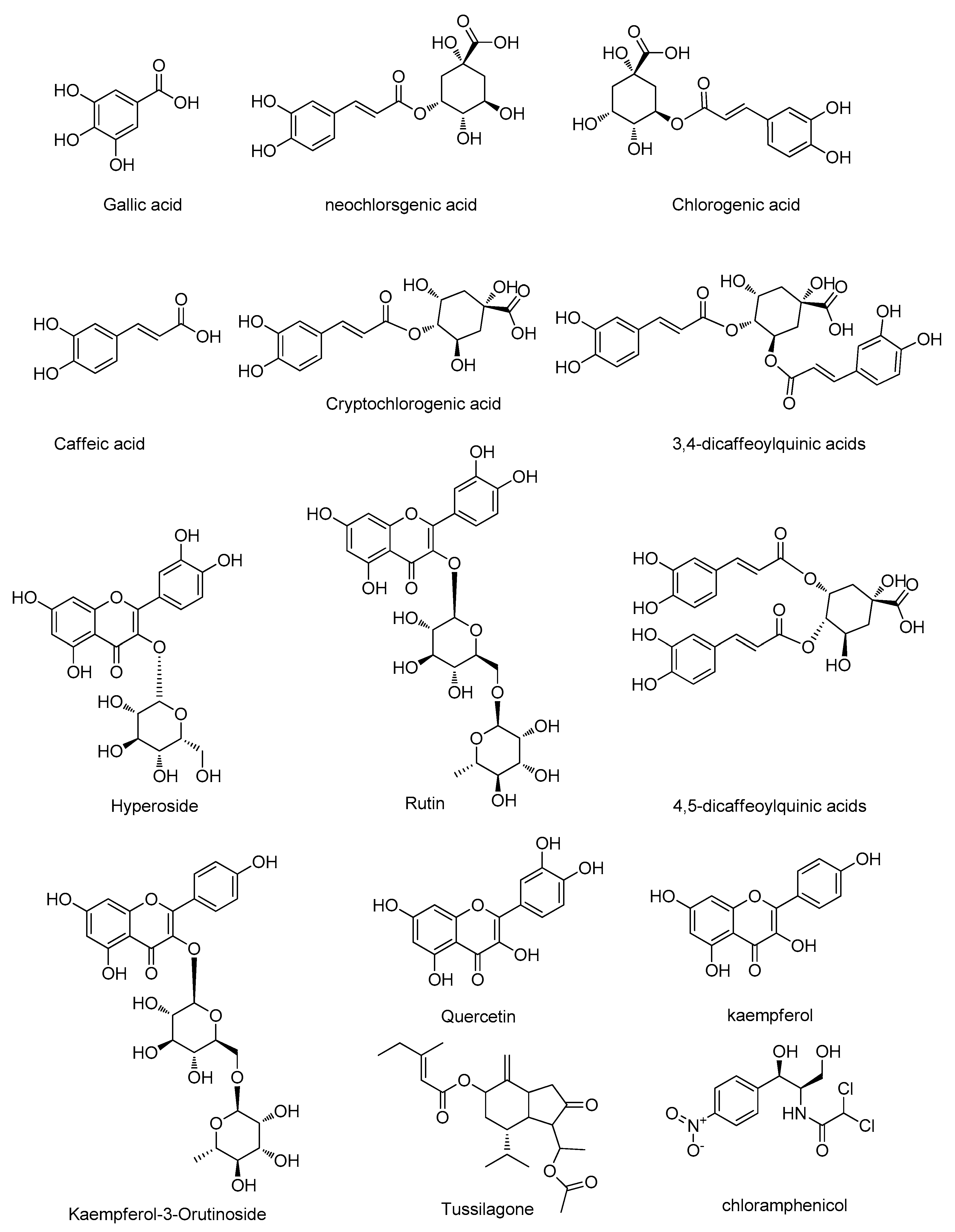
| Compound | Structure | Polarity | Retention Time (min) | Precursor (m/z) | Product (m/z) | Collision Energy (V) |
|---|---|---|---|---|---|---|
| Gallic acid | C7H6O5 | Negative | 1.48 | 169 | 124 | 14 |
| Neochlorogenic acid | C16H18O9 | Negative | 3.00 | 353 | 178 | 18 |
| Chlorogenic acid | C16H18O9 | Negative | 5.73 | 353 | 191 | 16 |
| Caffeic acid | C9H8O4 | Negative | 6.23 | 179 | 135 | 15 |
| Cryptochlorogenic acid | C16H18O9 | Negative | 6.26 | 353 | 173 | 16 |
| 3,4-Dicaffeoylquinic acids | C25H24O12 | Negative | 12.92 | 515 | 353 | 18 |
| Hyperoside | C21H20O12 | Positive | 13.25 | 465 | 302 | 13 |
| Rutin | C27H30O16 | Negative | 13.25 | 609 | 300 | 36 |
| 4,5-Dicaffeoylquinic acids | C25H24O12 | Negative | 14.42 | 515 | 191 | 30 |
| Kaempferol-3-O-rutinoside | C27H30O15 | Positive | 14.58 | 595 | 287 | 20 |
| Quercetin | C15H10O7 | Negative | 16.03 | 301 | 151 | 21 |
| Kaempferol | C15H10O6 | Negative | 17.65 | 285 | 186 | 29 |
| Tussilagone | C23H34O5 | Positive | 24.15 | 391 | 331 | 10 |
| Compound | Calibration Curves | R2 | Linear Range | LOD | LOQ | Precision | (RSD, %) | Stability |
|---|---|---|---|---|---|---|---|---|
| (μg/mL) | (μg/mL) | (μg/mL) | Intra-Day (n = 6) | Inter-Day (n = 3) | (RSD, %) | |||
| Gallic acid | y = 16.698x + 0.1611 | 0.9996 | 0.0101–1.0080 | 0.0018 | 0.0060 | 1.98 | 1.47 | 1.89 |
| Neochlorogenic acid | y = 31.38x + 0.108 | 0.9997 | 0.0105–8.3433 | 0.0020 | 0.0067 | 1.44 | 1.77 | 1.96 |
| Chlorogenic acid | y = 15.725x − 0.0613 | 0.9995 | 0.0119–67.9039 | 0.0017 | 0.0056 | 1.24 | 1.64 | 1.71 |
| Caffeic acid | y = 65.473x − 0.2765 | 0.9999 | 0.0111–6.6167 | 0.0017 | 0.0057 | 1.06 | 0.63 | 1.32 |
| Cryptochlorogenic acid | Y = 39.925x + 0.2561 | 0.9991 | 0.0102–9.9099 | 0.0012 | 0.0038 | 2.00 | 0.56 | 1.86 |
| 3,4-Dicaffeoylquinic acids | y = 28.66x − 0.1627 | 0.9993 | 0.0230–38.6226 | 0.0044 | 0.0146 | 1.50 | 0.94 | 1.94 |
| Hyperoside | y = 36.466x + 0.692 | 0.9994 | 0.0580–21.9959 | 0.0095 | 0.0316 | 1.19 | 1.82 | 1.90 |
| Rutin | y = 14.183x − 0.0769 | 0.9994 | 0.0218–44.6006 | 0.0045 | 0.0149 | 1.66 | 1.79 | 1.59 |
| 4,5-Dicaffeoylquinic acids | y = 46.665x − 0.4 | 0.9992 | 0.0115–45.9080 | 0.0025 | 0.0083 | 1.81 | 1.99 | 1.95 |
| Kaempferol-3-O-rutinoside | y = 46.092 − 0.1188 | 0.9999 | 0.0123–2.4550 | 0.0014 | 0.0049 | 1.94 | 0.26 | 1.97 |
| Quercetin | y = 1.3638x + 0.1916 | 0.9996 | 0.0131–2.6147 | 0.0036 | 0.0120 | 1.10 | 1.15 | 1.63 |
| Kaempferol | y = 2.2674x + 0.0698 | 0.9994 | 0.0194–0.9681 | 0.0030 | 0.0099 | 1.54 | 0.64 | 1.27 |
| Tussilagone | y = 5.0377x + 0.2941 | 0.9994 | 0.0202–18.1436 | 0.0043 | 0.0145 | 1.96 | 1.05 | 0.75 |
| Compound | Un-spiked (μg/mL) | Spiked (μg/mL) | Found (μg/mL) | Recovery (%) | RSD (%, n = 3) |
|---|---|---|---|---|---|
| Gallic acid | 0.0494 | 0.0247 0.0494 0.0707 | 0.0745 0.0965 0.1205 | 101.89 95.45 100.65 | 2.04 4.43 2.37 |
| Neochlorogenic acid | 1.0509 | 0.5252 1.0504 1.4630 | 1.5896 2.0834 2.5246 | 102.57 98.29 100.73 | 1.57 1.89 1.07 |
| Chlorogenic acid | 15.4059 | 7.7050 15.4100 23.0000 | 22.6848 30.2700 38.3895 | 94.47 96.46 99.93 | 1.39 4.18 1.33 |
| Caffeic acid | 0.4516 | 0.2261 0.4522 0.6630 | 0.6767 0.9065 1.1043 | 99.53 100.59 98.44 | 0.92 1.01 0.41 |
| Cryptochlorogenic acid | 1.8639 | 0.9622 1.8644 2.8420 | 2.8472 3.7046 4.6697 | 102.20 98.73 98.73 | 2.23 0.93 1.55 |
| 3,4-Dicaffeoylquinic acids | 12.0005 | 5.9993 11.9986 17.9350 | 17.8274 24.0563 29.7821 | 97.13 100.48 99.1448 | 1.26 1.45 2.82 |
| Hyperoside | 3.7520 | 1.8764 3.7528 5.5680 | 5.6739 7.5073 9.4219 | 102.42 100.07 101.83 | 1.01 2.04 1.30 |
| Rutin | 9.8986 | 4.9512 9.9024 14.8240 | 14.8921 19.6667 24.9019 | 100.85 98.64 101.21 | 1.82 2.20 1.47 |
| 4,5-Dicaffeoylquinic acids | 12.5749 | 6.2909 12.5818 18.9200 | 18.6837 25.3422 31.3067 | 97.11 101.47 99.01 | 1.19 1.49 2.06 |
| Kaempferol-3-O-rutinoside | 0.2668 | 0.1334 0.2668 0.3690 | 0.4056 0.5330 0.6251 | 104.06 99.76 97.08 | 0.16 4.21 1.06 |
| Quercetin | 0.8109 | 0.4050 0.8099 1.1790 | 1.2234 1.5868 1.9852 | 101.84 95.79 99.60 | 1.59 2.45 1.43 |
| Kaempferol | 0.0202 | 0.0101 0.0202 0.0291 | 0.0304 0.0400 0.0487 | 100.85 98.10 98.60 | 1.54 2.84 1.86 |
| Tussilagone | 4.5558 | 2.2802 4.5604 6.8680 | 6.8484 8.9890 11.4616 | 100.55 97.21 100.55 | 0.53 1.61 0.59 |
| NO. | Type | Source | Gallic Acid | Neochlorogenic Acid | Chlorogenic Acid | Caffeic Acid | Cryptochlorogenic Acid | 3,4-Dicaffeoylquinic acids | Hyperoside | Rutin | 4,5-Dicaffeoylquinic Acids | Kaempferol-3-O-rutinoside | Quercetin | Kaempferol | Tussilagone |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 | Raw | Anhui | 8.180.09 | 607.732.03 | 47.67 | 103.590.29 | 990.743.78 | 4360.7212.36 | 1726.837.82 | 4887.4628.20 | 5130.6820.70 | 151.410.48 | 198.530.99 | 16.240.07 | 3243.537.63 |
| S2 | Raw | Gansu | 2.690.07 | 234.590.47 | 4331.7722.01 | 59.280.12 | 370.290.56 | 1828.1728.98 | 672.191.2 | 1951.9519.89 | 2234.6123.73 | 51.210.68 | 153.222.76 | 17.750.08 | 1381.5912.14 |
| S3 | Raw | Gansu | 4.020.02 | 393.210.35 | 93904021.84 | 80.820.50 | 586.171.01 | 2734.772.88 | 1052.1514.79 | 3097.385.55 | 3070.067.04 | 86.871.12 | 173.012.76 | 14.710.02 | 1517.7814.60 |
| S4 | Raw | Gansu | 12.170.07 | 347.681.58 | 7793.0029.02 | 113.750.86 | 531.302.49 | 2600.5316.70 | 1066.5310.59 | 3267.1224.47 | 3075.6422.59 | 111.751.17 | 202.730.51 | 24.510.08 | 1489.647.65 |
| S5 | Raw | Hebei | 6.460.02 | 14.6.030.59 | 4203.9210.61 | 40.910.34 | 421.931.91 | 1370.877.38 | 476.983.49 | 1452.3111.23 | 1684.469.58 | 39.240.32 | 153.650.85 | 12.650.06 | 1268.356.72 |
| S6 | Raw | Jiangxi | 11.000.04 | 141.880.09 | 6068.6857.19 | 21.210.25 | 254.160.37 | 1178.5516.95 | 430.490.19 | 1184.7516.58 | 1463.334.88 | 41.320.36 | 143.531.20 | 14.170.02 | 1075.731.04 |
| S7 | Raw | Anhui | 7.920.01 | 585.792.54 | 9902.6326.02 | 214.140.96 | 1089.7224.08 | 4538.7732.61 | 1792.328.27 | 5048.7627.07 | 5714.8032.95 | 156.650.82 | 188.980.55 | 118.060.02 | 2813.9128.73 |
| S8 | Raw | Gansu | 10.950.08 | 958.488.36 | 13468.9727.20 | 1152.0010.30 | 1936.9918.15 | 7556.7745.95 | 4344.4743.47 | 8996.2954.20 | 9259.6425.92 | 443.816.09 | 393.015.18 | 10.000.02 | 3439.2813.47 |
| S9 | Raw | Hebei | 7.380.15 | 337.701.34 | 5948.2818.53 | 74.360.30 | 579.032.48 | 2539.196.82 | 839.864.42 | 2369.2018.98 | 2869.918.56 | 63.750.24 | 146.361.67 | 12040.06 | 158.439.12 |
| S10 | Raw | Henan | 10.730.01 | 728.211.60 | 12312.2831.65 | 180.961.81 | 1236.64 ± 4.92 | 5529.5120.33 | 2167.78.13 | 6110.418.52 | 5963.3622.59 | 206.870.62 | 209.440.10 | 37.890.07 | 2377.3613.95 |
| S11 | Raw | Hubei | 8.650.08 | 927.038.28 | 6292.1852.36 | 359.613.77 | 1141.20511.69 | 4345.7438.25 | 1056.355.68 | 2605.2715.17 | 5042.5515.07 | 84.860.75 | 242.770.99 | 28.100.18 | 831.422912.57 |
| S12 | Raw | Hunan | 8.380.06 | 543.961.60 | 10008.9146.12 | 170.360.73 | 851.022.67 | 38856012.41 | 1652.485.10 | 4188.7716.42 | 4542.4715.07 | 155.700.92 | 204.170.81 | 30.720.09 | 2419.2812.64 |
| S13 | Raw | Hunan | 9.540.11 | 487.714.11 | 8015.0348.09 | 134.590.99 | 797.606.38 | 3784.6128.35 | 1651.994.16 | 4202.3421.58 | 4505.7015.86 | 140.970.18 | 176.031.53 | 26.380.07 | 1791.418.66 |
| S14 | Raw | Shanxi | 3.830.04 | 425.464.72 | 8596.9276.78 | 358.542.53 | 724.243.67 | 3335.8820.81 | 1715.318.05 | 1611.002.85 | 3676.0323.38 | 36.510.07 | 176.021.77 | 11.530.01 | 1121.6911.36 |
| S15 | Raw | Sichuan | 5.840.04 | 381.734.64 | 8730.8561.76 | 247.743.58 | 479.233.98 | 2237.7715.14 | 706.658.75 | 1536.9910.82 | 2400.7714.58 | 41.360.46 | 166.451.12 | 10.180.01 | 1487.189.86 |
| P1 | Processed | Anhui | 8.470.02 | 715.192.55 | 8724.3726.62 | 62.940.25 | 1096.919.57 | 456.1.2747.51 | 2202.9417.89 | 5930.7024.53 | 4832.1049.77 | 213.802.35 | 284.982.35 | 39.320.02 | 3462.73 |
| P2 | Processed | Gansu | 13.430.11 | 319.781.73 | 3664.3412.29 | 81.410.89 | 457.584.19 | 2135.043.20 | 1252.226.09 | 2956.1915.76 | 2273.0934.22 | 89.770.66 | 252.500.66 | 34.760.09 | 2491.757.10 |
| P3 | Processed | Gansu | 12.720.06 | 598.073.88 | 5396.6710.40 | 150.981.31 | 854.986.13 | 4283.084.08 | 2056.8915.24 | 4522.434.05 | 4664.7040.76 | 160.541.18 | 280.931.18 | 38.010.11 | 3208.264.73 |
| P4 | Processed | Gansu | 9.770.03 | 612.471.96 | 6329.3937.03 | 80.541.77 | 849.834.38 | 3512.936.72 | 1753.091.08 | 4936.3016.36 | 3825.9911.42 | 184.883.31 | 422.040.62 | 52.520.04 | 3244.0914.47 |
| P5 | Processed | Hebei | 15.530.22 | 508.904.85 | 1918.0413.18 | 260.272.88 | 659.687.37 | 3010.423.49 | 1702.0116.02 | 4520.6412.05 | 3742.6410.51 | 167.642.18 | 388.340.07 | 54.580.31 | 3668.365.39 |
| P6 | Processed | Jiangxi | 12.170.23 | 501.964.43 | 3527.0215.90 | 99.81.56 | 734.749.01 | 2746.895.45 | 1492.105.20 | 3585.4722.56 | 3504.5537.80 | 116.711.25 | 305.001.25 | 15.810.01 | 1654.007.29 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Jiang, H.; Hou, A.; Guo, X.; Man, W.; Yan, M.; Xing, X.; Yang, B.; Wang, Q.; Kuang, H. Simultaneous Determination of Thirteen Q-Markers in Raw and Processed Tussilago farfara L. by UPLC-QQQ-MS/MS Coupled with Chemometrics. Molecules 2019, 24, 598. https://doi.org/10.3390/molecules24030598
Yang L, Jiang H, Hou A, Guo X, Man W, Yan M, Xing X, Yang B, Wang Q, Kuang H. Simultaneous Determination of Thirteen Q-Markers in Raw and Processed Tussilago farfara L. by UPLC-QQQ-MS/MS Coupled with Chemometrics. Molecules. 2019; 24(3):598. https://doi.org/10.3390/molecules24030598
Chicago/Turabian StyleYang, Liu, Hai Jiang, Ajiao Hou, Xinyue Guo, Wenjing Man, Meiling Yan, Xudong Xing, Bingyou Yang, Qiuhong Wang, and Haixue Kuang. 2019. "Simultaneous Determination of Thirteen Q-Markers in Raw and Processed Tussilago farfara L. by UPLC-QQQ-MS/MS Coupled with Chemometrics" Molecules 24, no. 3: 598. https://doi.org/10.3390/molecules24030598
APA StyleYang, L., Jiang, H., Hou, A., Guo, X., Man, W., Yan, M., Xing, X., Yang, B., Wang, Q., & Kuang, H. (2019). Simultaneous Determination of Thirteen Q-Markers in Raw and Processed Tussilago farfara L. by UPLC-QQQ-MS/MS Coupled with Chemometrics. Molecules, 24(3), 598. https://doi.org/10.3390/molecules24030598