
CCR5/CXCR4 Dual Antagonism for the Improvement of HIV Infection Therapy

 ,
, 
 ,
, 
 , ,
, , 

Abstract
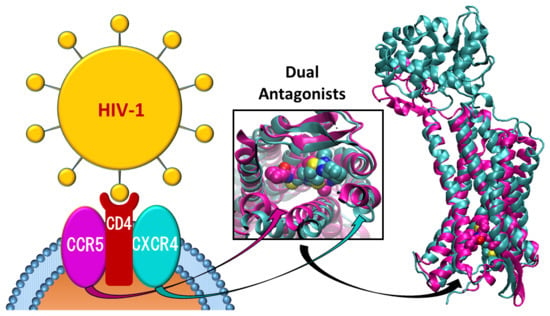
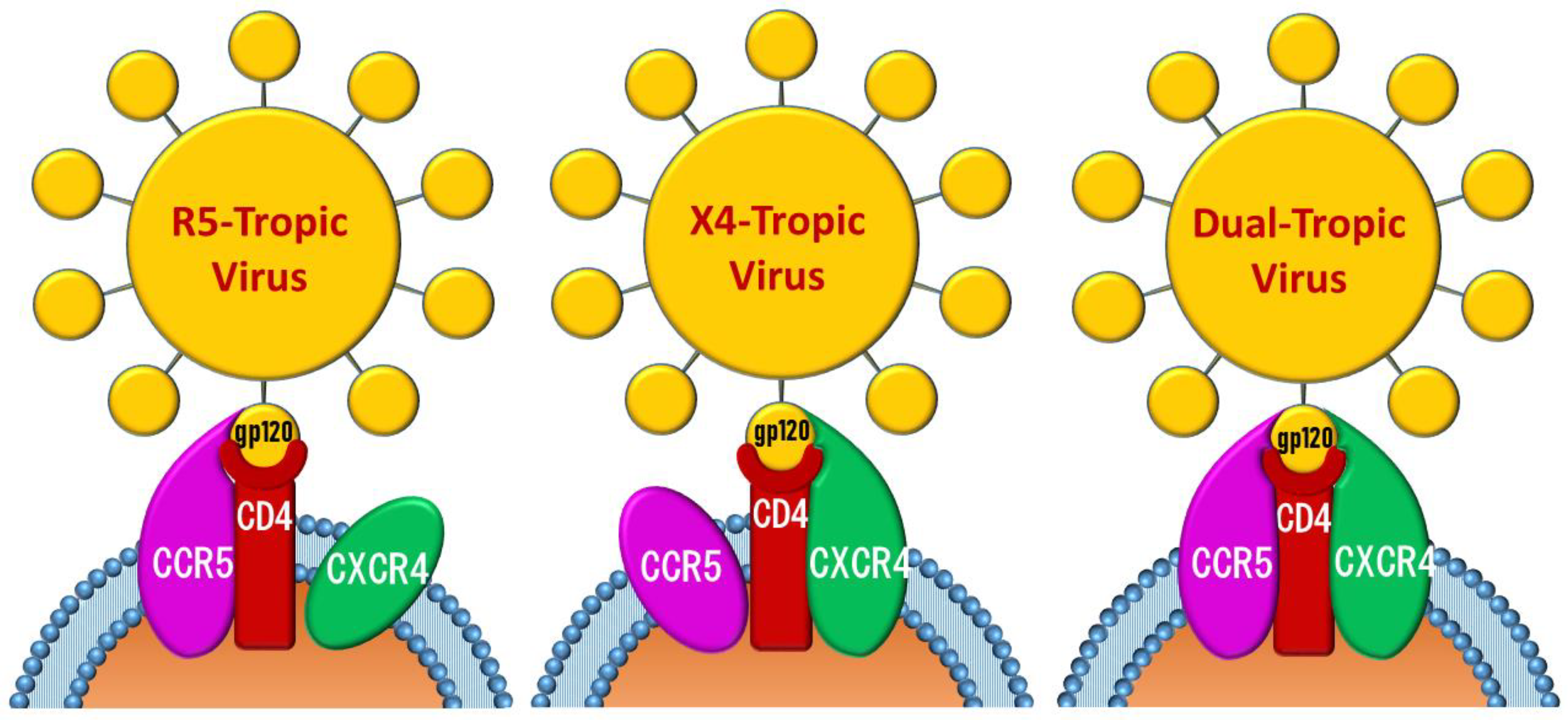
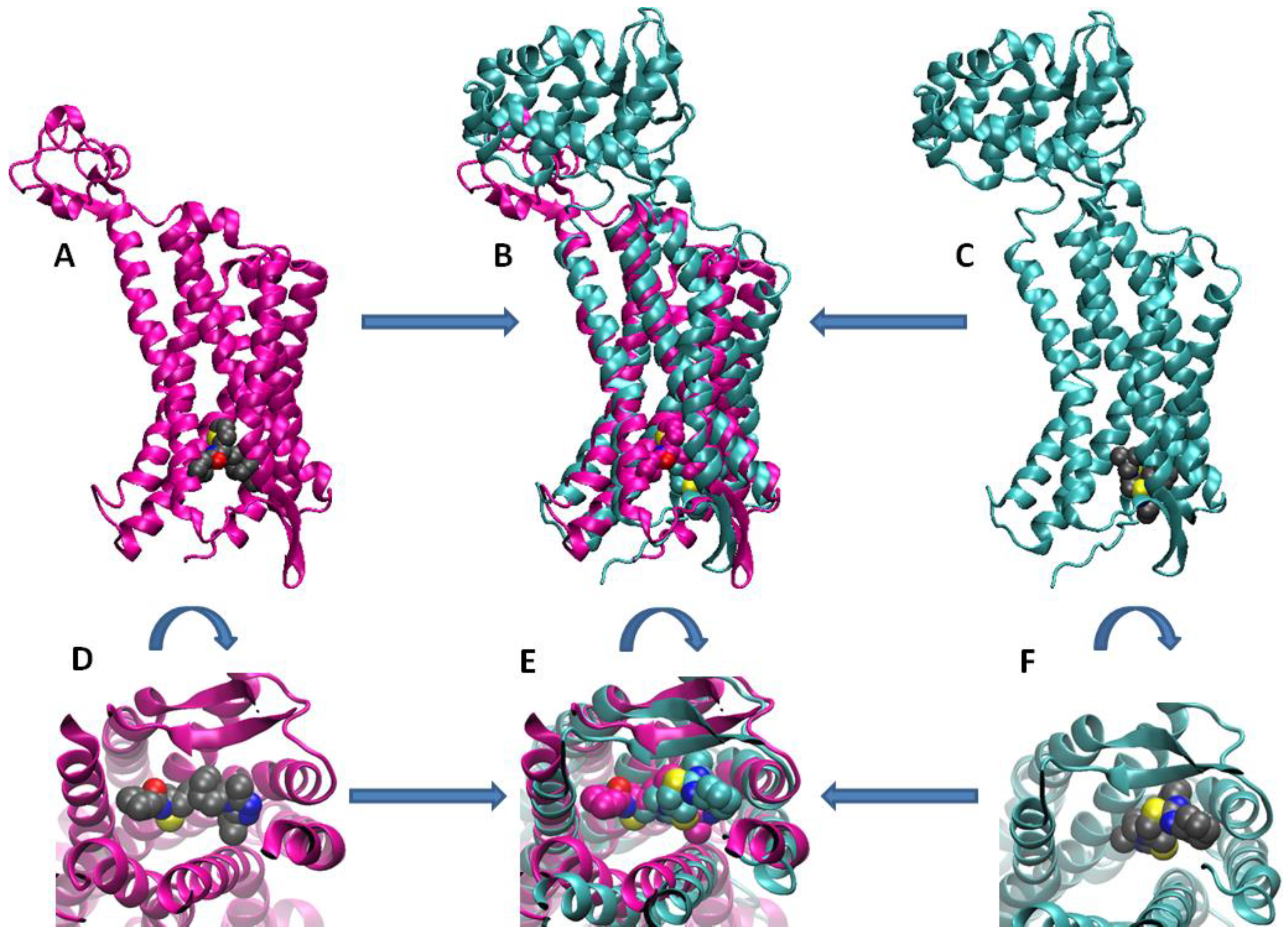
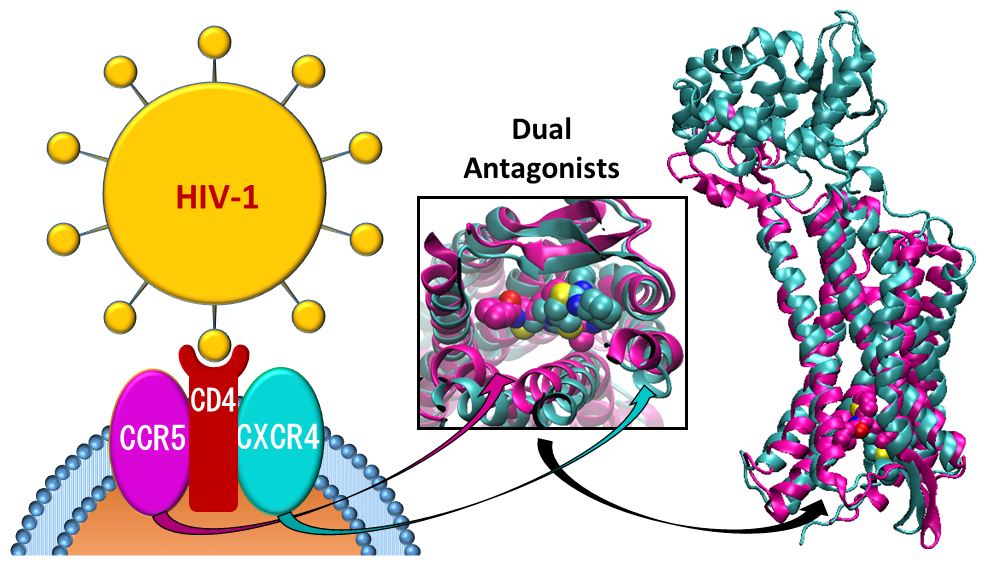{kind=link}
{kind=link}
{kind=link}
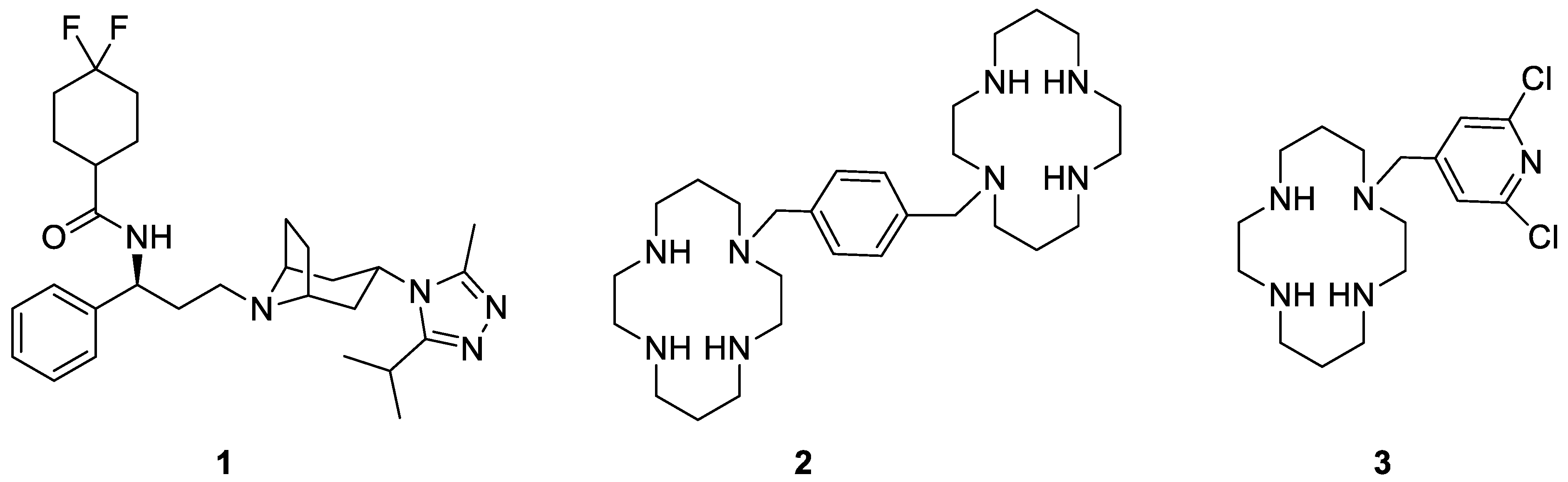{kind=link}
{kind=link}
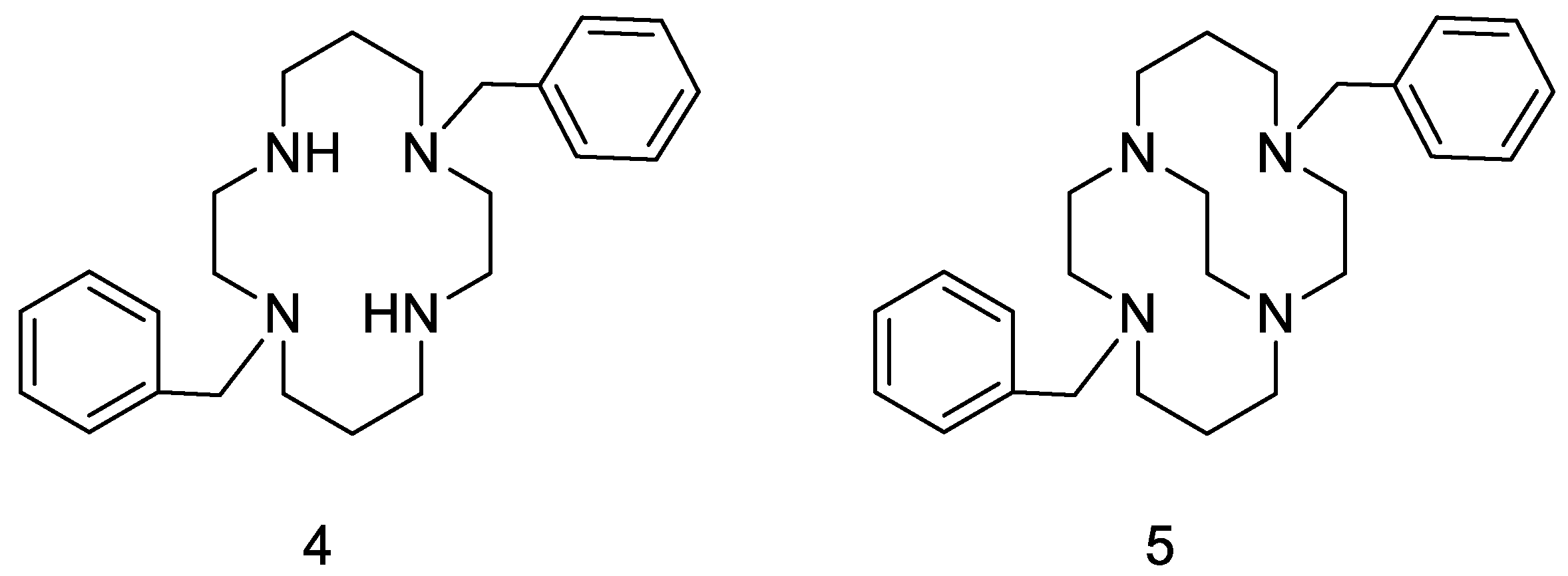
{kind=link}
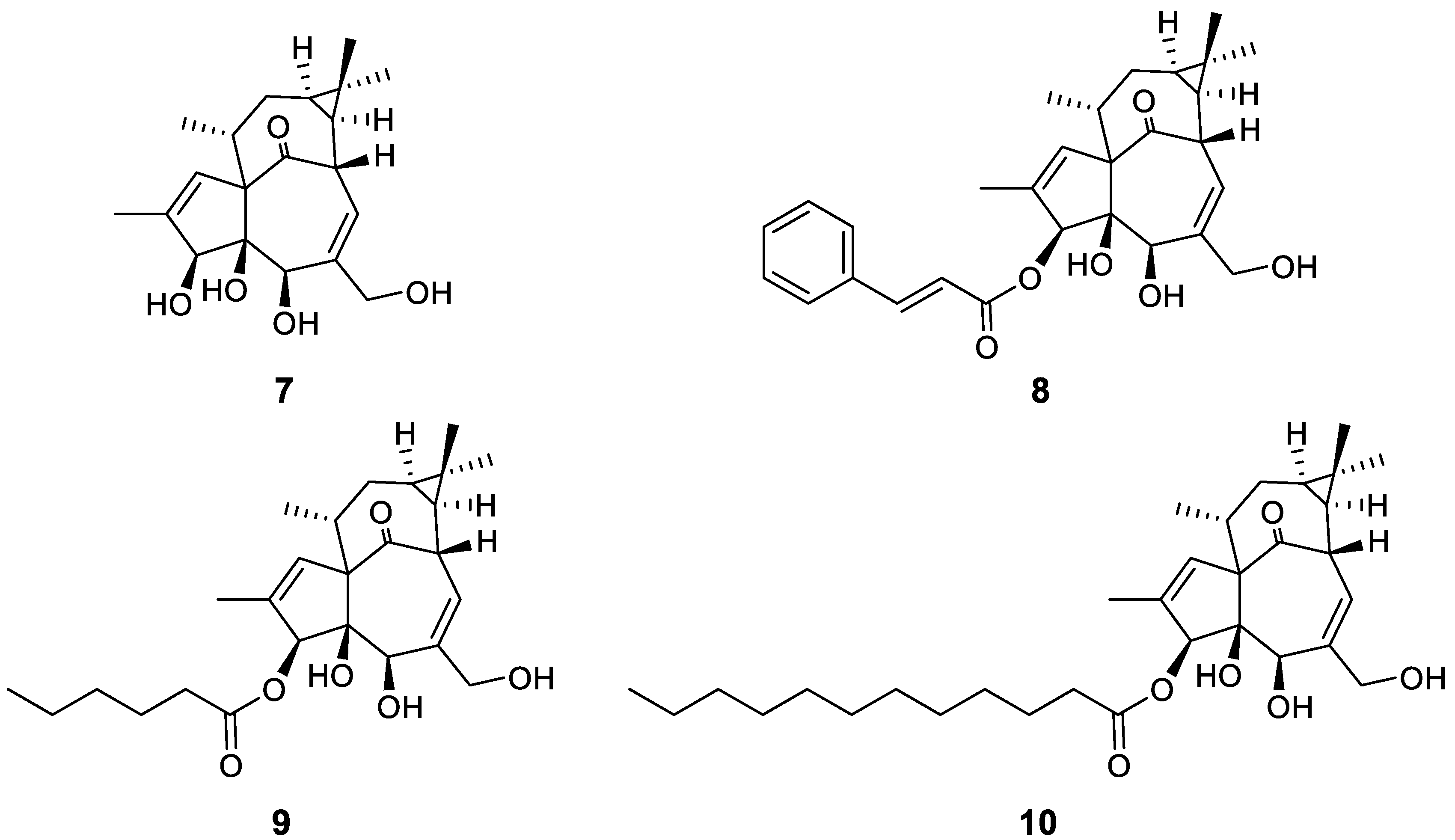{kind=link}
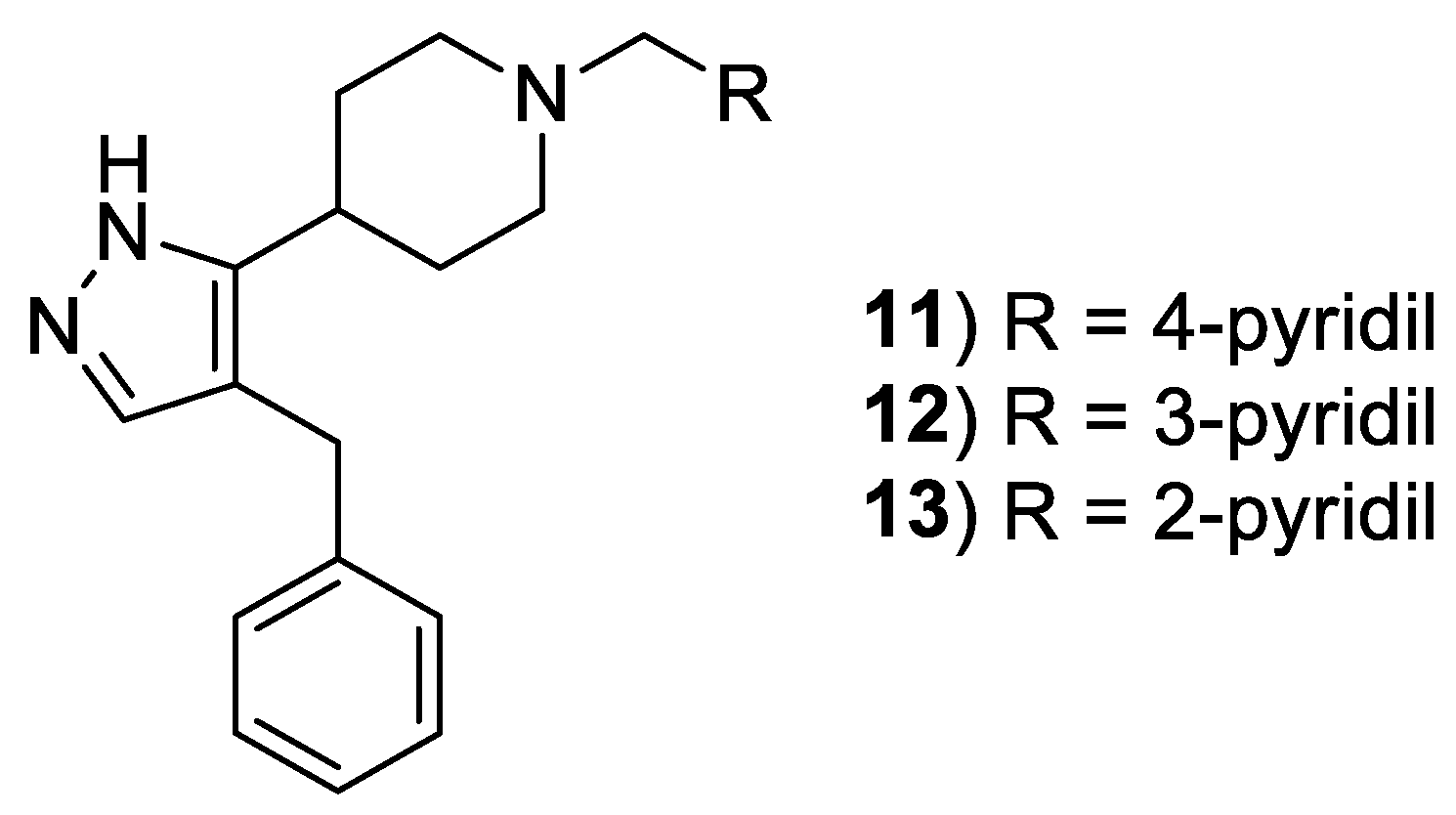{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. CCR5 and CXCR4 Antagonists
2.1. Earlier Co-Receptor Antagonists
2.2. Peptide-Based Antagonists
2.3. Diterpene Derivatives
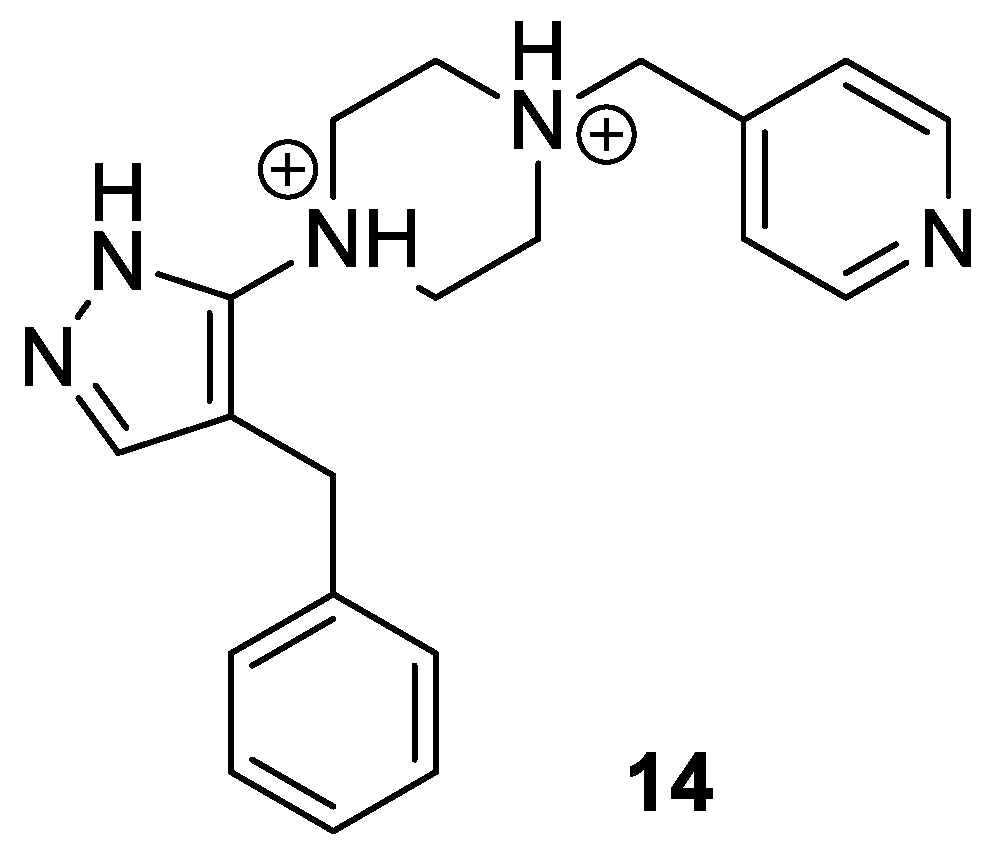
2.4. Pyrazole-Based Antagonists
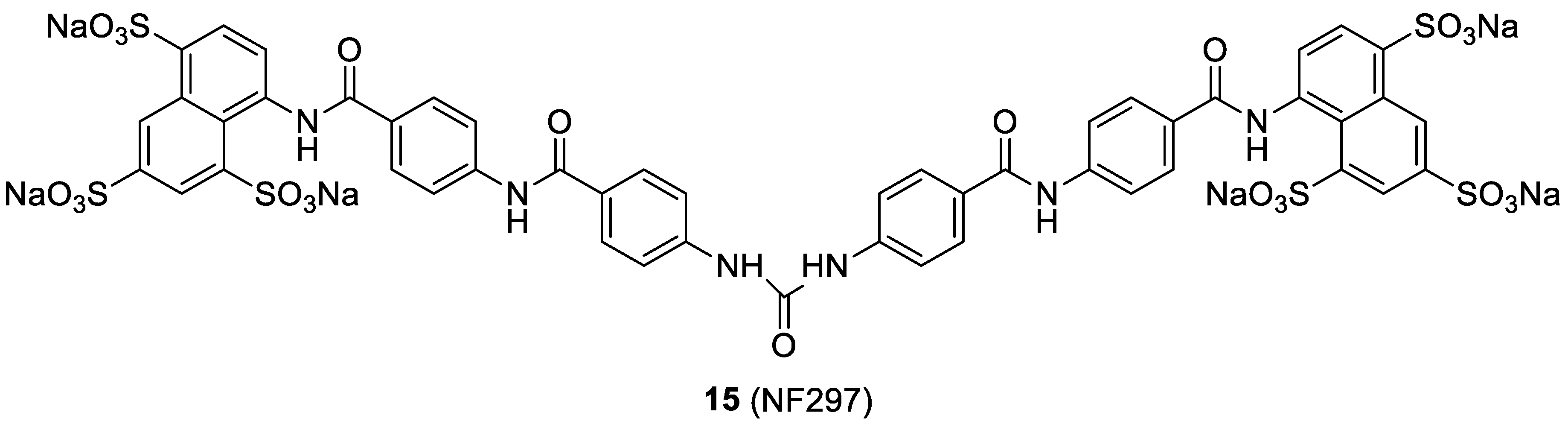
2.5. The Suramin Analog NF279
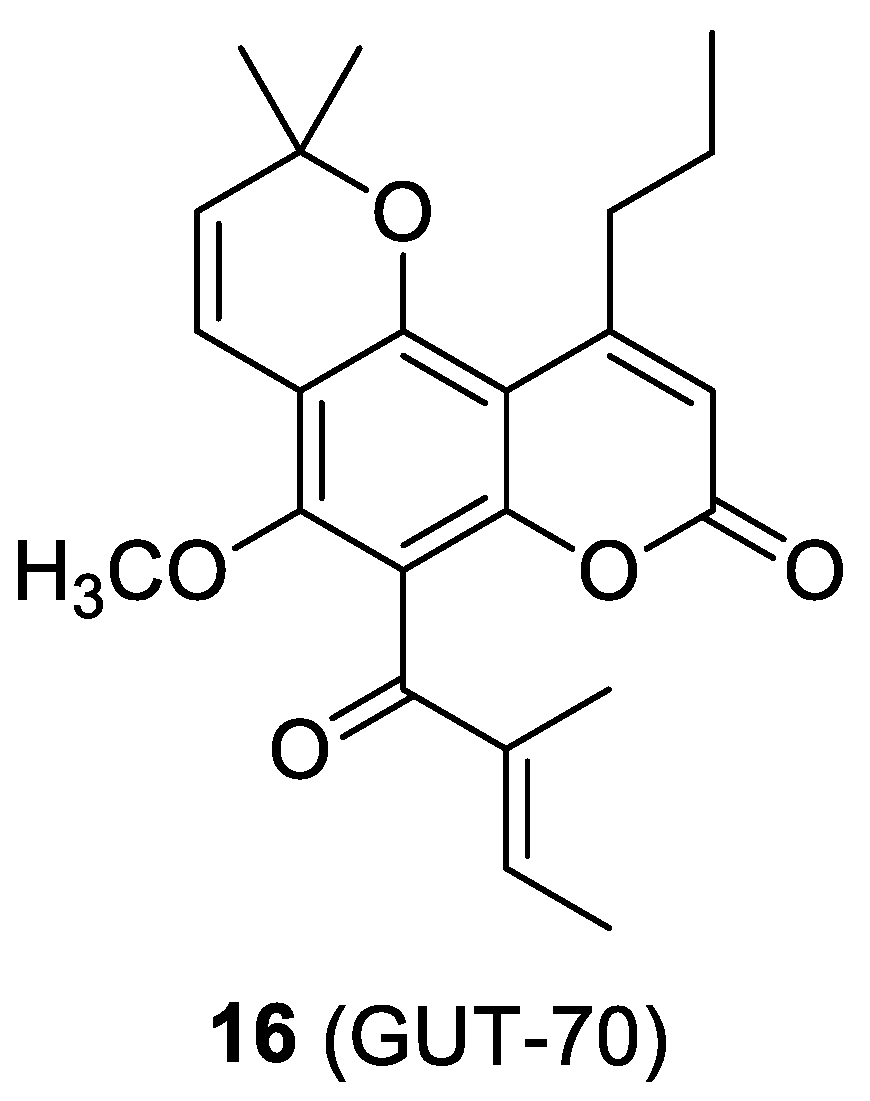
2.6. The Cumarin-Based Ligand GUT-70
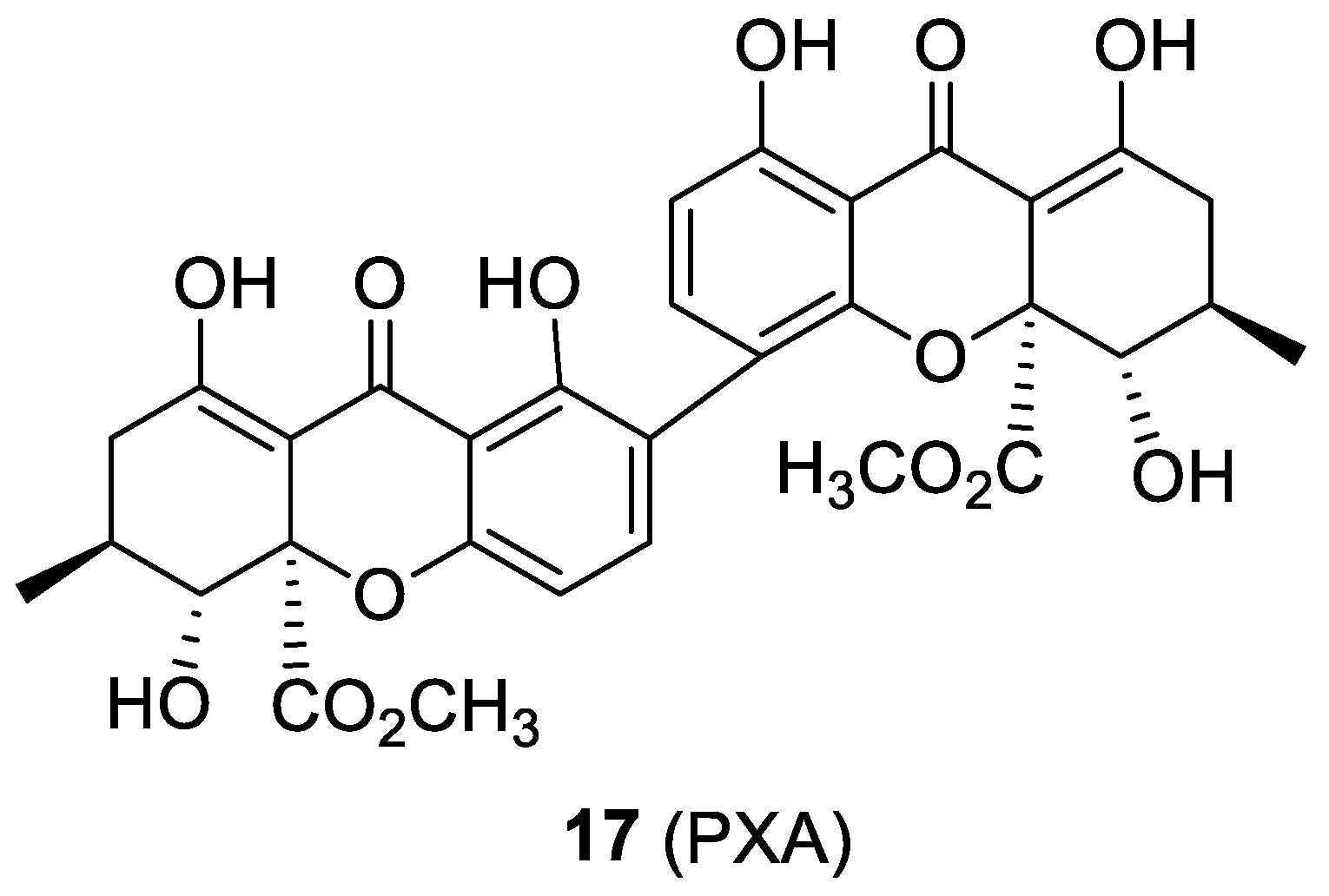
2.7. Penicillixanthone A
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cobucci, R.N.; Lima, P.H.; de Souza, P.C.; Costa, V.V.; Cornetta Mda, C.; Fernandes, J.V.; Goncalves, A.K. Assessing the impact of HAART on the incidence of defining and non-defining AIDS cancers among patients with HIV/AIDS: A systematic review. J. Infect. Public Health 2015, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Oliva-Moreno, J.; Trapero-Bertran, M. Economic Impact of HIV in the Highly Active Antiretroviral Therapy Era—Reflections Looking Forward. AIDS Rev. 2018, 20, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, A.J.; Loo, L.; Mohapatra, D.P. Chemokine co-receptor CCR5/CXCR4-dependent modulation of Kv2.1 channel confers acute neuroprotection to HIV-1 glycoprotein gp120 exposure. PLoS ONE 2013, 8, e76698. [Google Scholar] [CrossRef] [PubMed]
- Dragic, T.; Litwin, V.; Allaway, G.P.; Martin, S.R.; Huang, Y.; Nagashima, K.A.; Cayanan, C.; Maddon, P.J.; Koup, R.A.; Moore, J.P.; et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996, 381, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Scarlatti, G.; Tresoldi, E.; Bjorndal, A.; Fredriksson, R.; Colognesi, C.; Deng, H.K.; Malnati, M.S.; Plebani, A.; Siccardi, A.G.; Littman, D.R.; et al. In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat. Med. 1997, 3, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Grande, F.; Garofalo, A.; Neamati, N. Small molecules anti-HIV therapeutics targeting CXCR4. Curr. Pharm. Des. 2008, 14, 385–404. [Google Scholar] [PubMed]
- Singh, I.P.; Chauthe, S.K. Small molecule HIV entry inhibitors: Part II. Attachment and fusion inhibitors: 2004-2010. Expert Opin. Ther. Pat. 2011, 21, 399–416. [Google Scholar] [CrossRef]
- Oppermann, M. Chemokine receptor CCR5: Insights into structure, function, and regulation. Cell Signal. 2004, 16, 1201–1210. [Google Scholar] [CrossRef]
- Peng, P.; Chen, H.; Zhu, Y.; Wang, Z.; Li, J.; Luo, R.H.; Wang, J.; Chen, L.; Yang, L.M.; Jiang, H.; et al. Structure-Based Design of 1-Heteroaryl-1,3-propanediamine Derivatives as a Novel Series of CC-Chemokine Receptor 5 Antagonists. J. Med. Chem. 2018, 61, 9621–9636. [Google Scholar] [CrossRef]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef]
- Chien, H.C.; Chan, P.C.; Tu, C.C.; Day, Y.J.; Hung, L.M.; Juan, C.C.; Tian, Y.F.; Hsieh, P.S. Importance of PLC-Dependent PI3K/AKT and AMPK Signaling in RANTES/CCR5 Mediated Macrophage Chemotaxis. Chin. J. Physiol. 2018, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.; Pietrancosta, N.; Davidson, S.; Dutrieux, J.; Chauveau, L.; Cutolo, P.; Dy, M.; Scott-Algara, D.; Manoury, B.; Zirafi, O.; et al. Natural amines inhibit activation of human plasmacytoid dendritic cells through CXCR4 engagement. Nat. Commun. 2017, 8, 14253. [Google Scholar] [CrossRef] [PubMed]
- Rangel, H.R.; Bello, G.; Villalba, J.A.; Sulbaran, Y.F.; Garzaro, D.; Maes, M.; Loureiro, C.L.; de Waard, J.H.; Pujol, F.H. The Evolving HIV-1 Epidemic in Warao Amerindians Is Dominated by an Extremely High Frequency of CXCR4-Utilizing Strains. AIDS Res. Hum. Retrovir. 2015, 31, 1265–1268. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Prosser, A.R.; Cox, B.D.; Wilson, L.J.; Liotta, D. Design and synthesis of dual-tropic HIV entry inhibitors that utilize a homologous CCR5/CXCR4 binding site. In Proceedings of the 249th ACS National Meeting & Exposition, Denver, CO, USA, 22–26 March 2015. [Google Scholar]
- Gupta, S.; Prosser, A.R.; Cox, B.D.; Wilson, L.J.; Liotta, D.C. Design and synthesis of Dual Tropic HIV entry inhibitors that utilize a homologous CCR5/CXCR4 binding site. In Proceedings of the 66th Southeast Regional Meeting of the American Chemical Society, Nashville, TN, USA, 16–19 October 2014. [Google Scholar]
- Woollard, S.M.; Kanmogne, G.D. Maraviroc: A review of its use in HIV infection and beyond. Drug Des. Dev. Ther. 2015, 9, 5447–5468. [Google Scholar]
- Grande, F.; Giancotti, G.; Ioele, G.; Occhiuzzi, M.A.; Garofalo, A. An update on small molecules targeting CXCR4 as starting points for the development of anti-cancer therapeutics. Eur. J. Med. Chem. 2017, 139, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Schols, D.; Struyf, S.; Van Damme, J.; Este, J.A.; Henson, G.; De Clercq, E. Inhibition of T-tropic HIV strains by selective antagonization of the chemokine receptor CXCR4. J. Exp. Med. 1997, 186, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Aquaro, S.; Perno, C.F.; Balestra, E.; Balzarini, J.; Cenci, A.; Francesconi, M.; Panti, S.; Serra, F.; Villani, N.; Calio, R. Inhibition of replication of HIV in primary monocyte/macrophages by different antiviral drugs and comparative efficacy in lymphocytes. J. Leukoc. Biol. 1997, 62, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Donzella, G.A.; Schols, D.; Lin, S.W.; Este, J.A.; Nagashima, K.A.; Maddon, P.J.; Allaway, G.P.; Sakmar, T.P.; Henson, G.; De Clercq, E.; et al. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat. Med. 1998, 4, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Princen, K.; Hatse, S.; Vermeire, K.; Aquaro, S.; De Clercq, E.; Gerlach, L.O.; Rosenkilde, M.; Schwartz, T.W.; Skerlj, R.; Bridger, G.; et al. Inhibition of human immunodeficiency virus replication by a dual CCR5/CXCR4 antagonist. J. Virol. 2004, 78, 12996–13006. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.B.; Giesler, K.E.; Tahirovic, Y.A.; Truax, V.M.; Liotta, D.C.; Wilson, L.J. CCR5 receptor antagonists in preclinical to phase II clinical development for treatment of HIV. Expert Opin. Investig. Drugs 2016, 25, 1377–1392. [Google Scholar] [CrossRef] [PubMed]
- Surdo, M.; Balestra, E.; Saccomandi, P.; Di Santo, F.; Montano, M.; Di Carlo, D.; Sarmati, L.; Aquaro, S.; Andreoni, M.; Svicher, V.; et al. Inhibition of dual/mixed tropic HIV-1 isolates by CCR5-inhibitors in primary lymphocytes and macrophages. PLoS ONE 2013, 8, e68076. [Google Scholar] [CrossRef] [PubMed]
- Cavarelli, M.; Mainetti, L.; Pignataro, A.R.; Bigoloni, A.; Tolazzi, M.; Galli, A.; Nozza, S.; Castagna, A.; Sampaolo, M.; Boeri, E.; et al. Complexity and dynamics of HIV-1 chemokine receptor usage in a multidrug-resistant adolescent. AIDS Res. Hum. Retrovir. 2014, 30, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Surdo, M.; Alteri, C.; Puertas, M.C.; Saccomandi, P.; Parrotta, L.; Swenson, L.; Chapman, D.; Costa, G.; Artese, A.; Balestra, E.; et al. Effect of maraviroc on non-R5 tropic HIV-1: Refined analysis of subjects from the phase IIb study A4001029. Clin. Microbiol. Infect. 2015, 21, 103e1–103e6. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Villar, S.; Caruana, G.; Zlotnik, A.; Perez-Molina, J.A.; Moreno, S. Effects of Maraviroc versus Efavirenz in Combination with Zidovudine-Lamivudine on the CD4/CD8 Ratio in Treatment-Naive HIV-Infected Individuals. Antimicrob. Agents Chemother. 2017, 61, e01763-17. [Google Scholar] [CrossRef] [PubMed]
- Hubin, T.J.; Archibald, S.J.; Won, P.; Birdsong, O.C.; Epley, B.M.; Klassen, S.L.; Schols, D. Synthesis and evaluation of transition metal complex dual CXCR4/CCR5 antagonists. In Proceedings of the 245th ACS National Meeting & Exposition, New Orleans, LA, USA, 7–11 April 2013. [Google Scholar]
- Davilla, D.; Birdsong, O.; Schols, D.; Archibald, S.; Hubin, T. Bis- and pendant armed tetraazamacrocycle transition metal complex dual CXCR4/CCR5 antagonists. In Proceedings of the 251st ACS National Meeting & Exposition, San Diego, CA, USA, 13–17 March 2016. [Google Scholar]
- Available online: https://www.swosu.edu/academics/jur/docs/transitionmetals-v1.pdf (accessed on 31 January 2019).
- Tuzer, F.; Madani, N.; Kamanna, K.; Zentner, I.; LaLonde, J.; Holmes, A.; Upton, E.; Rajagopal, S.; McFadden, K.; Contarino, M.; et al. HIV-1 Env gp120 structural determinants for peptide triazole dual receptor site antagonism. Proteins 2013, 81, 271–290. [Google Scholar] [CrossRef] [PubMed]
- Abreu, C.M.; Price, S.L.; Shirk, E.N.; Cunha, R.D.; Pianowski, L.F.; Clements, J.E.; Tanuri, A.; Gama, L. Dual role of novel ingenol derivatives from Euphorbia tirucalli in HIV replication: Inhibition of de novo infection and activation of viral LTR. PLoS ONE 2014, 9, e97257. [Google Scholar] [CrossRef]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.D.; Prosser, A.R.; Sun, Y.; Li, Z.; Lee, S.; Huang, M.B.; Bond, V.C.; Snyder, J.P.; Krystal, M.; Wilson, L.J.; et al. Pyrazolo-Piperidines Exhibit Dual Inhibition of CCR5/CXCR4 HIV Entry and Reverse Transcriptase. ACS Med. Chem. Lett. 2015, 6, 753–757. [Google Scholar] [CrossRef]
- Taylor, C.A.; Miller, B.R., 3rd; Parish, C.A. Design and computational support for the binding stability of a new CCR5/CXCR4 dual tropic inhibitor: Computational design of a CCR5/CXCR4 drug. J. Mol. Graph. Model. 2017, 75, 71–79. [Google Scholar] [CrossRef]
- Marin, M.; Du, Y.; Giroud, C.; Kim, J.H.; Qui, M.; Fu, H.; Melikyan, G.B. High-Throughput HIV-Cell Fusion Assay for Discovery of Virus Entry Inhibitors. Assay Drug Dev. Technol. 2015, 13, 155–166. [Google Scholar] [CrossRef]
- Giroud, C.; Marin, M.; Hammonds, J.; Spearman, P.; Melikyan, G.B. P2X1 Receptor Antagonists Inhibit HIV-1 Fusion by Blocking Virus-Coreceptor Interactions. J. Virol. 2015, 89, 9368–9382. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N.; Rashmi, V.; Odhav, B. Review on natural coumarin lead compounds for their pharmacological activity. BioMed Res. Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Hattori, S.; Kariya, R.; Komizu, Y.; Kudo, E.; Goto, H.; Taura, M.; Ueoka, R.; Kimura, S.; Okada, S. Inhibition of HIV-1 entry by the tricyclic coumarin GUT-70 through the modification of membrane fluidity. Biochem. Biophys. Res. Commun. 2015, 457, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Kudo, E.; Taura, M.; Matsuda, K.; Shimamoto, M.; Kariya, R.; Goto, H.; Hattori, S.; Kimura, S.; Okada, S. Inhibition of HIV-1 replication by a tricyclic coumarin GUT-70 in acutely and chronically infected cells. Bioorg. Med. Chem. Lett. 2013, 23, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Tozser, J.; Blaha, I.; Copeland, T.D.; Wondrak, E.M.; Oroszlan, S. Comparison of the HIV-1 and HIV-2 proteinases using oligopeptide substrates representing cleavage sites in Gag and Gag-Pol polyproteins. FEBS Lett. 1991, 281, 77–80. [Google Scholar] [CrossRef]
- Gorelick, R.J.; Henderson, L.E. Human Retroviruses and AIDS 1994-Part III; Elsevier: Amsterdam, The Netherlands, 2017; p. 9. [Google Scholar]
- Leslie, G.J.; Wang, J.; Richardson, M.W.; Haggarty, B.S.; Hua, K.L.; Duong, J.; Secreto, A.J.; Jordon, A.P.; Romano, J.; Kumar, K.E.; et al. Potent and Broad Inhibition of HIV-1 by a Peptide from the gp41 Heptad Repeat-2 Domain Conjugated to the CXCR4 Amino Terminus. PLoS Pathog. 2016, 12, e1005983. [Google Scholar] [CrossRef]
- Tan, S.; Yang, B.; Liu, J.; Xun, T.; Liu, Y.; Zhou, X. Penicillixanthone A, a marine-derived dual-coreceptor antagonist as anti-HIV-1 agent. Nat. Prod. Res. 2017, 1–5. [Google Scholar] [CrossRef]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grande, F.; Occhiuzzi, M.A.; Rizzuti, B.; Ioele, G.; De Luca, M.; Tucci, P.; Svicher, V.; Aquaro, S.; Garofalo, A. CCR5/CXCR4 Dual Antagonism for the Improvement of HIV Infection Therapy. Molecules 2019, 24, 550. https://doi.org/10.3390/molecules24030550
Grande F, Occhiuzzi MA, Rizzuti B, Ioele G, De Luca M, Tucci P, Svicher V, Aquaro S, Garofalo A. CCR5/CXCR4 Dual Antagonism for the Improvement of HIV Infection Therapy. Molecules. 2019; 24(3):550. https://doi.org/10.3390/molecules24030550
Chicago/Turabian StyleGrande, Fedora, Maria Antonietta Occhiuzzi, Bruno Rizzuti, Giuseppina Ioele, Michele De Luca, Paola Tucci, Valentina Svicher, Stefano Aquaro, and Antonio Garofalo. 2019. "CCR5/CXCR4 Dual Antagonism for the Improvement of HIV Infection Therapy" Molecules 24, no. 3: 550. https://doi.org/10.3390/molecules24030550
APA StyleGrande, F., Occhiuzzi, M. A., Rizzuti, B., Ioele, G., De Luca, M., Tucci, P., Svicher, V., Aquaro, S., & Garofalo, A. (2019). CCR5/CXCR4 Dual Antagonism for the Improvement of HIV Infection Therapy. Molecules, 24(3), 550. https://doi.org/10.3390/molecules24030550