
Proof of an Outer Membrane Target of the Efflux Inhibitor Phe-Arg-β-Naphthylamide from Random Mutagenesis

 and
and
Abstract
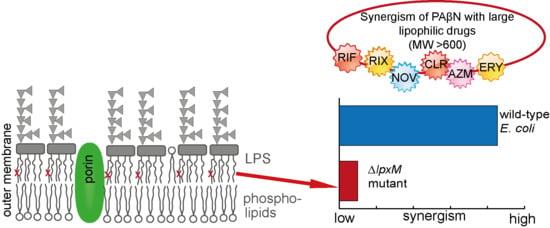
1. Introduction
2. Results and Discussion
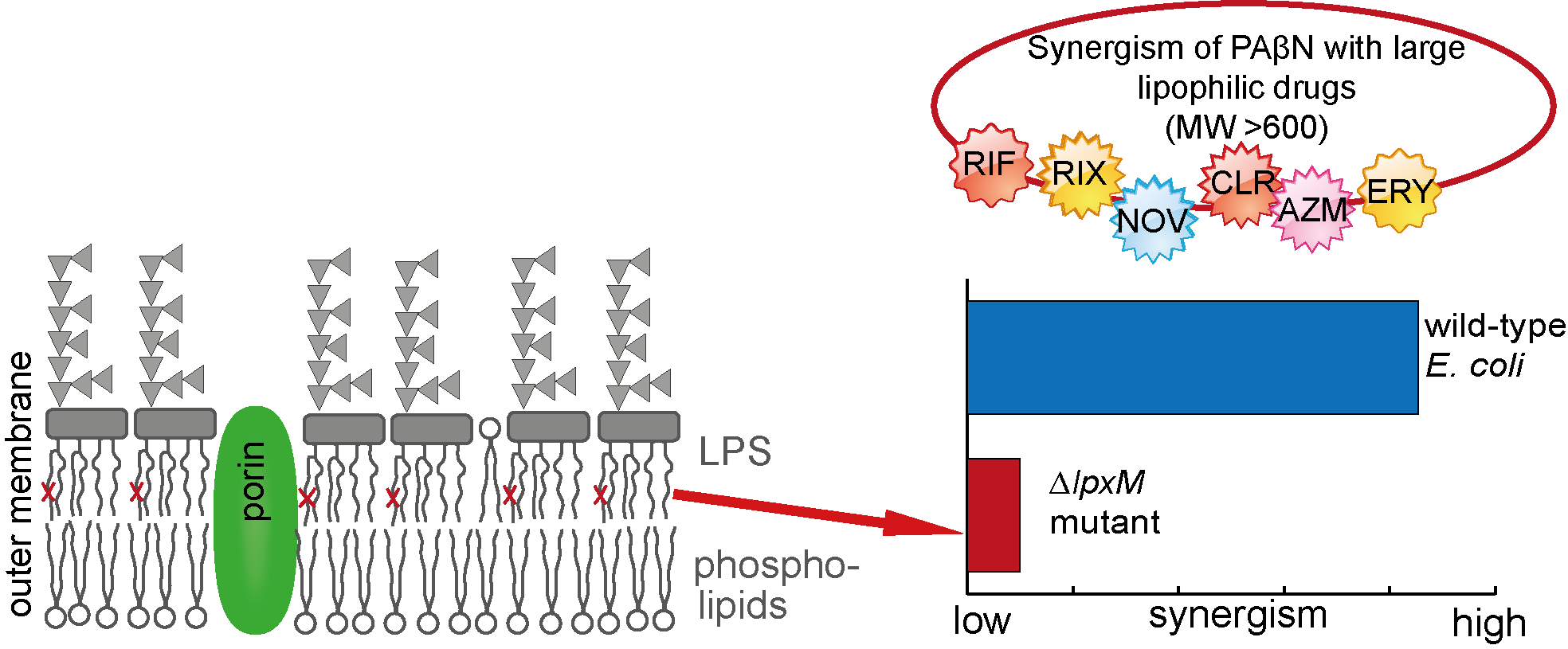
2.1. PAβN-Resistant Mutants from Random Mutagenesis Revealed Loss-of-Function Mutations in LpxM
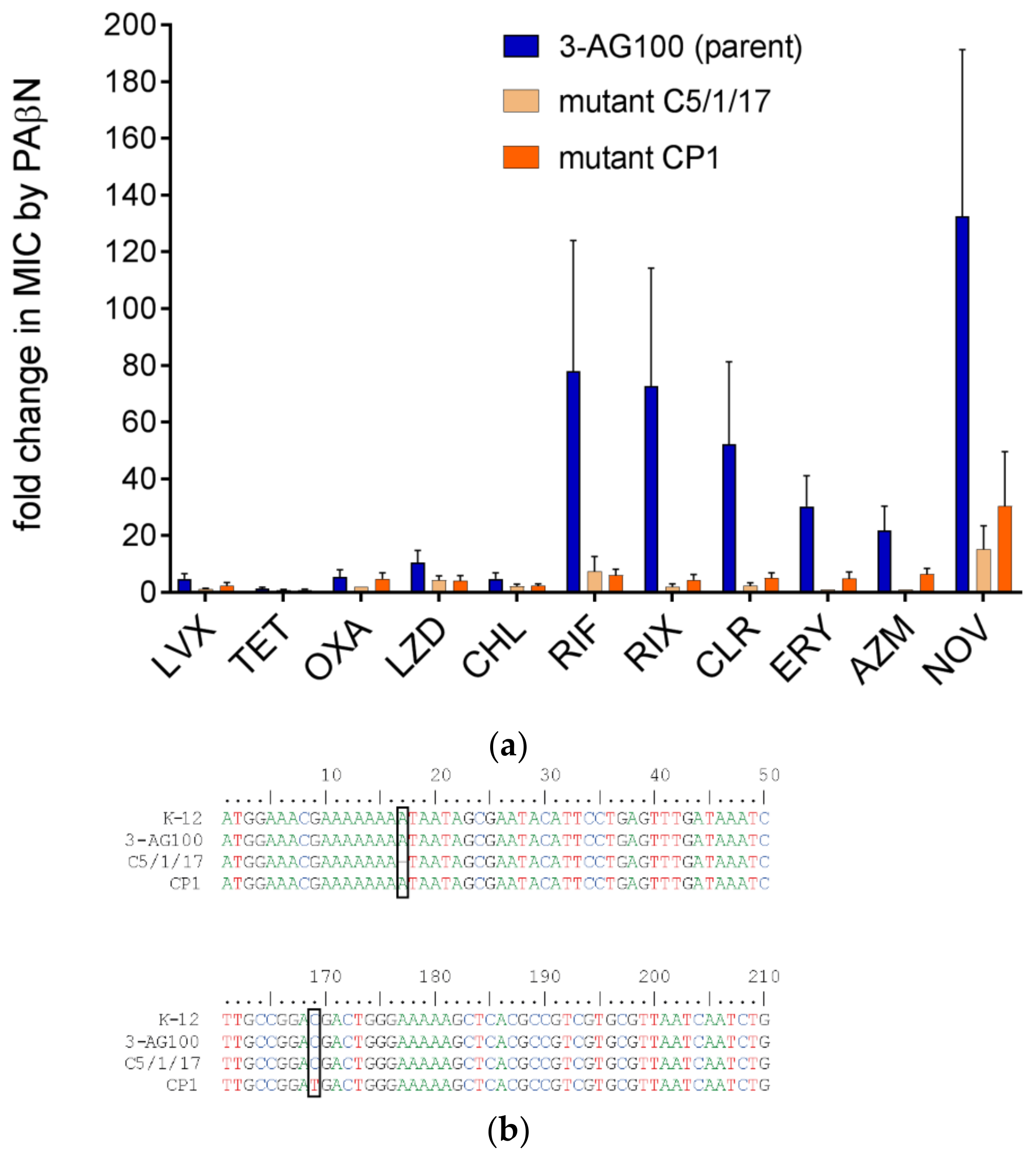
2.2. Proved Impact on PAβN Efficacy from an LpxM Knockout Mutant
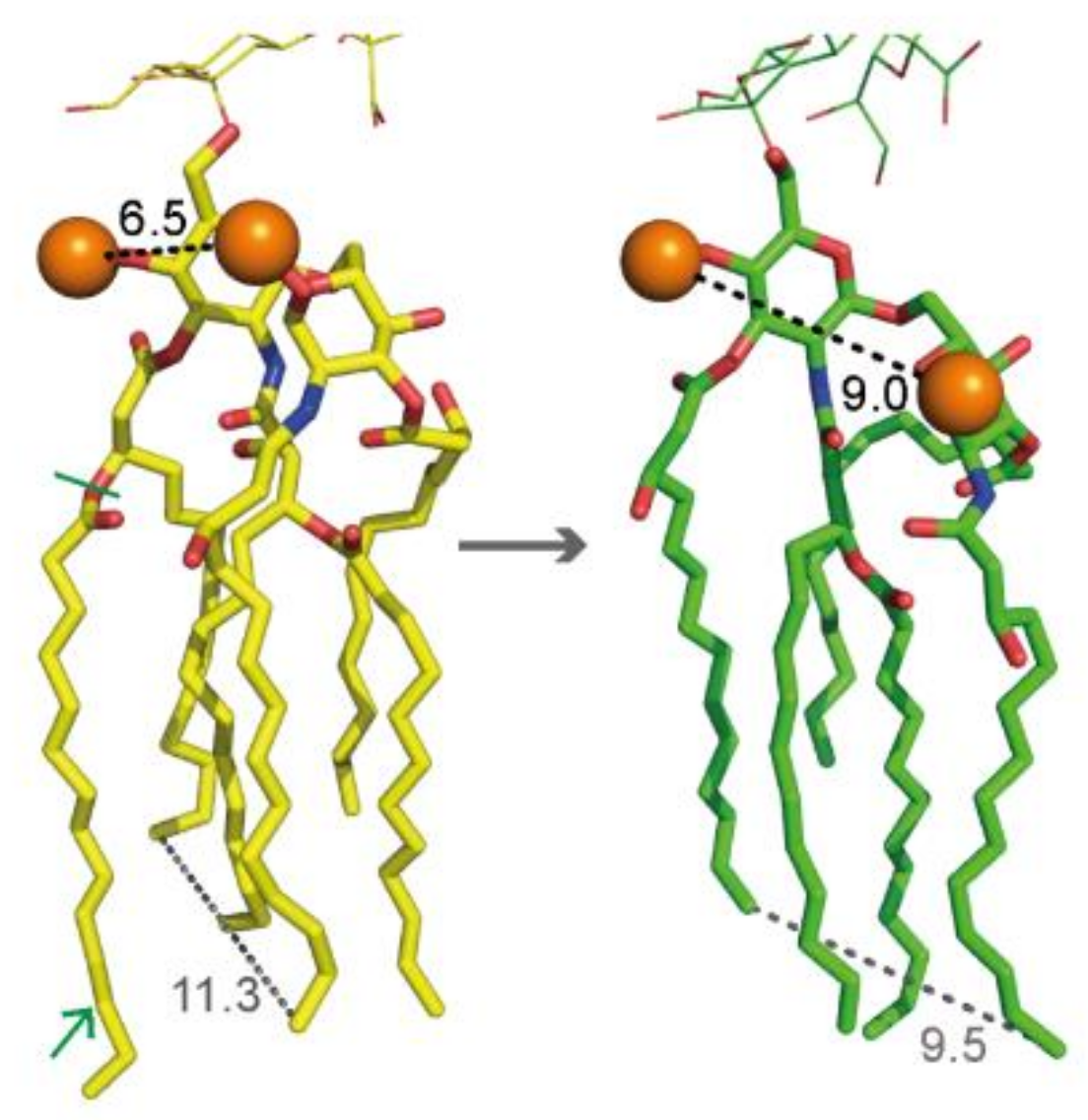
2.3. Hexa-Acylated Lipid A Structure from Wild-Type E. coli versus Penta-acylated from LpxM Mutants
2.4. Further Characterization of Mutant ∆lpxM
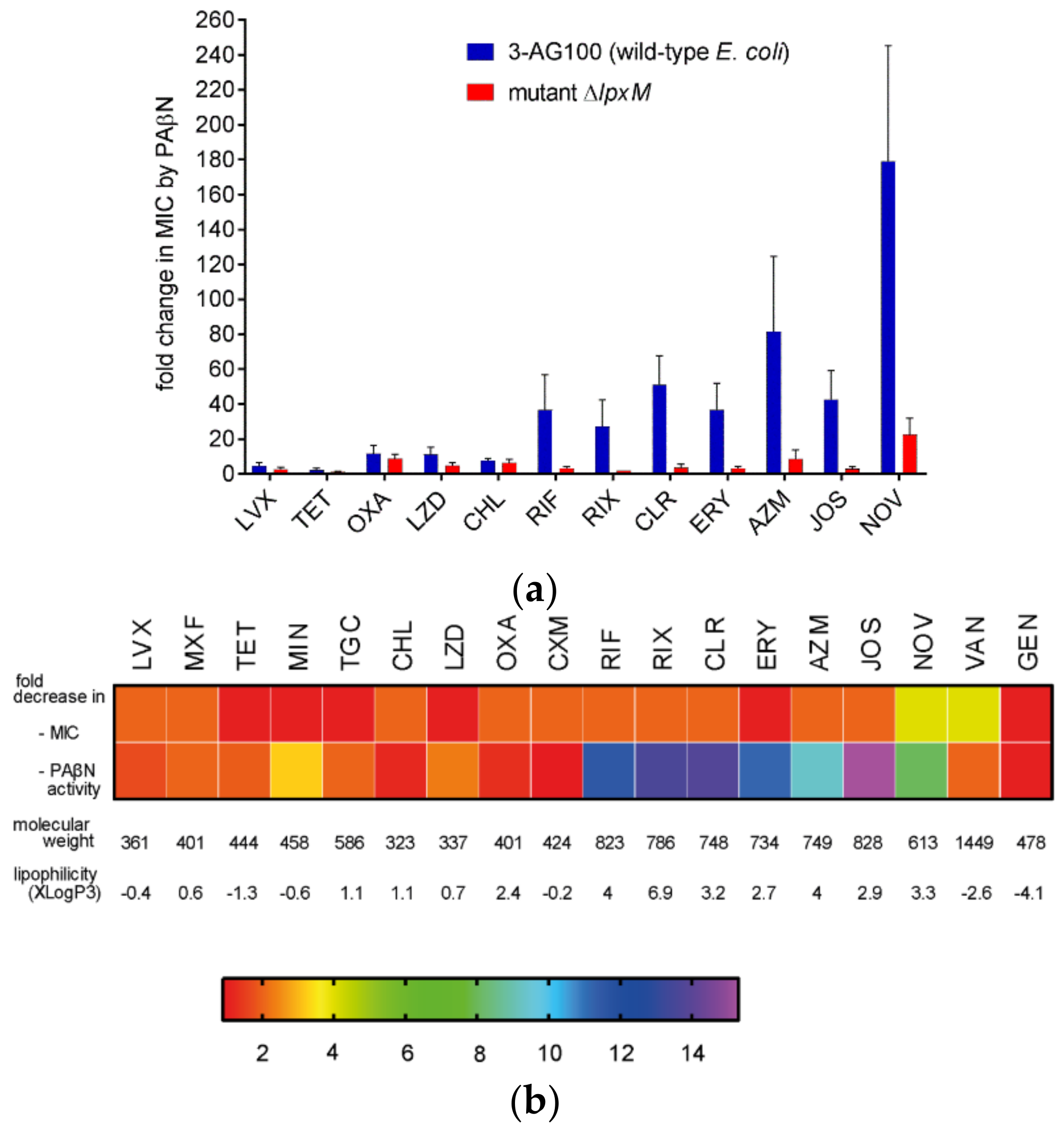
2.4.1. The Activity of other EPIs
2.4.2. The Activity of PMBN
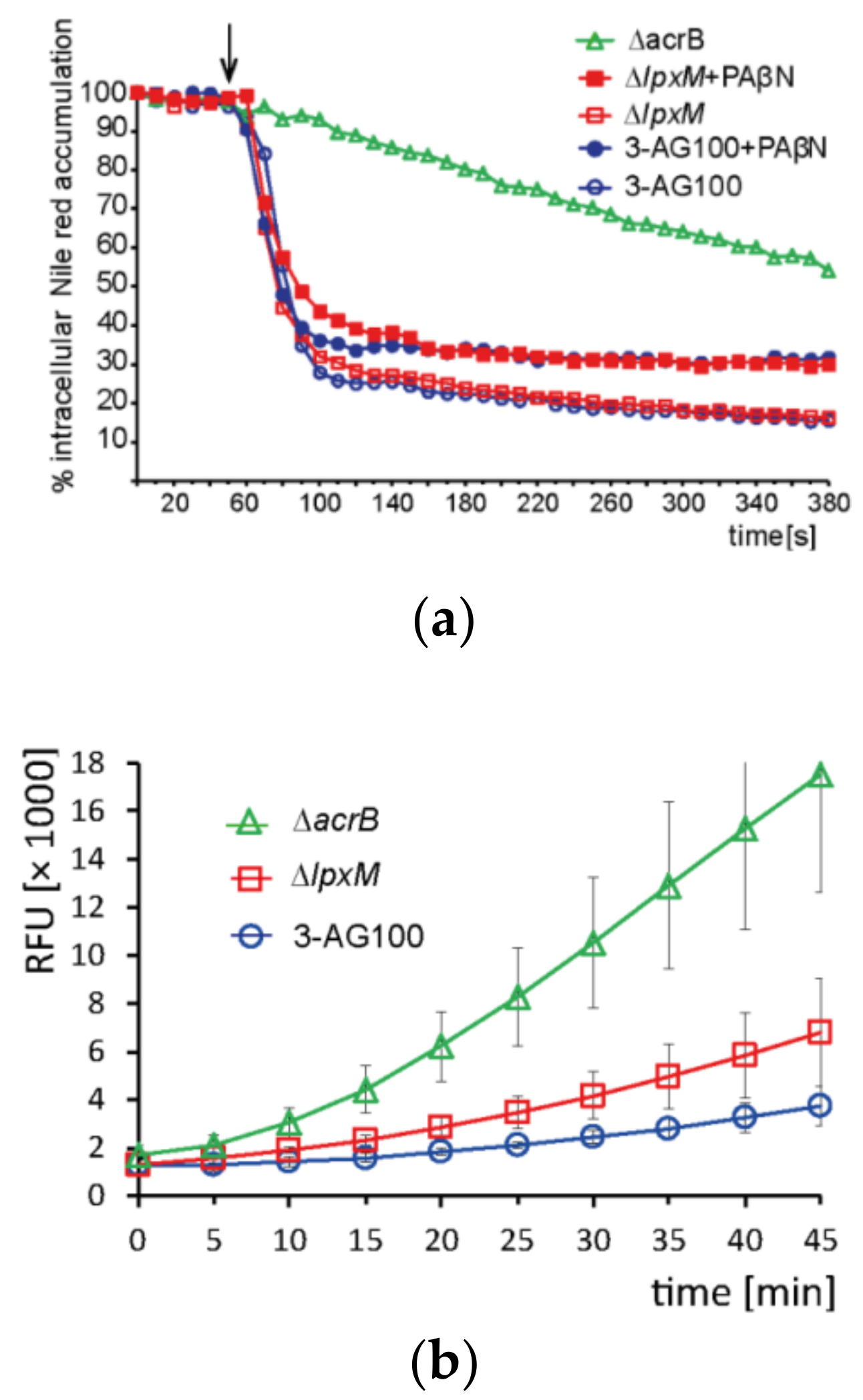
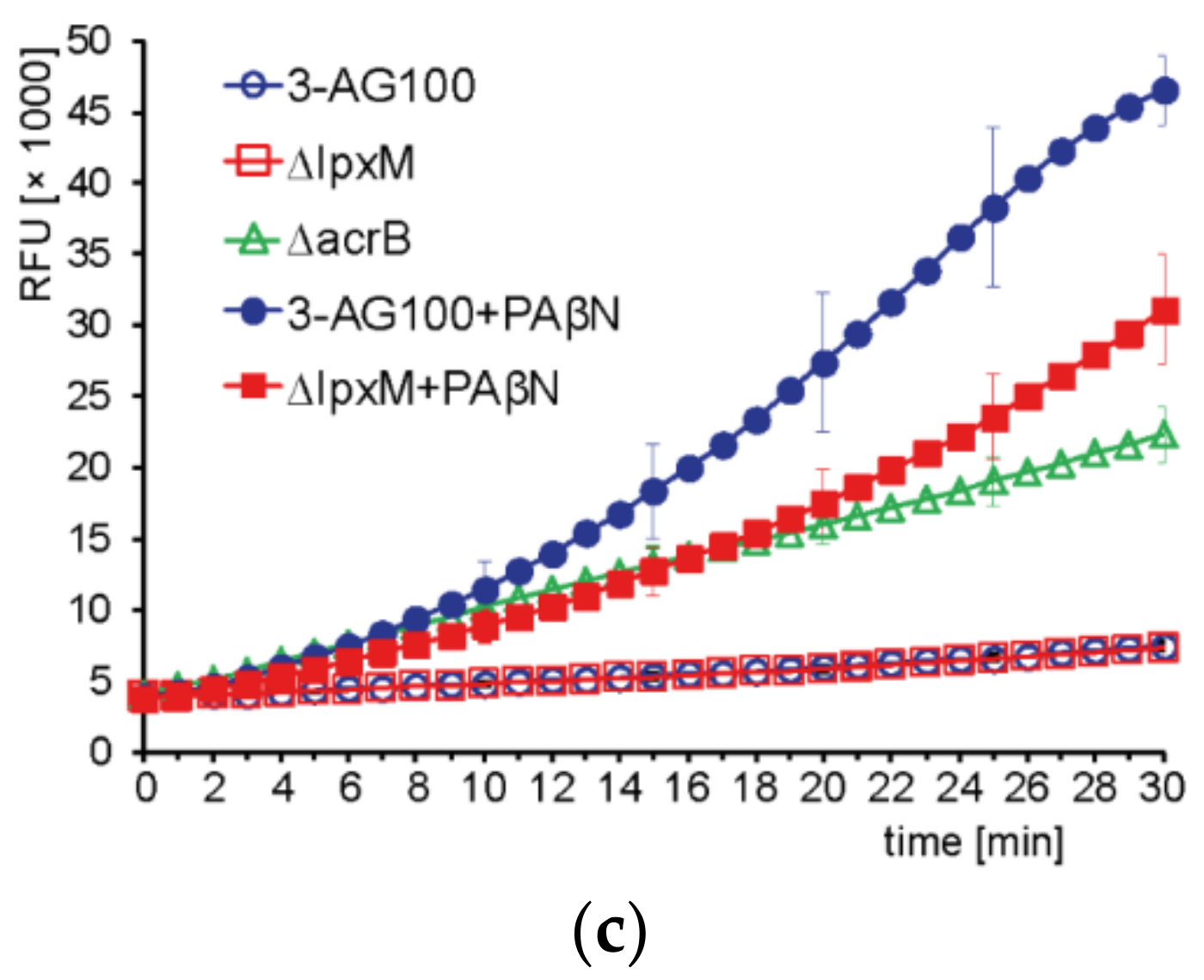
2.4.3. Intracellular Dye Accumulation
3. Materials and Methods
3.1. Bacterial Strains, Growth Conditions, and Chemicals
3.2. Susceptibility Testing
3.3. In Vitro Random Mutagenesis (Directed Evolution)
3.4. Sequencing
3.5. Site-Directed Reconstructions
3.6. Generation of Knockout Mutants
3.7. Intracellular Dye Accumulation Assays
3.8. LPS Modeling and Visualization
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vaara, M. Agents that increase the permeability of the outer membrane. Microbiol. Rev. 1992, 56, 395–411. [Google Scholar] [PubMed]
- Spengler, G.; Kincses, A.; Gajdacs, M.; Amaral, L. New Roads Leading to Old Destinations: Efflux Pumps as Targets to Reverse Multidrug Resistance in Bacteria. Molecules 2017, 22, 468. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H.; Pages, J.M. Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria. FEMS Microbiol. Rev. 2012, 36, 340–363. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Vavra, M.; Schweigger, T.M.; Rossen, J.W.A.; Matsumura, Y.; Kern, W.V. Contribution of AcrAB-TolC to multidrug resistance in an Escherichia coli sequence type 131 isolate. Int. J. Antimicrob. Agents 2017, 50, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Opperman, T.J.; Nguyen, S.T. Recent advances toward a molecular mechanism of efflux pump inhibition. Front. Microbiol. 2015, 6, 421. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Warren, M.S.; Lee, A.; Galazzo, J.; Fronko, R.; Lee, M.; Blais, J.; Cho, D.; Chamberland, S.; Renau, T.; et al. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: Novel agents for combination therapy. Antimicrob. Agents Chemother. 2001, 45, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Laudy, A.E.; Osinska, P.; Namyslowska, A.; Zajac, O.; Tyski, S. Modification of the susceptibility of gram-negative rods producing ESβLS to beta-lactams by the efflux phenomenon. PLoS ONE 2015, 10, e0119997. [Google Scholar] [CrossRef]
- Pages, J.M.; Lavigne, J.P.; Leflon-Guibout, V.; Marcon, E.; Bert, F.; Noussair, L.; Nicolas-Chanoine, M.H. Efflux pump, the masked side of beta-lactam resistance in Klebsiella pneumoniae clinical isolates. PLoS ONE 2009, 4, e4817. [Google Scholar] [CrossRef] [PubMed]
- Laudy, A.E.; Kulinska, E.; Tyski, S. The Impact of Efflux Pump Inhibitors on the Activity of Selected Non-Antibiotic Medicinal Products against Gram-Negative Bacteria. Molecules 2017, 22, 114. [Google Scholar] [CrossRef] [PubMed]
- Lamers, R.P.; Cavallari, J.F.; Burrows, L.L. The efflux inhibitor phenylalanine-arginine beta-naphthylamide (PAβN) permeabilizes the outer membrane of gram-negative bacteria. PLoS ONE 2013, 8, e60666. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Hayama, K.; Sakakihara, S.; Nishino, K.; Noji, H.; Iino, R.; Yamaguchi, A. Evaluation of multidrug efflux pump inhibitors by a new method using microfluidic channels. PLoS ONE 2011, 6, e18547. [Google Scholar] [CrossRef]
- Misra, R.; Morrison, K.D.; Cho, H.J.; Khuu, T. Importance of Real-Time Assays to Distinguish Multidrug Efflux Pump-Inhibiting and Outer Membrane-Destabilizing Activities in Escherichia coli. J. Bacteriol. 2015, 197, 2479–2488. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Aroca, F.; Meng, A.; Minz, T.; Page, M.G.; Dreier, J. Use of resazurin to detect mefloquine as an efflux-pump inhibitor in Pseudomonas aeruginosa and Escherichia coli. J. Microbiol. Methods 2009, 79, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, J.A.; Schuster, S.; Seeger, M.A.; Fähnrich, E.; Pos, K.M.; Kern, W.V. Site-directed mutagenesis reveals putative substrate binding residues in the Escherichia coli RND efflux pump AcrB. J. Bacteriol. 2008, 190, 8225–8229. [Google Scholar] [CrossRef]
- Yu, E.W.; Aires, J.R.; McDermott, G.; Nikaido, H. A periplasmic drug-binding site of the AcrB multidrug efflux pump: A crystallographic and site-directed mutagenesis study. J. Bacteriol. 2005, 187, 6804–6815. [Google Scholar] [CrossRef] [PubMed]
- Kinana, A.D.; Vargiu, A.V.; May, T.; Nikaido, H. Aminoacyl β-naphthylamides as substrates and modulators of AcrB multidrug efflux pump. Proc. Natl. Acad. Sci. USA 2016, 113, 1405–1410. [Google Scholar] [CrossRef]
- Vargiu, A.V.; Ruggerone, P.; Opperman, T.J.; Nguyen, S.T.; Nikaido, H. Molecular mechanism of MBX2319 inhibition of Escherichia coli AcrB multidrug efflux pump and comparison with other inhibitors. Antimicrob. Agents Chemother. 2014, 58, 6224–6234. [Google Scholar] [CrossRef]
- Nakashima, R.; Sakurai, K.; Yamasaki, S.; Nishino, K.; Yamaguchi, A. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 2011, 480, 565–569. [Google Scholar] [CrossRef]
- Iyer, R.; Ferrari, A.; Rijnbrand, R.; Erwin, A.L. A fluorescent microplate assay quantifies bacterial efflux and demonstrates two distinct compound binding sites in AcrB. Antimicrob. Agents Chemother. 2015, 59, 2388–2397. [Google Scholar] [CrossRef]
- Schuster, S.; Vavra, M.; Kern, W.V. Evidence of a Substrate-Discriminating Entrance Channel in the Lower Porter Domain of the Multidrug Resistance Efflux Pump AcrB. Antimicrob. Agents Chemother. 2016, 60, 4315–4323. [Google Scholar] [CrossRef]
- Schuster, S.; Kohler, S.; Buck, A.; Dambacher, C.; König, A.; Bohnert, J.A.; Kern, W.V. Random mutagenesis of the multidrug transporter AcrB from Escherichia coli for identification of putative target residues of efflux pump inhibitors. Antimicrob. Agents Chemother. 2014, 58, 6870–6878. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Warren, M.S.; Black, D.S.; Satou, T.; Murata, T.; Nishino, T.; Gotoh, N.; Lomovskaya, O. On the mechanism of substrate specificity by resistance nodulation division (RND)-type multidrug resistance pumps: The large periplasmic loops of MexD from Pseudomonas aeruginosa are involved in substrate recognition. Mol. Microbiol. 2002, 46, 889–901. [Google Scholar] [CrossRef]
- Wehmeier, C.; Schuster, S.; Fahnrich, E.; Kern, W.V.; Bohnert, J.A. Site-directed mutagenesis reveals amino acid residues in the Escherichia coli RND efflux pump AcrB that confer macrolide resistance. Antimicrob. Agents Chemother. 2009, 53, 329–330. [Google Scholar] [CrossRef]
- Bohnert, J.A.; Schuster, S.; Fähnrich, E.; Trittler, R.; Kern, W.V. Altered spectrum of multidrug resistance associated with a single point mutation in the Escherichia coli RND-type MDR efflux pump YhiV (MdtF). J. Antimicrob. Chemother. 2007, 59, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Somerville, J.E., Jr.; Cassiano, L.; Bainbridge, B.; Cunningham, M.D.; Darveau, R.P. A novel Escherichia coli lipid A mutant that produces an antiinflammatory lipopolysaccharide. J. Clin. Investig. 1996, 97, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Delcour, A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 2009, 1794, 808–816. [Google Scholar] [CrossRef]
- Karow, M.; Georgopoulos, C. Isolation and characterization of the Escherichia coli msbB gene, a multicopy suppressor of null mutations in the high-temperature requirement gene htrB. J. Bacteriol. 1992, 174, 702–710. [Google Scholar] [CrossRef]
- Clementz, T.; Zhou, Z.; Raetz, C.R. Function of the Escherichia coli msbB gene, a multicopy suppressor of htrB knockouts, in the acylation of lipid A. Acylation by MsbB follows laurate incorporation by HtrB. J. Biol. Chem. 1997, 272, 10353–10360. [Google Scholar] [CrossRef]
- Vaara, M.; Nurminen, M. Outer membrane permeability barrier in Escherichia coli mutants that are defective in the late acyltransferases of lipid A biosynthesis. Antimicrob. Agents Chemother. 1999, 43, 1459–1462. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Chen, J.; Ernst, R.K.; Wang, X. Influence of lipid A acylation pattern on membrane permeability and innate immune stimulation. Mar. Drugs 2013, 11, 3197–3208. [Google Scholar] [CrossRef]
- Xu, H.; Ling, J.; Gao, Q.; He, H.; Mu, X.; Yan, Z.; Gao, S.; Liu, X. Role of the lpxM lipid A biosynthesis pathway gene in pathogenicity of avian pathogenic Escherichia coli strain E058 in a chicken infection model. Vet. Microbiol. 2013, 166, 516–526. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Ingram, B.O.; Masoudi, A.; Raetz, C.R. Escherichia coli mutants that synthesize dephosphorylated lipid A molecules. Biochemistry 2010, 49, 8325–8337. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, J.A.; Kern, W.V. Selected arylpiperazines are capable of reversing multidrug resistance in Escherichia coli overexpressing RND efflux pumps. Antimicrob. Agents Chemother. 2005, 49, 849–852. [Google Scholar] [CrossRef] [PubMed]
- Opperman, T.J.; Kwasny, S.M.; Kim, H.S.; Nguyen, S.T.; Houseweart, C.; D’Souza, S.; Walker, G.C.; Peet, N.P.; Nikaido, H.; Bowlin, T.L. Characterization of a Novel Pyranopyridine Inhibitor of the AcrAB Efflux Pump of Escherichia coli. Antimicrob. Agents Chemother. 2014, 58, 722–733. [Google Scholar] [CrossRef]
- Hancock, R.E.; Chapple, D.S. Peptide antibiotics. Antimicrob. Agents Chemother. 1999, 43, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Kern, W.V.; Oethinger, M.; Jellen-Ritter, A.S.; Levy, S.B. Non-target gene mutations in the development of fluoroquinolone resistance in Escherichia coli. Antimicrob. Agents Chemother. 2000, 44, 814–820. [Google Scholar] [CrossRef]
- Ferdous, M.; Zhou, K.; Mellmann, A.; Morabito, S.; Croughs, P.D.; de Boer, R.F.; Kooistra-Smid, A.M.; Rossen, J.W.; Friedrich, A.W. Is Shiga Toxin-Negative Escherichia coli O157:H7 Enteropathogenic or Enterohemorrhagic Escherichia coli? Comprehensive Molecular Analysis Using Whole-Genome Sequencing. J. Clin. Microbiol. 2015, 53, 3530–3538. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % Dye Accumulation (Relative to ∆acrB Mutant without Adjuvants) 1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 3-AG100 | ∆lpxM | ∆acrB | |||||||
| Dye | +PAβN | +PMBN | +PAβN | +PMBN | +PAβN | +PMBN | |||
| Resazurin | 33.4 (±2) | 208.4 (±11) | 106.8 (±15) | 33.1 (±1) | 139.6 (±18) | 78.1 (±9) | 100 (±9) | 209.7 (±4) | 226.1 (±4) |
| Hoechst | 43.1 (±6) | 44.5 (±7) | 44.4 (±1) | 47.2 (±12) | 47.6 (±11) | 45.3 (±9) | 100 (±11) | 108.6 (±8) | 108.3 (±3) |
| Berberine | 31.5 (±7) | 32.6 (±8) | 18.7 (±0) | 22.2 (±1) | 22.3 (±1) | 19.6 (±1) | 100 (±14) | 117.8 (±14) | 90.9 (±1) |
| E. coli Strains and Mutants | Description | Source |
|---|---|---|
| 3-AG100 | E. coli K-12 AG100 derivative; overexpression of acrB. | Kern et al., 2000 [39] |
| C5/1/17 | PAβN-resistant serial selection mutant from 3-AG100. | Schuster et al., 2014 [21] |
| CP1 | PAβN-resistant in vitro random mutagenesis mutant from 3-AG100. | This study |
| 3-AG100acrBCP1 | Site-directed mutagenesis mutant from 3-AG100. | This study |
| CP1acrBWT | PAβN-resistant site-directed mutagenesis mutant from CP1. | This study |
| ∆lpxM 1 | PAβN-resistant lpxM knockout mutant from 3-AG100. | This study |
| ∆acrB 2 | Efflux-deficient acrB knockout mutant from 3-AG100. | Schuster et al., 2014 [21] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schuster, S.; Bohnert, J.A.; Vavra, M.; Rossen, J.W.; Kern, W.V. Proof of an Outer Membrane Target of the Efflux Inhibitor Phe-Arg-β-Naphthylamide from Random Mutagenesis. Molecules 2019, 24, 470. https://doi.org/10.3390/molecules24030470
Schuster S, Bohnert JA, Vavra M, Rossen JW, Kern WV. Proof of an Outer Membrane Target of the Efflux Inhibitor Phe-Arg-β-Naphthylamide from Random Mutagenesis. Molecules. 2019; 24(3):470. https://doi.org/10.3390/molecules24030470
Chicago/Turabian StyleSchuster, Sabine, Jürgen A. Bohnert, Martina Vavra, John W. Rossen, and Winfried V. Kern. 2019. "Proof of an Outer Membrane Target of the Efflux Inhibitor Phe-Arg-β-Naphthylamide from Random Mutagenesis" Molecules 24, no. 3: 470. https://doi.org/10.3390/molecules24030470
APA StyleSchuster, S., Bohnert, J. A., Vavra, M., Rossen, J. W., & Kern, W. V. (2019). Proof of an Outer Membrane Target of the Efflux Inhibitor Phe-Arg-β-Naphthylamide from Random Mutagenesis. Molecules, 24(3), 470. https://doi.org/10.3390/molecules24030470