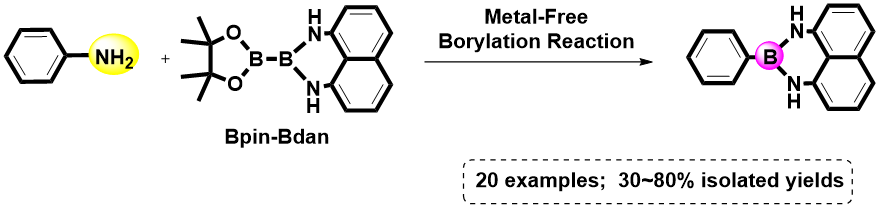
Direct Transformation from Arylamines to Aryl Naphthalene-1,8-diamino Boronamides: A Metal-Free Sandmeyer-Type Process


Abstract

1. Introduction
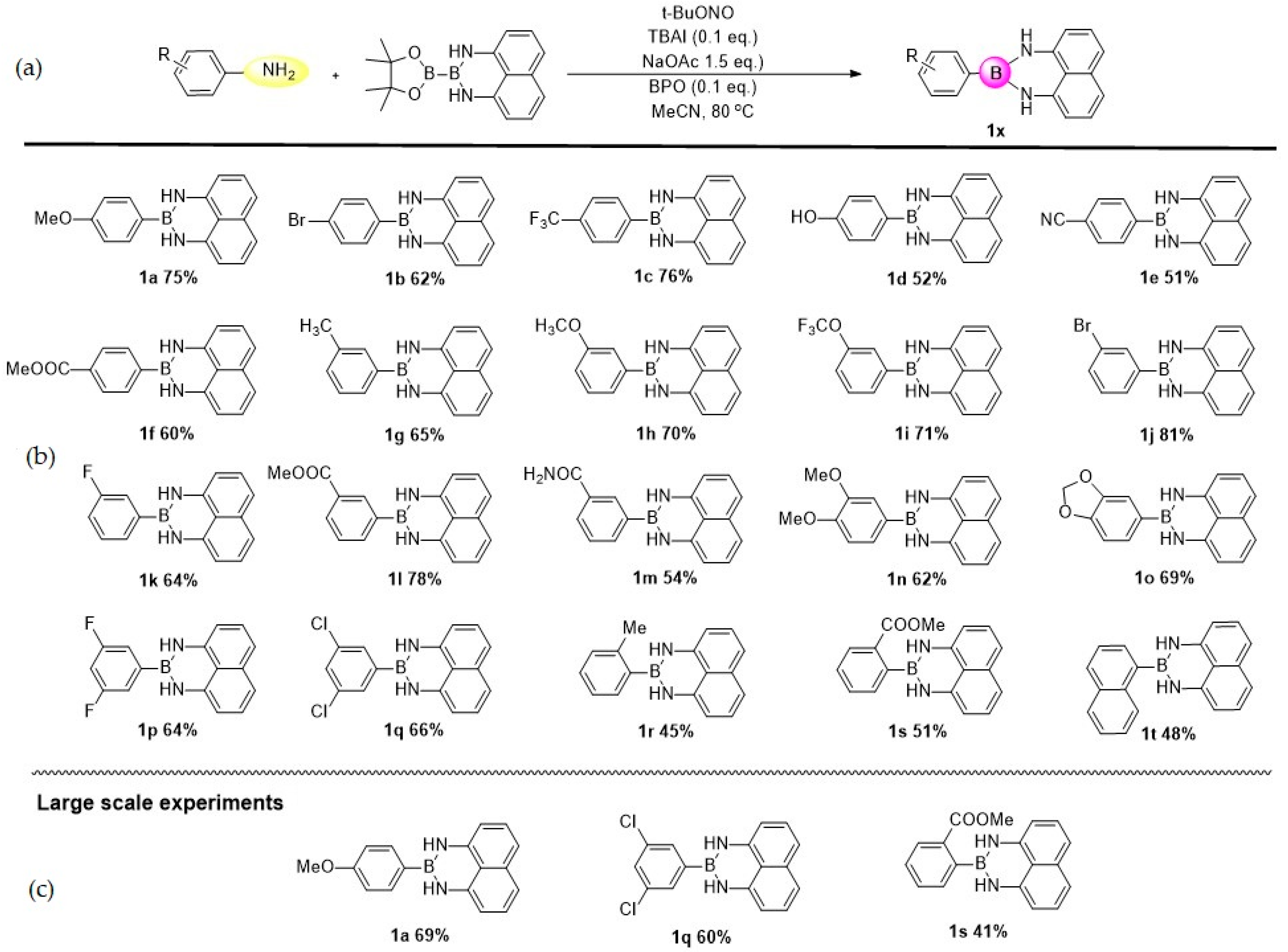
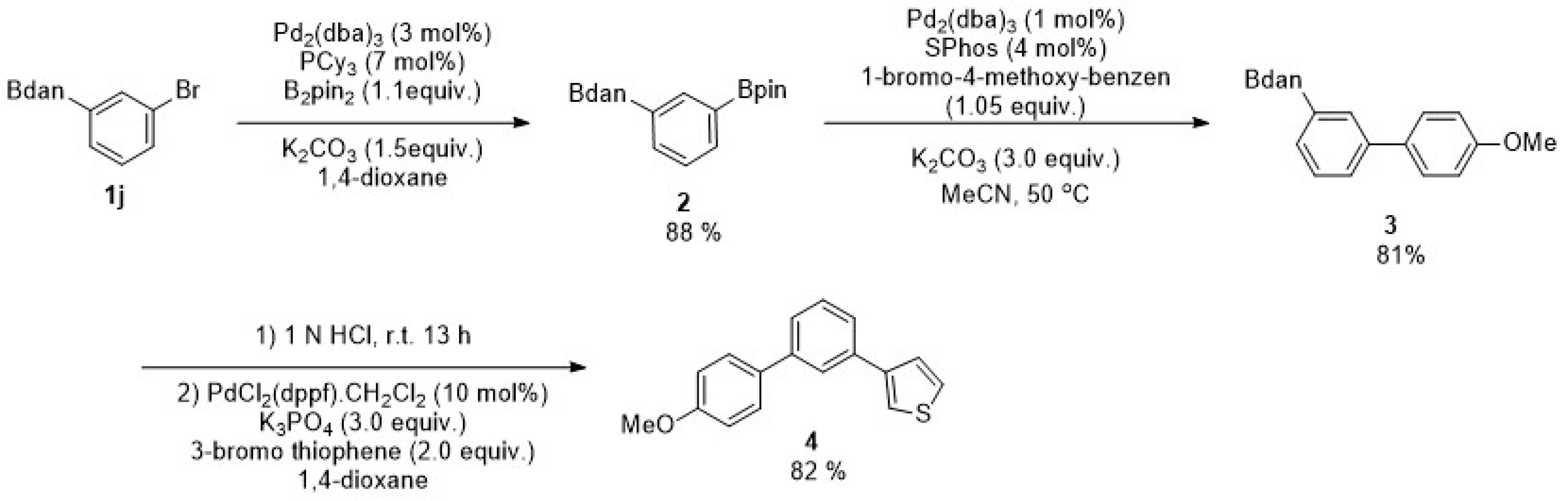
2. Results
3. Discussion
4. Materials and Methods
4.1. Methods and Material
4.1.1. General Information
4.1.2. Structural Analysis
4.1.3. Materials
4.2. General Procedure for the Direct Transformation from Arylamines to Aryl Naphthalene-1,8-diamino Boronamides
4.3. Analytical Data of Products 1a–1t
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hall, D.J. Boronic Acids; Wiley: Weinheim, Germany, 2011. [Google Scholar]
- Lennox, A.J.; Lloyd-Jones, G.C. Selection of Boron Reagents for Suzuki-Miyaura Coupling. Chem. Soc. Rev. 2014, 43, 412–443. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, J.W.B.; Watson, A.J.B. Recent Developments in Organoboron Chemistry: Old Dogs, New Tricks. Chem 2017, 3, 31–55. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, S.; Li, P. Boron-Selective Reactions as Powerful Tools for Modular Synthesis of Diverse Complex Molecules. Chem. Soc. Rev. 2015, 44, 8848–8858. [Google Scholar] [CrossRef] [PubMed]
- Neeve, E.C.; Geier, S.J.; Mkhalid, I.A.I.; Westcott, S.A.; Marder, T.B. Diboron(4) Compounds: From Structural Curiosity to Synthetic Workhorse. Chem. Rev. 2016, 116, 9091–9161. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, G.; Zhang, S.; Wang, H.; Wang, L.; Liu, L.; Jiao, J.; Li, P. Recent Advances in Catalytic C−H Borylation Reactions. Tetrahedron 2017, 73, 7123–7157. [Google Scholar] [CrossRef]
- Takaya, J.; Iwasawa, N. Catalytic, Direct Synthesis of Bis(boronate) Compounds. ACS Catal. 2012, 2, 1993–2006. [Google Scholar] [CrossRef]
- Xu, L.; Li, P. Differentiated Di- and Polyboron Compounds: Synthesis and Application in Successive Suzuki–Miyaura Coupling. Synlett 2014, 25, 1799–1802. [Google Scholar] [CrossRef]
- Noguchi, H.; Hojo, K.; Suginome, M. Boron-Masking Strategy for the Selective Synthesis of Oligoarenes via Iterative Suzuki−Miyaura Coupling. J. Am. Chem. Soc. 2007, 129, 758–759. [Google Scholar] [CrossRef]
- Iwadate, N.; Suginome, M. Synthesis of B-Protected β-Styrylboronic Acids via Iridium-Catalyzed Hydroboration of Alkynes with 1,8-Naphthalenediaminatoborane Leading to Iterative Synthesis of Oligo(phenylenevinylene)s. Org. Lett. 2009, 11, 1899–1902. [Google Scholar] [CrossRef]
- Wang, C.; Glorius, F. Controlled Iterative Cross-Coupling: On the Way to the Automation of Organic Synthesis. Angew. Chem. Int. Ed. 2009, 48, 5240–5244. [Google Scholar] [CrossRef]
- Gillis, E.P.; Burke, M.D. A Simple and Modular Strategy for Small Molecule Synthesis: Iterative Suzuki−Miyaura Coupling of B-Protected Haloboronic Acid Building Blocks. J. Am. Chem. Soc. 2007, 129, 6716–6717. [Google Scholar] [CrossRef] [PubMed]
- Molander, G.A.; Sandrock, D.L. Orthogonal Reactivity in Boryl-Substituted Organotrifluoroborates. J. Am. Chem. Soc. 2008, 130, 15792–15793. [Google Scholar] [CrossRef]
- Noguchi, H.; Shioda, T.; Chou, C.-M.; Suginome, M. Differentially Protected Benzenediboronic Acids: Divalent Cross-Coupling Modules for the Efficient Synthesis of Boron-Substituted Oligoarenes. Org. Lett. 2008, 10, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Tobisu, M.; Chatani, N. Devising Boron Reagents for Orthogonal Functionalization through Suzuki–Miyaura Cross-Coupling. Angew. Chem. Int. Ed. 2009, 48, 3565–3568. [Google Scholar] [CrossRef] [PubMed]
- Iwadate, N.; Suginome, M. Differentially Protected Diboron for Regioselective Diboration of Alkynes: Internal-Selective Cross-Coupling of 1-Alkene-1,2-diboronic Acid Derivatives. J. Am. Chem. Soc. 2010, 132, 2548–2549. [Google Scholar] [CrossRef] [PubMed]
- Hyodo, K.; Suetsugu, M.; Nishihara, Y. Diborylation of Alkynyl MIDA Boronates and Sequential Chemoselective Suzuki–Miyaura Couplings: A Formal Carboborylation of Alkynes. Org. Lett. 2013, 16, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Ding, S.; Li, P. Site-Differentiated Polyboron Arenes Prepared by Direct C–H Borylation and Their Highly Selective Suzuki–Miyaura Cross-Coupling Reactions. Angew. Chem. Int. Ed. 2014, 53, 1822–1826. [Google Scholar] [CrossRef]
- Ishiyama, T.; Murata, M.; Miyaura, N. Palladium(0)-Catalyzed Cross-Coupling Reaction of Alkoxydiboron with Haloarenes: A Direct Procedure for Arylboronic Esters. J. Org. Chem. 1995, 60, 7508–7510. [Google Scholar] [CrossRef]
- Ishiyama, T.; Itoh, Y.; Kitano, T.; Miyaura, N. Synthesis of Arylboronates via the Palladium(0)-Catalyzed Cross-Coupling Reaction of Tetra(alkoxo)diborons with Aryl Triflates. Tetrahedron Lett. 1997, 38, 3447–3450. [Google Scholar] [CrossRef]
- Kleeberg, C.; Dang, L.; Lin, Z.; Marder, T.B. A Facile Route to Aryl Boronates: Room-Temperature, Copper-Catalyzed Borylation of Aryl Halides with Alkoxy Diboron Reagents. Angew. Chem. Int. Ed. 2009, 48, 5350–5354. [Google Scholar] [CrossRef]
- Yamamoto, T.; Morita, T.; Takagi, J.; Yamakawa, T. NiCl2(PMe3)2-Catalyzed Borylation of Aryl Chlorides. Org. Lett. 2011, 13, 5766–5769. [Google Scholar] [CrossRef] [PubMed]
- Molander, G.A.; Cavalcanti, L.N.; García-García, C. Nickel-Catalyzed Borylation of Halides and Pseudohalides with Tetrahydroxydiboron [B2(OH)4]. J. Org. Chem. 2013, 78, 6427–6439. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.L.; Marciasini, L.D.; Vaultier, M.; Pucheault, M. A Synergy for Refunctionalization of Boron. Synlett 2014, 25, 0551–0555. [Google Scholar] [CrossRef]
- Xu, L.; Li, P. Direct Introduction of a Naphthalene-1,8-Diamino Boryl [B(dan)] Group by a Pd-Catalysed Selective Boryl Transfer Reaction. Chem. Commun. 2015, 51, 5656–5659. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Takemoto, Y.; Kamio, S.; Osaka, I.; Takaki, K. Copper-Catalyzed Direct Borylation of Alkyl, Alkenyl and aryl Halides with B(dan). Org. Chem. Front. 2017, 4, 1215–1219. [Google Scholar] [CrossRef]
- Yoshida, H.; Kamio, S.; Osaka, I. Copper-Catalyzed Borylation of Bromoaryl Triflates with Diborons: Chemoselective Replacement of an Ar–Br Bond. Chem. Lett. 2018, 47, 957–959. [Google Scholar] [CrossRef]
- Dang, L.; Lin, Z.; Marder, T.B. Boryl Ligands and Their Roles in Metal-Catalysed Borylation Reactions. Chem. Commun. 2009, 3987–3995. [Google Scholar] [CrossRef]
- Hodgson, H.H. The Sandmeyer Reaction. Chem. Rev. 1947, 40, 251–277. [Google Scholar] [CrossRef]
- Galli, C. Radical Reactions of Arenediazonium Ions: An Easy Entry into the Chemistry of the Aryl Radical. Chem. Rev. 1988, 88, 765–792. [Google Scholar] [CrossRef]
- Mo, F.; Qiu, D.; Zhang, Y.; Wang, J. Renaissance of Sandmeyer-Type Reactions: Conversion of Aromatic C–N Bonds into C–X Bonds (X = B, Sn, P, or CF3). Acc. Chem. Res. 2018, 51, 496–506. [Google Scholar] [CrossRef]
- Mo, F.; Dong, G.; Zhang, Y.; Wang, J. Recent Applications of Arene Diazonium Salts in Organic Synthesis. Org. Biomol. Chem. 2013, 11, 1582–1593. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Jiang, Y.; Qiu, D.; Zhang, Y.; Wang, J. Direct Conversion of Arylamines to Pinacol Boronates: A Metal-Free Borylation Process. Angew. Chem. Int. Ed. 2010, 49, 1846–1849. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Jin, L.; Zheng, Z.; Meng, H.; Mo, F.; Wang, X.; Zhang, Y.; Wang, J. Synthesis of Pinacol Arylboronates from Aromatic Amines: A Metal-Free Transformation. J. Org. Chem. 2013, 78, 1923–1933. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Wang, S.; Tang, S.; Meng, H.; Jin, L.; Mo, F.; Zhang, Y.; Wang, J. Synthesis of Trimethylstannyl Arylboronate Compounds by Sandmeyer-Type Transformations and Their Applications in Chemoselective Cross-Coupling Reactions. J. Org. Chem. 2014, 79, 1979–1988. [Google Scholar] [CrossRef]
- Qiu, D.; Zhang, Y.; Wang, J. Direct Synthesis of Arylboronic Pinacol Esters from Arylamines. Org. Chem. Front. 2014, 1, 422–425. [Google Scholar] [CrossRef]
- Erb, W.; Albini, M.; Rouden, J.; Blanchet, J. Sequential One-Pot Access to Molecular Diversity through Aniline Aqueous Borylation. J. Org. Chem. 2014, 79, 10568–10580. [Google Scholar] [CrossRef]
- Qi, X.; Li, H.-P.; Peng, J.-B.; Wu, X.-F. Borylation of Aryldiazonium Salts at Room Temperature in an Aqueous Solution under Catalyst-Free Conditions. Tetrahedron Lett. 2017, 58, 3851–3853. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, X.; Fang, H. Additive- and Photocatalyst-Free Borylation of Arylazo Sulfones under Visible Light. J. Org. Chem. 2018, 83, 12831–12837. [Google Scholar] [CrossRef]
- Cid, J.; Carbó, J.J.; Fernández, E. A Clear-Cut Example of Selective Bpin-Bdan Activation and Precise Bdan Transfer on Boron Conjugate Addition. Chem. Eur. J. 2014, 20, 3616–3620. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Takemoto, Y.; Takaki, K. A Masked Diboron in Cu-Catalysed Borylation Reaction: Highly Regioselective Formal Hydroboration of Alkynes for Synthesis of Branched Alkenylborons. Chem. Commun. 2014, 50, 8299–8302. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Takemoto, Y.; Takaki, K. Direct Synthesis of Boron-Protected Alkenyl- and Alkylborons via Copper-Catalyzed Formal Hydroboration of Alkynes and Alkenes. Asian J. Org. Chem. 2014, 3, 1204–1209. [Google Scholar] [CrossRef]
- Miralles, N.; Cid, J.; Cuenca, A.B.; Carbo, J.J.; Fernandez, E. Mixed Diboration of Alkenes in a Metal-Free Context. Chem. Commun. 2015, 51, 1693–1696. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Takemoto, Y.; Takaki, K. Borylstannylation of Alkynes with Inverse Regioselectivity: Copper-Catalyzed Three-Component Coupling using a Masked Diboron. Chem. Commun. 2015, 51, 6297–6300. [Google Scholar] [CrossRef] [PubMed]
- Kageyuki, I.; Osaka, I.; Takaki, K.; Yoshida, H. Copper-Catalyzed B(dan)-Installing Carboboration of Alkenes. Org. Lett. 2017, 19, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Snead, R.F.; Dai, Y.; Slebodnick, C.; Yang, Y.; Yu, H.; Yao, F.; Santos, W.L. Substrate-Assisted, Transition-Metal-Free Diboration of Alkynamides with Mixed Diboron: Regio- and Stereoselective Access to trans-1,2-Vinyldiboronates. Angew. Chem. Int. Ed. 2017, 56, 5111–5115. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Huang, D.; Wu, X. Recent Advances in C–B Bond Formation through a Free Radical Pathway. Adv. Synth. Catal. 2018, 360, 1040–1053. [Google Scholar] [CrossRef]
- Chen, K.; Wang, L.; Meng, G.; Li, P. Recent Advances in Transition-Metal-Free Aryl C–B Bond Formation. Synthesis 2017, 49, 4719–4730. [Google Scholar] [CrossRef]
- Chen, K.; Cheung, M.S.; Lin, Z.; Li, P. Metal-Free Borylation of Electron-Rich Aryl (Pseudo)Halides under Continuous-Flow Photolytic Conditions. Org. Chem. Front. 2016, 3, 875–879. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, S.; He, P.; Li, P. Efficient Metal-Free Photochemical Borylation of Aryl Halides under Batch and Continuous-Flow Conditions. Chem. Sci. 2016, 7, 3676–3680. [Google Scholar] [CrossRef]
- Jiang, M.; Yang, H.; Fu, H. Visible-Light Photoredox Borylation of Aryl Halides and Subsequent Aerobic Oxidative Hydroxylation. Org. Lett. 2016, 18, 5248–5251. [Google Scholar] [CrossRef]
- Liu, W.; Yang, X.; Gao, Y.; Li, C.-J. Simple and Efficient Generation of Aryl Radicals from Aryl Triflates: Synthesis of Aryl Boronates and Aryl Iodides at Room Temperature. J. Am. Chem. Soc. 2017, 139, 8621–8627. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiao, L. Pyridine-Catalyzed Radical Borylation of Aryl Halides. J. Am. Chem. Soc. 2017, 139, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Yamane, M. Transition-Metal-Free Borylation of Aryltriazene Mediated by BF3·OEt2. Org. Lett. 2012, 14, 4560–4563. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, L.; Yan, G.-B. Metal-Free, Visible Light-Induced Borykation of Aryldiazonium Salts: A Simple and Green Synthetic Route to Arylboronates. Adv. Synth. Catal. 2012, 354, 2625–2628. [Google Scholar] [CrossRef]
- Zhang, J.-M.; Wu, H.-H.; Zhang, J.-L. Cesium Carbonate Mediated Borylation of Aryl Iodides with Diboron in Methanol. Eur. J. Org. Chem. 2013, 6263–6266. [Google Scholar] [CrossRef]
- Erb, W.; Hellal, A.; Albini, M.; Rouden, J.; Blanchet, J. An Easy Route to (Hetero)arylboronic Acids. Chem. Eur. J. 2014, 20, 6608–6612. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.-J.; Xue, D.; Jia, Z.-H.; Wang, C.; Xiao, J.-L. Methanol-Promoted Borylation of Arylamines: A Simple and Green Synthetic Method to Arylboronic Acid and Arylboronates. Synlett 2014, 25, 1577–1584. [Google Scholar] [CrossRef]
- Ahammed, S.; Nandi, S.; Kundu, D.; Ranu, B.-C. One-pot Suzuki coupling of aromatic amines via visible light photocatalyzed metal free borylation using t-BuONO at room temperature. Tetrahedron Lett. 2016, 57, 1551–1557. [Google Scholar] [CrossRef]
- Pinet, S.; Liautard, V.; Debiais, M.; Pucheault, M. Radical Metal-Free Borylation of Aryl Iodides. Synthesis 2017, 49, 4759–4768. [Google Scholar] [CrossRef]
- Mfuh, A.-M.; Nuyen, V.-T.; Chhetri, B.; Burch, J.-E.; Doyle, J.-D.; Nesterov, V.-N.; Arman, H.-D.; Larionov, O.-V. Additive- and Metal-Free, Predictably 1,2- and 1,3-Regioselective, Photoinduced Dual C-H/C-X-Borylation of Haloarenes. J. Am. Chem. Soc. 2016, 138, 8408–8411. [Google Scholar] [CrossRef]
- Mfuh, A.-M.; Doyle, J.-D.; Chhetri, B.; Arman, H.-D.; Larionov, O.-V. Scalable, Metal- and Additive-Free, Photoinduced Borylation of Haloarenes and Quaternary Arylammonium Salts. J. Am. Chem. Soc. 2016, 138, 2985–2988. [Google Scholar] [CrossRef] [PubMed]
- Iwadate, N.; Suginome, M. Synthesis of masked haloareneboronic acids via iridium-catalyzed aromatic C–H borylation with 1,8-naphthalenediaminatoborane (danBH). J. Orgomet. Chem. 2009, 694, 1713–1717. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds (1a–1t, 2, 3 and 4) are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry 1 | ArNH2 | Base | Additive | T (°C) | Yield 2 (%) |
|---|---|---|---|---|---|
| 1 | 2.0 eq. | - | - | RT | - |
| 2 | 2.0 eq. | - | - | 80 | 28 |
| 3 | 2.0 eq. | - | BPO (benzoyl peroxide) (0.1 eq.) | RT | 13 |
| 4 | 2.0 eq. | - | BPO (0.1eq.) | 80 | 31 |
| 5 | 2.0 eq. | NaOAc (1.5 eq.) | BPO (0.1 eq.) | 80 | 68 |
| 6 | 2.0 eq. | NaOAc (1.5 eq.) | - | 80 | - |
| 7 | 2.0 eq. | - | BPO (0.1 eq.), TBAI (tetrabutylammonium iodide) (0.1 eq.) | 80 | 64 |
| 8 | 2.0 eq. | - | TBAI (0.1 eq.) | 80 | 22 |
| 93 | 2.0 eq. | NaOAc (1.5 eq.) | BPO (0.1 eq.), TBAI (0.1 eq.) | 80 | 75 |
| 10 | 2.0 eq. | NaOAc (1.5 eq.) | BPO (0.1 eq.), TBAI (0.1 eq.) | RT | 10 |
| 11 | 2.0 eq. | NaOAc (1.5 eq.) | BPO (0.1 eq.), TBAI (0.1 eq.) | 50 | 35 |
| 12 | 2.0 eq. | NaOAc (1.5 eq.) | BPO (0.1 eq.), TBAI (0.1 eq.) | 100 | 50 |
| 13 | 2.0 eq. | NaOAc (1.5 eq.) | BPO (0.1 eq.), TBAI (0.1 eq.) | 120 | 20 |
| 14 | 1.0 eq. | NaOAc (1.5 eq.) | BPO (0.1 eq.), TBAI (0.1 eq.) | 80 | 33 |
| 15 | 1.2 eq. | NaOAc (1.5 eq.) | BPO (0.1 eq.), TBAI (0.1 eq.) | 80 | 41 |
| 16 | 1.5 eq. | NaOAc (1.5 eq.) | BPO (0.1 eq.), TBAI (0.1 eq.) | 80 | 52 |
| 17 | 1.8 eq. | NaOAc (1.5 eq.) | BPO (0.1 eq.), TBAI (0.1 eq.) | 80 | 58 |
| 18 | 2.0 eq. | t-BuOK (1.5 eq.) | BPO (0.1 eq.), TBAI (0.1 eq.) | 80 | 29 |
| 19 | 2.0 eq. | CsCO3 (1.5 eq.) | BPO (0.1 eq.), TBAI (0.1 eq.) | 80 | trace |
| 20 | 2.0 eq. | NaOCH3 (1.5 eq.) | BPO (0.1 eq.), TBAI (0.1 eq.) | 80 | 32 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, S.; Ma, Q.; Zhu, M.; Ren, H.; Tian, S.; Zhao, Y.; Miao, Z. Direct Transformation from Arylamines to Aryl Naphthalene-1,8-diamino Boronamides: A Metal-Free Sandmeyer-Type Process. Molecules 2019, 24, 377. https://doi.org/10.3390/molecules24030377
Ding S, Ma Q, Zhu M, Ren H, Tian S, Zhao Y, Miao Z. Direct Transformation from Arylamines to Aryl Naphthalene-1,8-diamino Boronamides: A Metal-Free Sandmeyer-Type Process. Molecules. 2019; 24(3):377. https://doi.org/10.3390/molecules24030377
Chicago/Turabian StyleDing, Siyi, Qiang Ma, Min Zhu, Huaping Ren, Shaopeng Tian, Yuzhen Zhao, and Zongcheng Miao. 2019. "Direct Transformation from Arylamines to Aryl Naphthalene-1,8-diamino Boronamides: A Metal-Free Sandmeyer-Type Process" Molecules 24, no. 3: 377. https://doi.org/10.3390/molecules24030377
APA StyleDing, S., Ma, Q., Zhu, M., Ren, H., Tian, S., Zhao, Y., & Miao, Z. (2019). Direct Transformation from Arylamines to Aryl Naphthalene-1,8-diamino Boronamides: A Metal-Free Sandmeyer-Type Process. Molecules, 24(3), 377. https://doi.org/10.3390/molecules24030377