
Ultrasound-Assisted Solvent Extraction of a Porous Membrane Packed Sample for the Determination of Tobacco-Specific Nitrosamines in the Replacement Liquids for E-Cigarettes


Abstract
:1. Introduction
2. Results
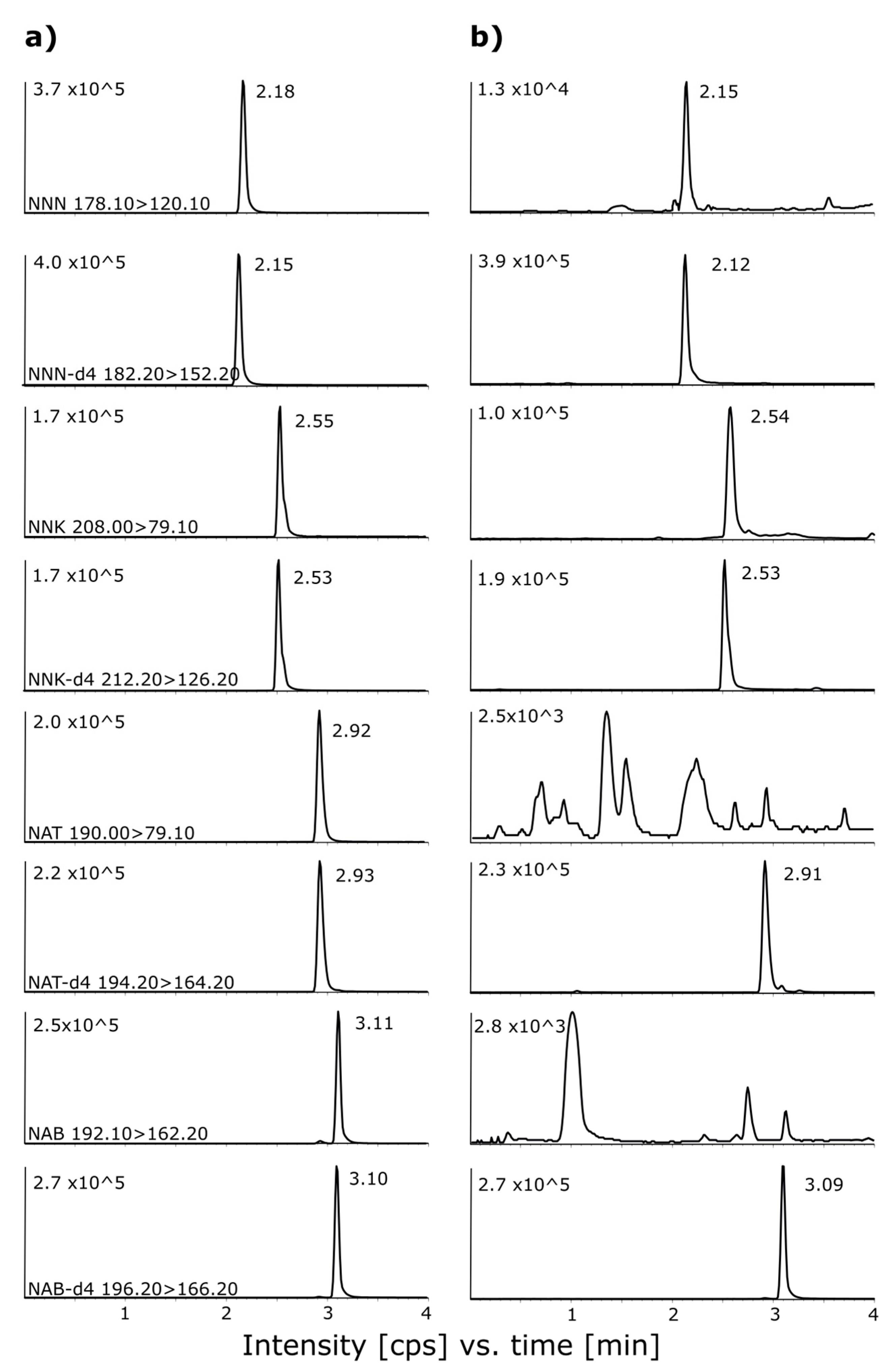
2.1. Detection Parameters and Chromatographic Separation
2.2. Optimization of the Extraction Procedure
2.3. Method Validation
2.4. Analysis of Real Samples
3. Discussion
4. Materials and Methods
4.1. Standards and Solutions Preparations
4.2. Instruments and Analytical Conditions
4.3. Sample Preparation of the Replacement Liquids and Spiked Samples
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Cheng, T. Chemical evaluation of electronic cigarettes. Tob. Control 2014, 23, ii11–ii17. [Google Scholar] [CrossRef] [PubMed]
- Aszyk, J.; Kubica, P.; Woźniak, M.K.; Namieśnik, J.; Wasik, A.; Kot-Wasik, A. Evaluation of flavour profiles in e-cigarette refill solutions using gas chromatography–tandem mass spectrometry. J. Chromatogr. A 2018, 1547, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Kubica, P.; Kot-Wasik, A.; Wasik, A.; Namieśnik, J. “Dilute & Shoot” approach for rapid determination of trace amounts of nicotine in zero-level e-liquids by reversed phase liquid chromatography and hydrophilic interactions liquid chromatography coupled with tandem mass spectrometry-electrospray ionization. J. Chromatogr. A 2013, 1289, 13–18. [Google Scholar] [PubMed]
- Farsalinos, K.E.; Gillman, G.; Poulas, K.; Voudris, V. Tobacco-Specific Nitrosamines in Electronic Cigarettes: Comparison between liquid and aerosol levels. Int. J. Environ. Res. Public Health 2015, 12, 9046–9053. [Google Scholar] [CrossRef] [Green Version]
- Farsalinos, K.E.; Polosa, R. Safety evaluation and risk assessment of electronic cigarettes as tobacco cigarette substitutes: A systematic review. Ther. Adv. Drug Saf. 2014, 5, 67–86. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S. Biochemistry, biology, and carcinogenicity of tobacco-specific N- nitrosamines. Chem. Res. Toxicol. 1998, 11, 559–603. [Google Scholar] [CrossRef]
- Wagner, K.A.; Finkel, N.H.; Fossett, J.E.; Gillman, I.G. Development of a quantitative method for the analysis of tobacco-specific nitrosamines in mainstream cigarette smoke using isotope dilution liquid chromatography/electrospray ionization tandem mass spectrometry. Anal. Chem. 2005, 77, 1001–1006. [Google Scholar] [CrossRef]
- Xiong, W.; Hou, H.; Jiang, X.; Tang, G.; Hu, Q. Simultaneous determination of four tobacco-specific N-nitrosamines in mainstream smoke for Chinese Virginia cigarettes by liquid chromatography-tandem mass spectrometry and validation under ISO and “Canadian intense” machine smoking regimes. Anal. Chim. Acta 2010, 674, 71–78. [Google Scholar] [CrossRef]
- Kim, H.J.; Shin, H.S. Determination of tobacco-specific nitrosamines in replacement liquids of electronic cigarettes by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2013, 1291, 48–55. [Google Scholar] [CrossRef]
- Farsalinos, K.E.; Gene Gillman, I.; Melvin, M.S.; Paolantonio, A.R.; Gardow, W.J.; Humphries, K.E.; Brown, S.E.; Poulas, K.; Voudris, V. Nicotine levels and presence of selected tobacco-derived toxins in tobacco flavoured electronic cigarette refill liquids. Int. J. Environ. Res. Public Health 2015, 12, 3439–3452. [Google Scholar] [CrossRef]
- World Health Organization International Agency for research on cancer. Proceedings of the IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, Lyon, France, 11–18 June 2002.
- Famele, M.; Ferranti, C.; Abenavoli, C.; Palleschi, L.; Mancinelli, R.; Draisci, R. The chemical components of electronic cigarette cartridges and refill fluids: Review of analytical methods. Nicotine Tob. Res. 2015, 17, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Jo, S.-H.; Kim, K.-H.; Lee, S.; Brown, R.; Jo, S.-H. Analytical Method for Measurement of Tobacco-Specific Nitrosamines in E-Cigarette Liquid and Aerosol. Appl. Sci. 2018, 8, 2699. [Google Scholar] [CrossRef] [Green Version]
- Ishizaki, A.; Kataoka, H. A sensitive method for the determination of tobacco-specific nitrosamines in mainstream and sidestream smokes of combustion cigarettes and heated tobacco products by online in-tube solid-phase microextraction coupled with liquid chromatography-tandem mass. Anal. Chim. Acta 2019, 1075, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Determination of Tobacco Specific Nitrosamines in Mainstream Cigarette Smoke by LC-MS/MS. Available online: https://www.coresta.org/determination-tobacco-specific-nitrosamines-mainstream-cigarette-smoke-lc-msms-29198.html (accessed on 17 December 2019).
- Wu, M.J.; Dai, Y.H.; Tuo, S.X.; Hu, N.N.; Li, Y.; Chen, X.M. Analysis of four tobacco-specific nitrosamines in mainstream cigarette smoke of Virginia cigarettes by LC-MS/MS. J. Cent. South Univ. Technol. (English Ed.) 2008, 15, 627–631. [Google Scholar] [CrossRef]
- Kavvadias, D.; Scherer, G.; Urban, M.; Cheung, F.; Errington, G.; Shepperd, J.; McEwan, M. Simultaneous determination of four tobacco-specific N-nitrosamines (TSNA) in human urine. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Wang, C.; Lin, S.; Hsieh, D.P.H. Liquid chromatography/tandem mass spectrometric method for the simultaneous determination of tobacco-specific nitrosamine NNK and its five metabolites. Talanta 2007, 73, 76–80. [Google Scholar] [CrossRef]
- Hu, C.W.; Hsu, Y.W.; Chen, J.L.; Tam, L.M.; Chao, M.R. Direct analysis of tobacco-specific nitrosamine NNK and its metabolite NNAL in human urine by LC-MS/MS: Evidence of linkage to methylated DNA lesions. Arch. Toxicol. 2014, 88, 291–299. [Google Scholar] [CrossRef]
- Wu, W.; Ashley, D.L.; Watson, C.H. Simultaneous determination of five tobacco-specific nitrosamines in mainstream cigarette smoke by isotope dilution liquid chromatography/electrospray ionization tandem mass spectrometry. Anal. Chem. 2003, 75, 4827–4832. [Google Scholar] [CrossRef]
- Kavvadias, D.; Scherer, G.; Cheung, F.; Errington, G.; Shepperd, J.; McEwan, M. Determination of tobacco-specific N-nitrosamines in urine of smokers and non-smokers Tobacco-specific N-nitrosamines in urine of smokers and non-smokers D. Kavvadias et al. Biomarkers 2009, 14, 547–553. [Google Scholar] [CrossRef]
- Brunnemann, K.D.; Hoffmann, D. Analytical studies on tobacco-specific n-nitrosamines in tobacco and tobacco smoke. Crit. Rev. Toxicol. 1991, 21, 235–240. [Google Scholar] [CrossRef]
- Stepanov, I.; Carmella, S.G.; Hecht, S.S.; Duca, G. Analysis of tobacco-specific nitrosamines in Moldovan cigarette tobacco. J. Agric. Food Chem. 2002, 50, 2793–2797. [Google Scholar] [CrossRef] [PubMed]
- Carmella, S.G.; McIntee, E.J.; Chen, M.; Hecht, S.S. Enantiomeric composition of N’-nitrosonornicotine and N’-nitrosoanatabine in tobacco. Carcinogenesis 2000, 21, 839–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Determination of Tobacco Specific Nitrosamines in Cigarette Mainstream Smoke—GC-TEA Method. Available online: https://www.coresta.org/determination-tobacco-specific-nitrosamines-cigarette-mainstream-smoke-gc-tea-method-29186.html (accessed on 17 December 2019).
- Determination of Tobacco-Specific Nitrosamines in Tobacco and Tobacco Products by LC-MS/MS. Available online: https://www.coresta.org/sites/default/files/technical_documents/main/CRM_72-July2017.pdf (accessed on 17 December 2019).
- Goniewicz, M.L.; Knysak, J.; Gawron, M.; Kosmider, L.; Sobczak, A.; Kurek, J.; Prokopowicz, A.; Jablonska-Czapla, M.; Rosik-Dulewska, C.; Havel, C.; et al. Levels of selected carcinogens and toxicants in vapour from electronic cigarettes. Tob. Control 2014, 23, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laugesen, M. Safety Report on the Ruyan® e-cigarette Cartridge and Inhaled Aerosol. Available online: http://www.healthnz.co.nz/RuyanCartridgeReport30-Oct-08.pdf (accessed on 17 December 2019).
- Płotka-Wasylka, J.; Szczepańska, N.; de la Guardia, M.; Namieśnik, J. Miniaturized solid-phase extraction techniques. TrAC-Trends Anal. Chem. 2015, 73, 19–38. [Google Scholar] [CrossRef]
- Sajid, M.; Woźniak, M.K.; Płotka-Wasylka, J. Ultrasound-assisted solvent extraction of porous membrane packed solid samples: A new approach for extraction of target analytes from solid samples. Microchem. J. 2019, 144, 117–123. [Google Scholar] [CrossRef]
- Robles, A.D.; Fabjanowicz, M.; Płotka-Wasylka, J.; Konieczka, P. Organic Acids and Polyphenols Determination in Polish Wines by Ultrasound-Assisted Solvent. Molecules 2019, 24, 4376. [Google Scholar] [CrossRef] [Green Version]
- Konieczka, P.; Namieśnik, J. Quality Assurance and Quality Control in the Analytical Chemical Laboratory; Taylor&Francis Group: Boca Raton, FL, USA, 2018. [Google Scholar]
Sample Availability: Samples of the compounds are available from the author after personal communication. |


{kind=link}
{kind=link}
| Analyte | Precursor Ion [M + H]+ [m/z] | Product Ions [m/z] | Collision Energy [V] | Q1 Prerod [V] | Q3 Prerod [V] |
|---|---|---|---|---|---|
| NNK | 208.00 | 79.10 1 | −26 | −40 | −29 |
| 148.20 | −11 | −12 | −13 | ||
| NNN | 178.10 | 120.10 1 | −14 | −19 | −25 |
| 148.20 | −21 | −13 | −30 | ||
| NAT | 190.00 | 79.10 1 | −14 | −29 | −13 |
| 160.20 | −14 | −11 | −16 | ||
| NAB | 192.10 | 162.20 1 | −10 | −14 | −29 |
| 133.20 | −14 | −19 | −29 | ||
| NNK-d4 | 212.20 | 126.20 1 | −10 | −12 | −22 |
| 152.15 | −10 | −13 | −30 | ||
| NNN-d4 | 182.20 | 152.20 1 | −14 | −12 | −14 |
| 122.10 | −12 | −29 | −12 | ||
| NAT-d4 | 194.20 | 164.20 1 | −20 | −12 | −16 |
| 110.20 | −10 | −17 | −23 | ||
| NAB-d4 | 196.20 | 166.20 1 | −14 | −13 | −30 |
| 137.20 | −19 | −23 | −23 |
| Analyte | Calibration Curve Equation (6 Points, n = 3) | Sa | Sb | LOD [ng/g] | LOQ [ng/g] | R2 |
|---|---|---|---|---|---|---|
| NNK | y = 0.01211x + 0.0045 | 0.00029 | 0.0031 | 0.9 | 2.6 | 0.9931 |
| NNN | y = 0.08552x − 0.0014 | 0.00070 | 0.0075 | 0.3 | 0.9 | 0.9992 |
| NAT | y = 0.05133x − 0.0016 | 0.00041 | 0.0044 | 0.3 | 0.9 | 0.9992 |
| NAB | y = 0.05865x + 0.00022 | 0.00073 | 0.00791 | 0.4 | 1.3 | 0.9981 |
| Analyte | Spiking Level | Mean Recovery [ng/g] (%), n = 6 | SD [ng/g] | CV [%] | Repeatability, n = 6 | |||
|---|---|---|---|---|---|---|---|---|
| Day | Mean Recovery [ng/g] (%) | SD [ng/g] | CV [%] | |||||
| NNK | 5 | 4.7 (93) | 0.2 | 3.3 | 1 | 19.0 (95) | 0.7 | 3.6 |
| 20 | 19.0 (95) | 0.7 | 3.6 | 2 | 19.3 (96) | 0.6 | 2.9 | |
| 40 | 41.5 (104) | 0.9 | 2.1 | 3 | 18.5 (92) | 0.8 | 4.2 | |
| NNN | 5 | 4.9 (97) | 0.2 | 4.0 | 1 | 20.9 (105) | 0.8 | 3.7 |
| 20 | 20.9 (105) | 0.8 | 3.7 | 2 | 21.2 (106) | 0.9 | 4.1 | |
| 40 | 41.4 (104) | 0.9 | 2.2 | 3 | 20.5 (103) | 0.7 | 3.2 | |
| NAT | 5 | 4.6 (92) | 0.2 | 3.2 | 1 | 18.0 (90) | 0.5 | 2.5 |
| 20 | 18.0 (90) | 0.5 | 2.5 | 2 | 17.7 (88) | 0.3 | 1.8 | |
| 40 | 34.8 (87) | 0.7 | 1.9 | 3 | 18.6 (93) | 0.6 | 3.2 | |
| NAB | 5 | 4.9 (97) | 0.2 | 4.9 | 1 | 19.4 (97) | 0.4 | 2.2 |
| 20 | 19.4 (97) | 0.4 | 2.2 | 2 | 18.9 (95) | 0.8 | 4.1 | |
| 40 | 37.5 (94) | 0.7 | 1.9 | 3 | 18.7 (93) | 0.6 | 3.3 | |
| No. Sample and Flavour | Amount of Sample [g] | PG/VG Ratio; (Density) [g/cm3] | NNK ± SD [ng/mL] | NNN ± SD [ng/mL] | NAT ± SD [ng/mL] | NAB ± SD [ng/mL] |
|---|---|---|---|---|---|---|
| Producer 1 | 50/50; (1.15) | |||||
| 1. Cherry | 1.059 | 1.20 ± 0.05 | - | - | - | |
| Producer 2 | 60/40; (1.13) | |||||
| 2. Fruit mix | 1.102 | 2.6 ± 0.3 | 7.5 ± 0.2 | - | - | |
| 3. Tobacco | 1.092 | 7.6 ± 0.4 | - | 1.7 ± 0.1 | - | |
| 4. Vanilla | 1.088 | 7.5 ± 0.6 | 4.1 ± 0.6 | - | - | |
| 5. Mint | 1.095 | 2.9 ± 0.3 | 15.6 ± 1.3 | - | - | |
| Producer 3 | 60/40; (1.13) | |||||
| 6. Apple | 1.066 | 54.3 ± 3.0 | 4.2 ± 0.2 | - | - | |
| 7. Mint | 1.057 | - | 4.6 ± 0.2 | - | - | |
| Producer 4 | 80/20; (1.08) | |||||
| 8. Strawberry | 1.047 | 16.1 ± 0.9 | - | - | - | |
| 9. Tobacco | 1.031 | 35.8 ± 2.0 | 30.2 ± 1.1 | 2.5 ± 0.1 | - |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubica, P. Ultrasound-Assisted Solvent Extraction of a Porous Membrane Packed Sample for the Determination of Tobacco-Specific Nitrosamines in the Replacement Liquids for E-Cigarettes. Molecules 2019, 24, 4618. https://doi.org/10.3390/molecules24244618
Kubica P. Ultrasound-Assisted Solvent Extraction of a Porous Membrane Packed Sample for the Determination of Tobacco-Specific Nitrosamines in the Replacement Liquids for E-Cigarettes. Molecules. 2019; 24(24):4618. https://doi.org/10.3390/molecules24244618
Chicago/Turabian StyleKubica, Paweł. 2019. "Ultrasound-Assisted Solvent Extraction of a Porous Membrane Packed Sample for the Determination of Tobacco-Specific Nitrosamines in the Replacement Liquids for E-Cigarettes" Molecules 24, no. 24: 4618. https://doi.org/10.3390/molecules24244618