
Postmortem Quantitative Analysis of Prion Seeding Activity in the Digestive System

 , ,
, , 
Abstract
:1. Introduction
2. Results
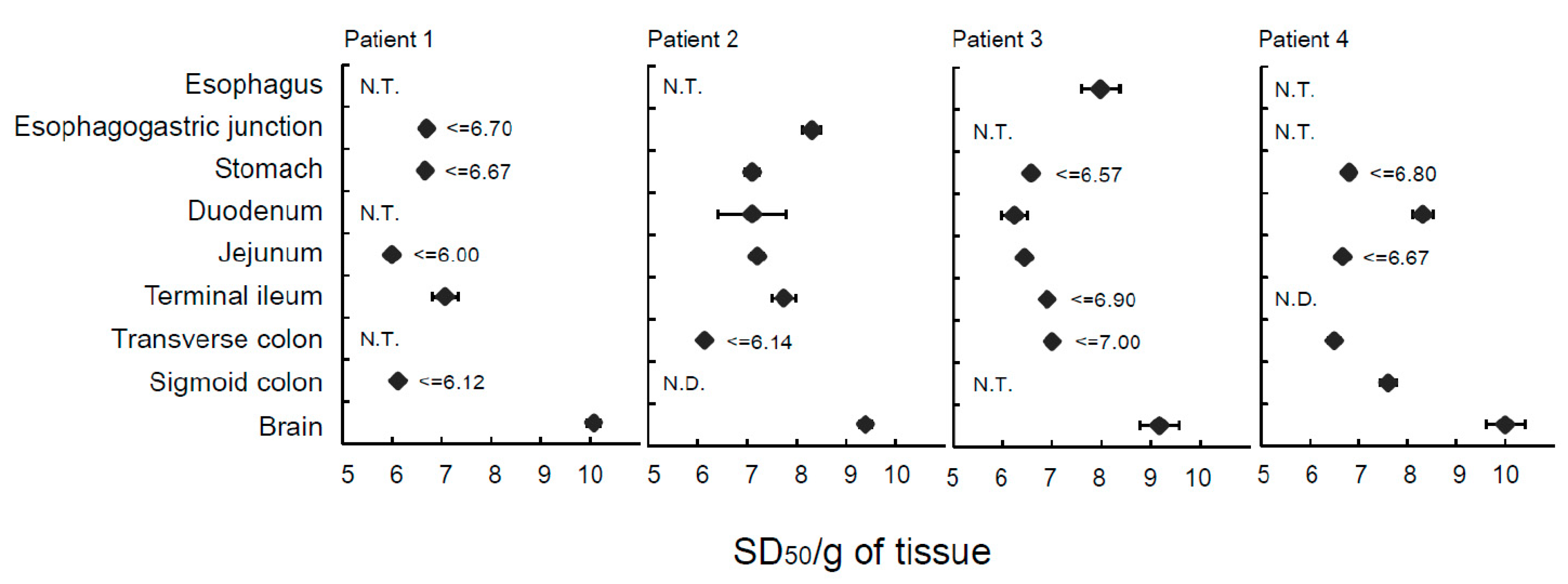
2.1. Analysis of Extraneural Tissues from Patients with sCJD and Healthy Subjects Using Endpoint RT-QuIC Assay
2.2. Analysis of Extraneural Tissues from a Patient with GSS and a Patient with Genetic HPD Using Endpoint RT-QuIC Assay
2.3. Immunohistochemical Staining of the Gut
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Tissue Homogenate Preparation
4.3. Endpoint RT-QuIC
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thadani, V.; Penar, P.L.; Partington, J.; Kalb, R.; Janssen, R.; Schonberger, L.B.; Rabkin, C.S.; Prichard, J.W. Creutzfeldt-Jakob disease probably acquired from a cadaveric dura mater graft. Case Rep. J. Neurosurgery 1988, 69, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Bernoulli, C.; Siegfried, J.; Baumgartner, G.; Regli, F.; Rabinowicz, T.; Gajdusek, D.C.; Gibbs, C.J., Jr. Danger of accidental person-to-person transmission of Creutzfeldt-Jakob disease by surgery. Lancet 1977, 1, 478–479. [Google Scholar] [CrossRef]
- Will, R.G.; Matthews, W.B. Evidence for case-to-case transmission of Creutzfeldt-Jakob disease. J. Neurol. Neurosurg. Psychiatry 1982, 45, 235–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubenstein, R.; Chang, B. Re-assessment of PrP(Sc) distribution in sporadic and variant CJD. PLoS ONE 2013, 8, e66352. [Google Scholar] [CrossRef] [PubMed]
- Glatzel, M.; Abela, E.; Maissen, M.; Aguzzi, A. Extraneural pathologic prion protein in sporadic Creutzfeldt-Jakob disease. N. Engl. J. Med. 2003, 349, 1812–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atarashi, R.; Satoh, K.; Sano, K.; Fuse, T.; Yamaguchi, N.; Ishibashi, D.; Matsubara, T.; Nakagaki, T.; Yamanaka, H.; Shirabe, S.; et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat. Med. 2011, 17, 175–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takatsuki, H.; Satoh, K.; Sano, K.; Fuse, T.; Nakagaki, T.; Mori, T.; Ishibashi, D.; Mihara, B.; Takao, M.; Iwasaki, Y.; et al. Rapid and Quantitative Assay of Amyloid-Seeding Activity in Human Brains Affected with Prion Diseases. PLoS ONE 2015, 10, e0126930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.; Mitteregger-Kretzschmar, G.; Giese, A.; Kretzschmar, H.A. Establishing quantitative real-time quaking-induced conversion (qRT-QuIC) for highly sensitive detection and quantification of PrPSc in prion-infected tissues. Acta Neuropathol. Commun. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orrú, C.D.; Bongianni, M.; Tonoli, G.; Ferrari, S.; Hughson, A.G.; Groveman, B.R.; Fiorini, M.; Pocchiari, M.; Monaco, S.; Caughey, B.; et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N. Engl. J. Med. 2014, 371, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Manca, M.; Foutz, A.; Camacho, M.V.; Raymond, G.J.; Race, B.; Orru, C.D.; Yuan, J.; Shen, P.; Li, B.; et al. Early preclinical detection of prions in the skin of prion-infected animals. Nat. Commun. 2019, 10, 247. [Google Scholar] [CrossRef] [PubMed]
- Baiardi, S.; Redaelli, V.; Ripellino, P.; Rossi, M.; Franceschini, A.; Moggio, M.; Sola, P.; Ladogana, A.; Fociani, P.; Magherini, A.; et al. Prion-related peripheral neuropathy in sporadic Creutzfeldt-Jakob disease. J. Neurol. Neurosurg. Psychiatry 2019, 90, 424–442. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N. The Genotype-Tissue Expression (GTEx) project. Nat. Genetics. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Mardinoglu, A.; Kampf, C.; Sjöstedt, E.; Asplund, A. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Mori, K.; Ito, M.; Kawai, Y.; Hoshino, K.I.; Kawabata, Y.; Mimuro, M.; Yoshida, M. Gastrostomy in patients with prion disease. Prion. 2017, 11, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilham, J.M.; Orru, C.D.; Bessen, R.A.; Atarashi, R.; Sano, K.; Race, B.; Meade-White, K.D.; Taubner, L.M.; Timmes, A.; Caughey, B. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathogens. 2010, 6, e1001217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, Y.; Mohri, S.; Ironside, J.W.; Muramoto, T.; Kitamoto, T. Humanized knock-in mice expressing chimeric prion protein showed varied susceptibility to different human prions. Am. J. Pathol. 2003, 163, 2585–2593. [Google Scholar] [CrossRef] [Green Version]
- Collinge, J.; Whitfield, J.; McKintosh, E.; Beck, J.; Mead, S.; Thomas, D.J.; Alpers, M.P. Kuru in the 21st century—An acquired human prion disease with very long incubation periods. Lancet 2006, 367, 2068–2074. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
| Tissue | Patient 1 | Patient 2 | Patient 3 | Patient 4 | ||||
|---|---|---|---|---|---|---|---|---|
| Mean #3 | ±S.D. | Mean | ±S.D. | Mean | ±S.D. | Mean | ±S.D. | |
| Esophagus | ≤6.70 | 8.38 | ±0.16 | 7.98 | ±0.39 | N.E. | ||
| Stomach | ≤6.50 | 7.10 | ±0.14 | ≤6.57 | ≤6.80 | |||
| Duodenum | N.E | 7.10 | ±0.70 | 6.24 | ±0.25 | 8.31 | ±0.20 | |
| Jejunum | ≤6.67 | 7.20 | ±0.12 | 6.44 | ±0.10 | ≤6.67 | ||
| Terminal ileum | 7.07 | ±0.26 | 7.74 | ±0.25 | ≤6.90 | N.D. | ||
| Transverse colon | N.E. | ≤6.14 | ≤7.00 | 6.50 | ±0.11 | |||
| Sigmoid colon | ≤6.12 | N.D. | N.E. | 7.60 | ±0.17 | |||
| Brain #3 | 10.08 | ±0.12 | 9.42 | ±0.12 | 9.17 | ±0.42 | 10.00 | ±0.35 |
| Tissue | Patient 5 | |
|---|---|---|
| Mean | ±S.D. | |
| Esophagus | 6.93 | ±0.32 |
| Stomach | 7.34 | ±0.20 |
| Duodenum | ≤6.57 | |
| Jejunum | 7.22 | ±0.30 |
| Appendix | 8.33 | ±0.67 |
| Transverse colon | 8.46 | ±0.44 |
| Sigmoid colon | N.E. | |
| Brain | 10.18 | ±0.34 |
| Tissue | Patient 6 | |
|---|---|---|
| Mean | ±S.D. | |
| Esophagus | 8.5 | ±0.00 |
| Stomach | 8.21 | ±0.34 |
| Duodenum | 6.81 | ±0.31 |
| Jejunum | ≤6.33 | ±0.00 |
| Ileum | 7.17 | ±0.94 |
| Appendix | 7.23 | ±0.20 |
| Cecum | ≤6.64 | |
| Transverse colon | ≤5.80 | |
| Sigmoid colon | 7.47 | ±0.50 |
| Brain | 10.3 | ±0.35 |
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | |
|---|---|---|---|---|---|---|
| Sporadic CJD | Sporadic CJD | Sporadic CJD | Sporadic CJD | GSS (P102L) | Genetic CJD (E200K) | |
| sex | male | male | female | male | male | male |
| codon 129 | MV | MM | MM | MM | MM | MM |
| Typing of PrP-res | type 2 | type 1 | type 1 | type 1 | - | type 1 |
| Age at onset (years) | 69 | 70 | 59 | 62 | 43 | 66 |
| Period from onset to death (months) | 27 | 18 | 78 | 60 | 39 | 6 |
| Period from onset to akinetic mutism (months) | 3 | 2 | 3 | 4 | 25 | 3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Satoh, K.; Fuse, T.; Nonaka, T.; Dong, T.; Takao, M.; Nakagaki, T.; Ishibashi, D.; Taguchi, Y.; Mihara, B.; Iwasaki, Y.; et al. Postmortem Quantitative Analysis of Prion Seeding Activity in the Digestive System. Molecules 2019, 24, 4601. https://doi.org/10.3390/molecules24244601
Satoh K, Fuse T, Nonaka T, Dong T, Takao M, Nakagaki T, Ishibashi D, Taguchi Y, Mihara B, Iwasaki Y, et al. Postmortem Quantitative Analysis of Prion Seeding Activity in the Digestive System. Molecules. 2019; 24(24):4601. https://doi.org/10.3390/molecules24244601
Chicago/Turabian StyleSatoh, Katsuya, Takayuki Fuse, Toshiaki Nonaka, Trong Dong, Masaki Takao, Takehiro Nakagaki, Daisuke Ishibashi, Yuzuru Taguchi, Ban Mihara, Yasushi Iwasaki, and et al. 2019. "Postmortem Quantitative Analysis of Prion Seeding Activity in the Digestive System" Molecules 24, no. 24: 4601. https://doi.org/10.3390/molecules24244601
APA StyleSatoh, K., Fuse, T., Nonaka, T., Dong, T., Takao, M., Nakagaki, T., Ishibashi, D., Taguchi, Y., Mihara, B., Iwasaki, Y., Yoshida, M., & Nishida, N. (2019). Postmortem Quantitative Analysis of Prion Seeding Activity in the Digestive System. Molecules, 24(24), 4601. https://doi.org/10.3390/molecules24244601