
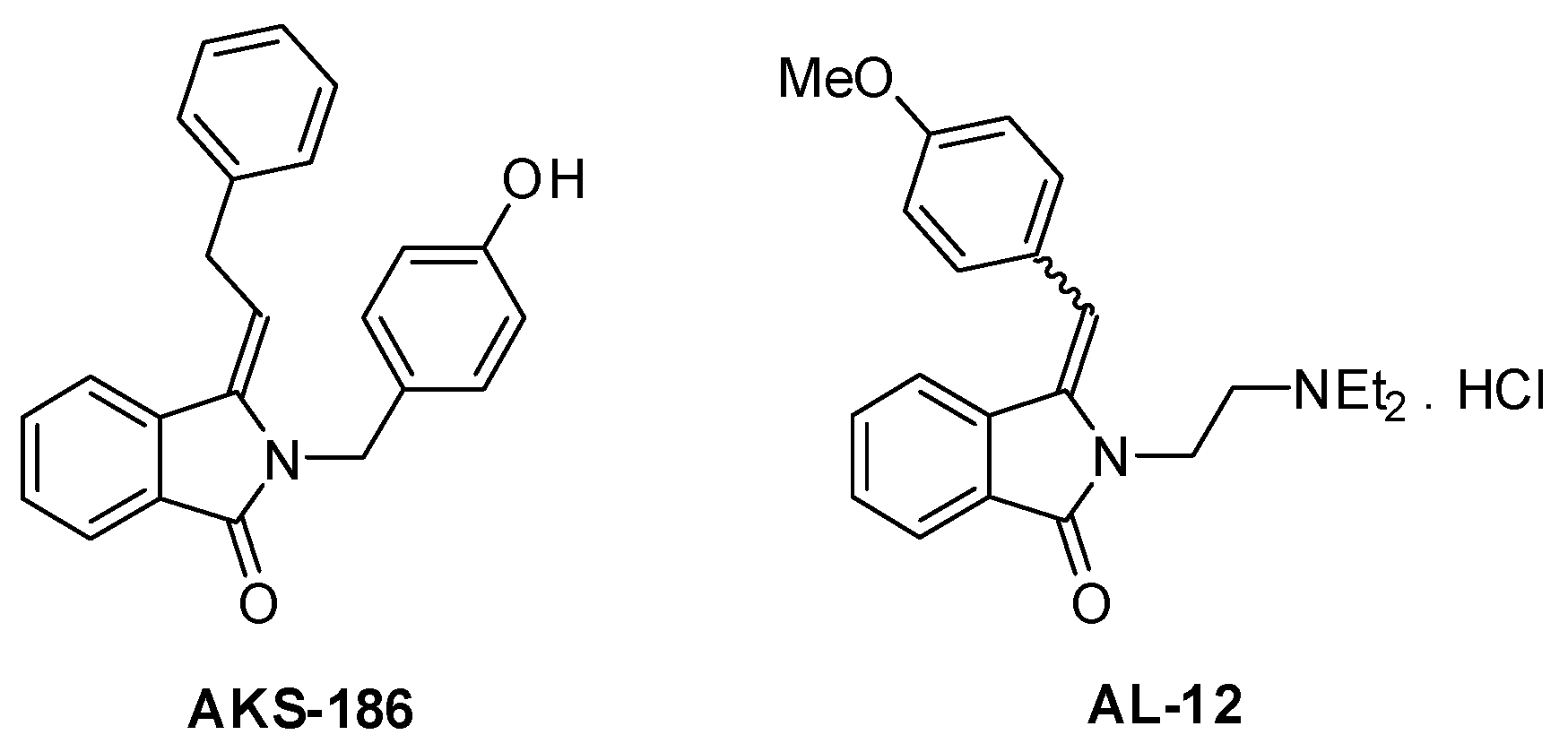
Continuous Flow Photochemical and Thermal Multi-Step Synthesis of Bioactive 3-Arylmethylene-2,3-Dihydro-1H-Isoindolin-1-Ones



Abstract
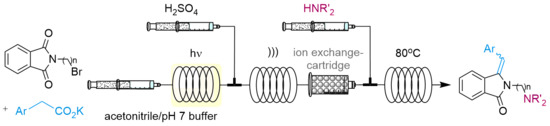
:
1. Introduction
2. Results
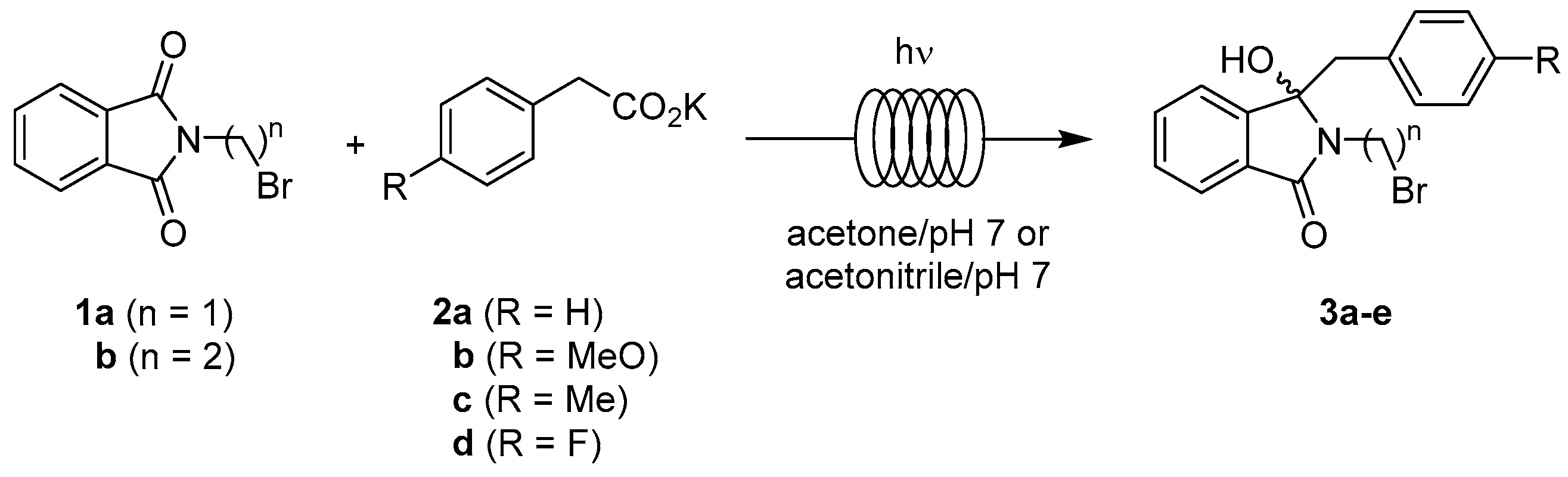
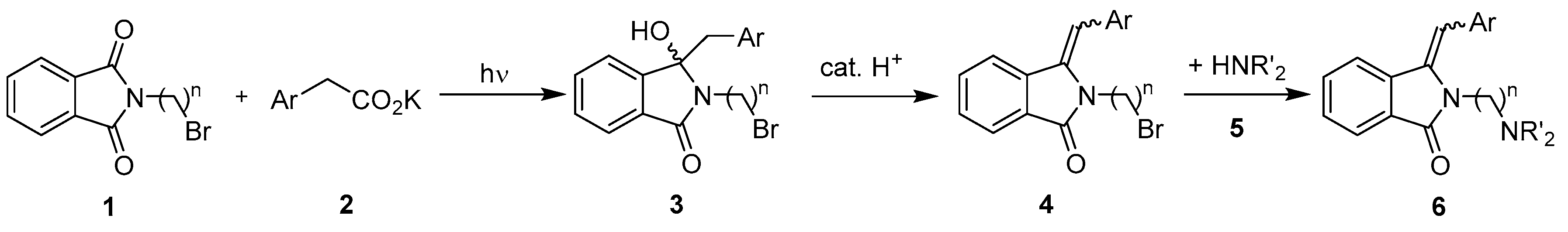
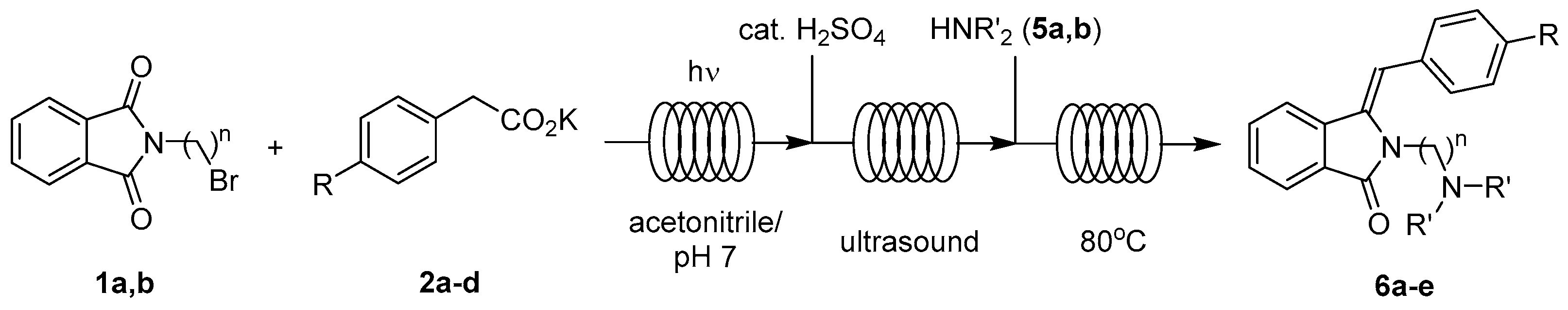
2.1. Synthesis Optimization
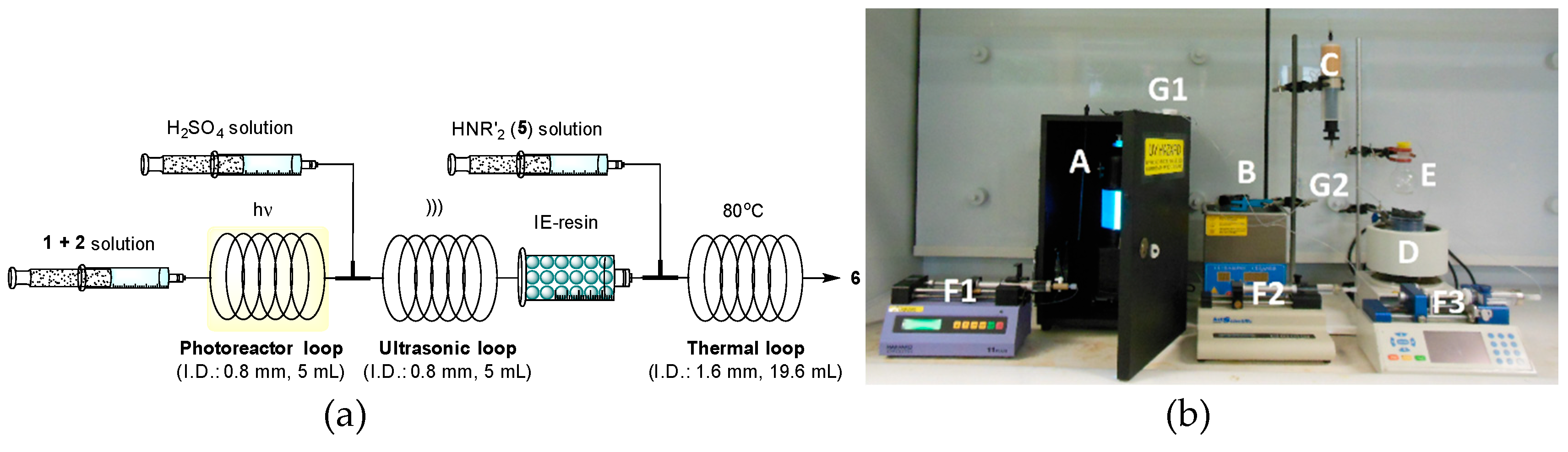
2.2. Flow Reactor Design
2.3. Continuous-Flow Operations
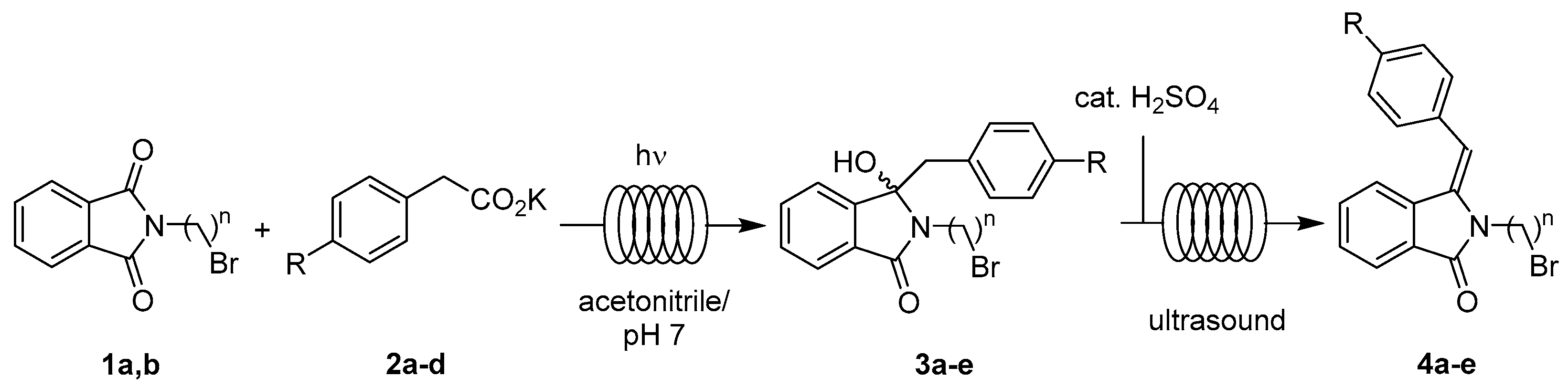
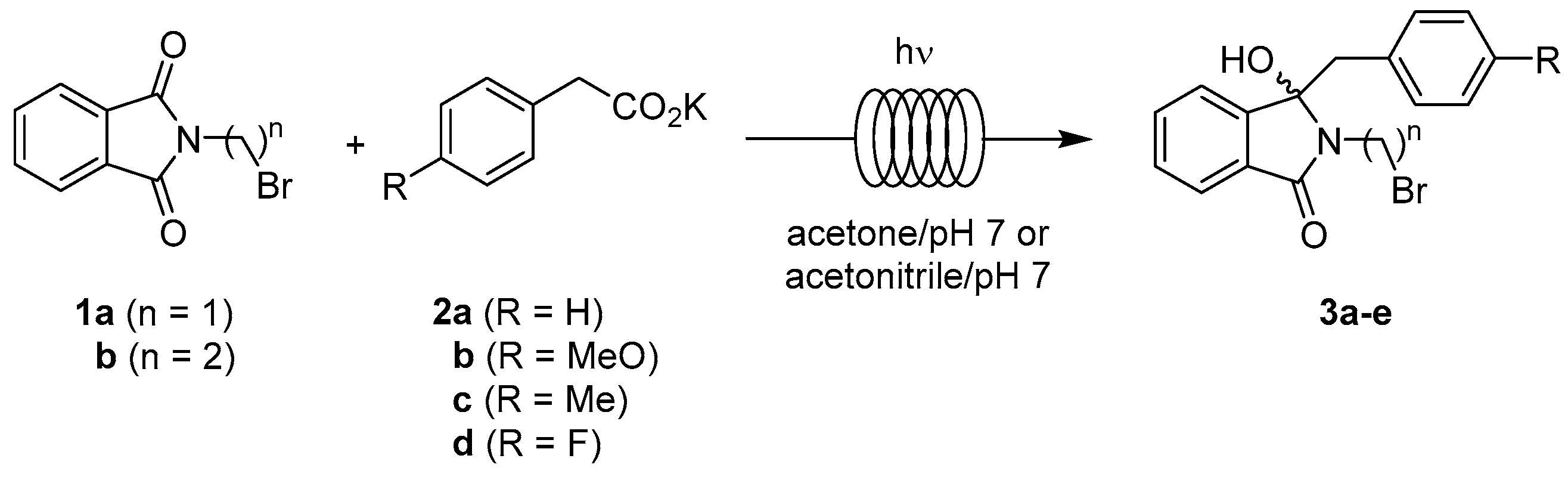
2.3.1. Photodecarboxylation
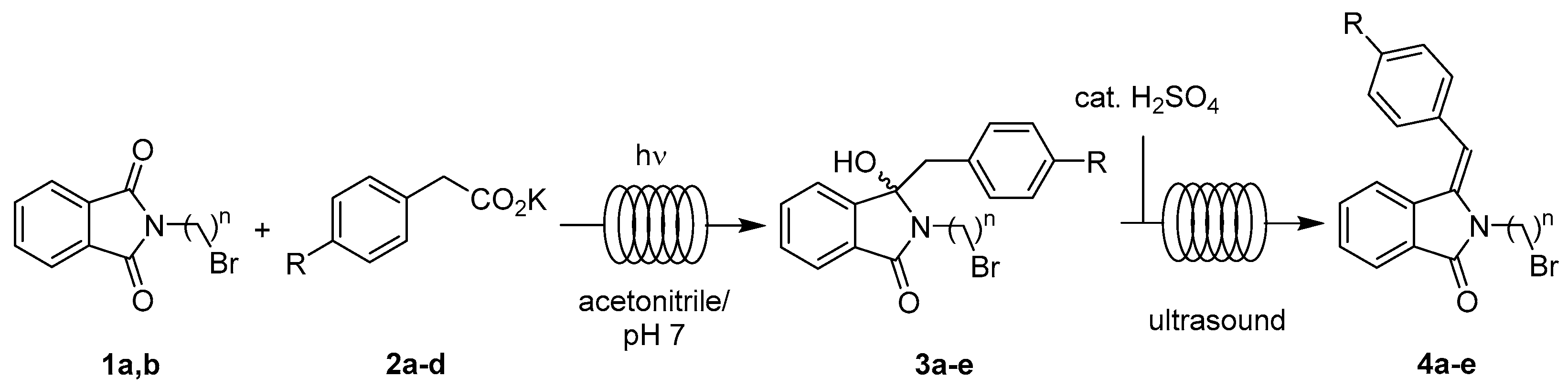
2.3.2. Photodecarboxylation-Dehydration Coupling
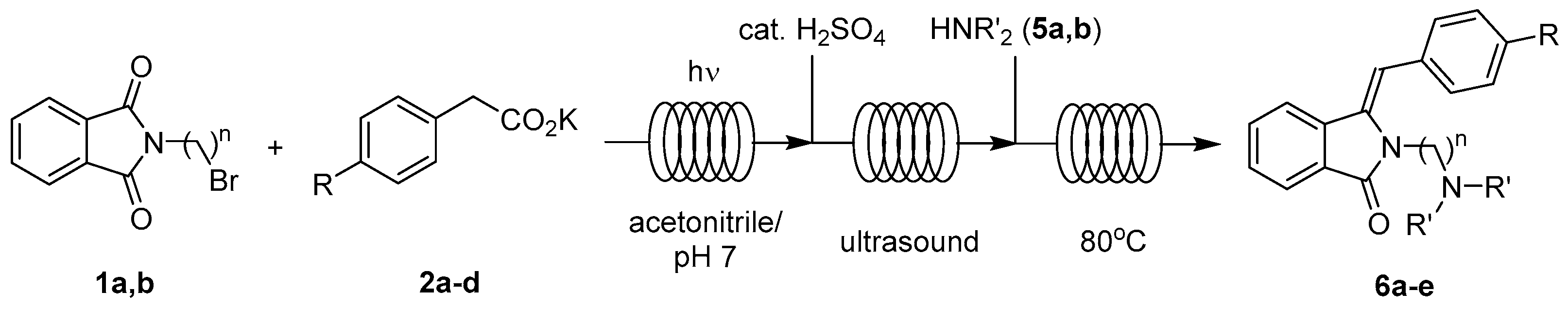
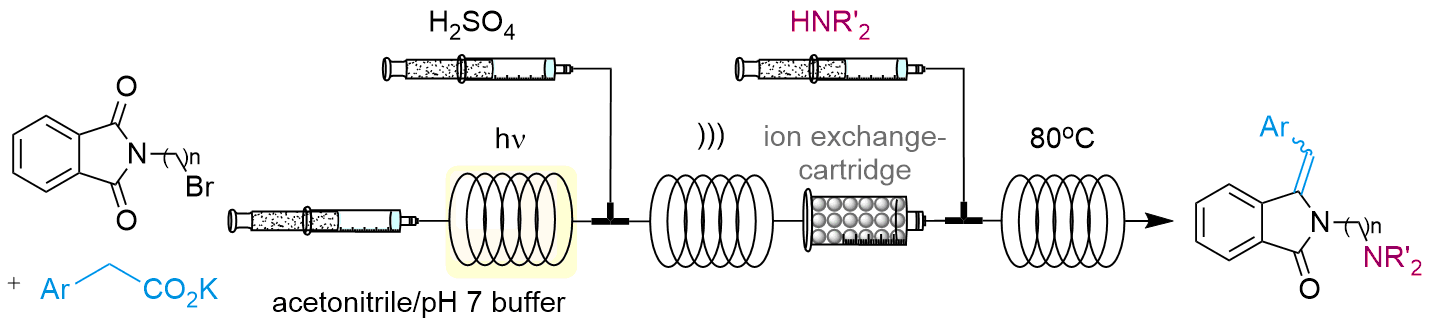
2.3.3. In Series Multistep Flow Operation
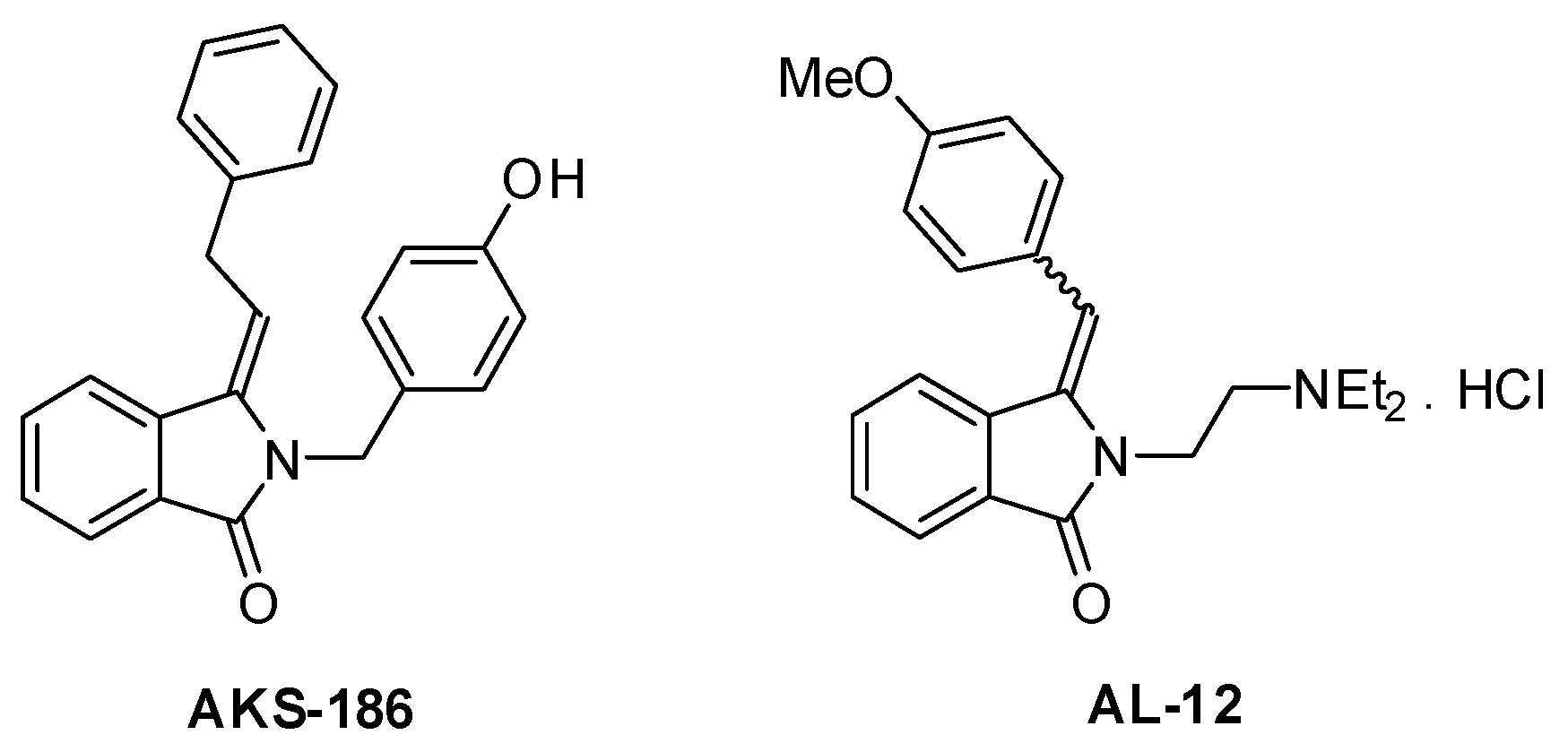
3. Discussion
4. Materials and Methods
4.1. Experimental Procedures
4.1.1. Photodecarboxylations under Flow Conditions
4.1.2. Coupled Photodecarboxylative Addition and Dehydration under Flow Conditions
4.1.3. In Series Photodecarboxylative Addition with Thermal Dehydration and Amination under Flow Conditions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bently, K.W. β-Phenylethylamines and the isoquinoline alkaloids. Nat. Prod. Rep. 2006, 23, 444–463. [Google Scholar] [CrossRef] [PubMed]
- Csende, F.; Stájer, G. Approaches to the formation of condensed isoindolones. Curr. Org. Chem. 2005, 9, 1261–1276. [Google Scholar] [CrossRef]
- Belluau, V.; Noeureuil, P.; Ratzke, E.; Skvortsov, A.; Gallagher, S.; Motti, C.A.; Oelgemöller, M. Photodecarboxylative benzylations of phthalimide in pH 7 buffer: A simple access to 3-arylmethyleneisoindolin-1-ones. Tetrahedron Lett. 2010, 51, 4738–4741. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Cygon, P.; Lex, J.; Griesbeck, A.G. The photodecarboxylative addition of carboxylates to phthalimides: Scope and limitations. Heterocycles 2003, 59, 669–684. [Google Scholar] [CrossRef]
- Hatoum, F.; Engler, J.; Zelmer, C.; Wißen, J.; Motti, C.A.; Lex, J.; Oelgemöller, M. Photodecarboxylative addition of carboxylates to phthalimides: A concise access to biologically active 3-(alkyl and aryl)methylene-1H-isoindolin-1-ones. Tetrahedron Lett. 2012, 53, 5573–5577. [Google Scholar] [CrossRef]
- Anamimoghadam, O.; Mumtaz, S.; Nietsch, A.; Saya, G.; Motti, C.A.; Junk, P.C.; Qureshi, A.M.; Oelgemöller, M. The photodecarboxylative addition of carboxylates to phthalimides as a key-step in the synthesis of biologically active 3-arylmethylene-2,3-dihydro-1H-isoindolin-1-ones. Beilstein. J. Org. Chem. 2017, 13, 2833–2841. [Google Scholar] [CrossRef] [Green Version]
- Griesbeck, A.G.; Maptue, N.; Bondock, S.; Oelgemöller, M. The excimer radiation system: A powerful tool for preparative organic photochemistry. A technical note. Photochem. Photobiol. Sci. 2003, 2, 450–451. [Google Scholar] [CrossRef]
- Josland, S.; Mumtaz, S.; Oelgemöller, M. Photodecarboxylations in an advanced meso-scale continuous-flow photoreactor. Chem. Eng. Technol. 2016, 39, 81–87. [Google Scholar] [CrossRef]
- Plutschack, M.B.; Pieber, B.; Gilmore, K.; Seeberger, P.H. The Hitchhiker’s Guide to Flow Chemistry. Chem. Rev. 2017, 117, 11796–11893. [Google Scholar] [CrossRef]
- Britton, J.; Jamison, T.F. The assembly and use of continuous flow systems for chemical synthesis. Nat. Protoc. 2017, 12, 2423–2446. [Google Scholar] [CrossRef]
- Mizuno, K.; Nishiyama, Y.; Ogaki, T.; Terao, K.; Ikeda, H.; Kakiuchi, K.J. Utilization of microflow reactors to carry out synthetically useful organic photochemical reactions. Photochem. Photobiol. C Photochem. Rev. 2016, 29, 107–147. [Google Scholar] [CrossRef]
- Gilmore, K.; Seeberger, P.H. Continuous flow photochemistry. Chem. Rec. 2014, 14, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Coyle, E.E.; Oelgemöller, M. Micro-photochemistry: Photochemistry in microstructured reactors. The new photochemistry of the future? Photochem. Photobiol. Sci. 2008, 7, 1313–1428. [Google Scholar] [CrossRef] [PubMed]
- Britton, J.; Raston, C.L. Multi-step continuous-flow synthesis. Chem. Soc. Rev. 2017, 46, 1250–1271. [Google Scholar] [CrossRef] [Green Version]
- Otake, Y.; Nakamura, H.; Fuse, S. Recent advances in the integrated micro-flow synthesis containing photochemical reactions. Tetrahedron Lett. 2018, 59, 1691–1697. [Google Scholar] [CrossRef]
- Ang, W.S.; Halton, B. The configuration of alkylidenephthalimidine derivatives. Aust. J. Chem. 1971, 24, 851–856. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, C.-A.; Lu, K.; Daniewska, I.; Leon, J.D.; Kong, R.; Forray, C.; Li, B.; Hegde, L.G.; Wolinsky, T.D.; et al. Synthesis and SAR investigations for novel melanin-concentrating hormone 1 receptor (MCH1) antagonists part 1. The discovery of arylacetamides as viable replacements for the dihydropyrimidinone moiety of an HTS hit. J. Med. Chem. 2007, 50, 3870–3882. [Google Scholar] [CrossRef]
- Ploskonka, A.M.; Marzen, S.E.; DeCosta, J.B. Facile synthesis and direct activation of zirconium based metal−organic frameworks from acetone. Ind. Eng. Chem. Res. 2017, 56, 1478–1484. [Google Scholar] [CrossRef]
- Aida, S.; Terao, K.; Nishiyama, Y.; Kakiuchi, K.; Oelgemöller, M. Microflow photochemistry—a reactor comparison study using the photochemical synthesis of terebic acid as a model reaction. Tetrahedron Lett. 2012, 53, 5578–5581. [Google Scholar] [CrossRef]
- Horie, T.; Sumino, M.; Tanaka, T.; Matsushita, Y.; Ichimura, T.; Yoshida, J.-i. Photodimerization of maleic anhydride in a microreactor without clogging. Org. Proc. Res. Devel. 2010, 14, 405–410. [Google Scholar] [CrossRef]
- DeLaney, E.N.; Lee, D.S.; Elliott, L.D.; Jin, J.; Booker-Milburn, K.I.; Poliakoff, M.; George, M.W. A laboratory-scale annular continuous flow reactor for UV photochemistry using excimer lamps for discrete wavelength excitation and its use in a wavelength study of a photodecarboxlyative cyclisation. Green Chem. 2017, 19, 1431–1438. [Google Scholar] [CrossRef] [Green Version]
- Mumtaz, S.; Robertson, M.J.; Oelgemöller, M. Recent advances in photodecarboxylations involving phthalimides. Aust. J. Chem. 2018, 71, 634–648. [Google Scholar] [CrossRef]
- Görner, H.; Oelgemöller, M.; Griesbeck, A.G. Photodecarboxylation study of carboxy-substituted N-alkylphthalimides in aqueous solution: Time resolved UV-Vis spectroscopy and conductometry. J. Phys. Chem. A 2002, 106, 1458–1464. [Google Scholar] [CrossRef]
- Shvydkiv, O.; Yavorskyy, A.; Tan, S.B.; Nolan, K.; Hoffmann, N.; Youssef, A.; Oelgemöller, M. Microphotochemistry: A reactor comparison study using the photosensitized addition of isopropanol to furanones as a model reaction. Photochem. Photobiol. Sci. 2011, 10, 1399–1404. [Google Scholar] [CrossRef]
- Braun, A.M.; Maurette, M.; Oliveros, E. Photochemical Technology; Wiley: Chichester, UK, 1991. [Google Scholar]
- Kise, N.; Kawano, Y.; Sakurai, T. Reductive coupling of phthalimides with ketones and aldehydes by low-valent titanium: One-pot synthesis of alkylideneisoindolin-1-ones. J. Org. Chem. 2013, 78, 12453–12459. [Google Scholar] [CrossRef]
- Li, L.; Janesko, B.G. 3-Methyleneisoindolin-1-one assembly via base- and CuI/L-proline-catalyzed domino reaction: Mechanism of regioselective anionic cyclization. J. Org. Chem. 2016, 81, 10802–10808. [Google Scholar] [CrossRef]
- Chen, Y.; Sabio, J.C.; Hartman, R.L. When solids stop flow chemistry in commercial tubing. J. Flow Chem. 2015, 5, 166–171. [Google Scholar] [CrossRef] [Green Version]
- Drewry, D.H.; Coe, D.M.; Poon, S. Solid-supported reagents in organic synthesis. Med. Res. Rev. 1999, 19, 97–148. [Google Scholar] [CrossRef]
- Eames, J.; Watkinson, M. Polymeric scavenger reagents in organic synthesis. Eur. J. Org. Chem. 2001, 1213–1224. [Google Scholar] [CrossRef]
- van Male, P.; de Croon, M.H.J.M.; Tiggelaar, R.M.; van den Berg, A. Heat and mass transfer in a square microchannel with asymmetric heating. Internat. J. Heat Mass Transf. 2004, 47, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Maryanoff, B.E.; Zhang, H.-C.; Cohen, J.H.; Turchi, I.J.; Maryanoff, C.A. Cyclizations of N-acyliminium ions. Chem. Rev. 2004, 104, 1431–1628. [Google Scholar] [CrossRef]
- Wernerova, M.; Hudlicky, T. On the practical limits of determining isolated product yields and ratios of stereoisomers: Reflections, analysis, and redemption. Synlett 2010, 2701–2707. [Google Scholar] [CrossRef]
- Hunter, R.; Moore, J.; Guthrie, D.; Robertson, M.J.; Oelgemöller, M. Rapid photochemical reaction studies under continuous-flow conditions in the Vapourtec UV-150 reactor—A technical note. Curr. Org. Chem. 2018, 22, 2501–2508. [Google Scholar] [CrossRef]
- Yang, C.; Sheng, X.; Zhang, L.; Yu, J.; Huang, D. Arylacetic acids in organic synthesis. Asian J. Org. Chem. 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.; König, B. Decarboxylative reactions with and without light – a comparison. Green Chem. 2018, 20, 323–361. [Google Scholar] [CrossRef]
- Yoshimi, Y. Photoinduced electron transfer-promoted decarboxylative radical reactions of aliphatic carboxylic acids by organic photoredox system. J. Photochem. Photobiol. A Chem. 2017, 342, 116–130. [Google Scholar] [CrossRef]
- Budac, D.; Wan, P. Photodecarboxylation: Mechanism and synthetic utility. J. Photochem. Photobiol. A Chem. 1992, 67, 135–166. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 3a–e, 4a–e, and 6a–e are available from the authors. |

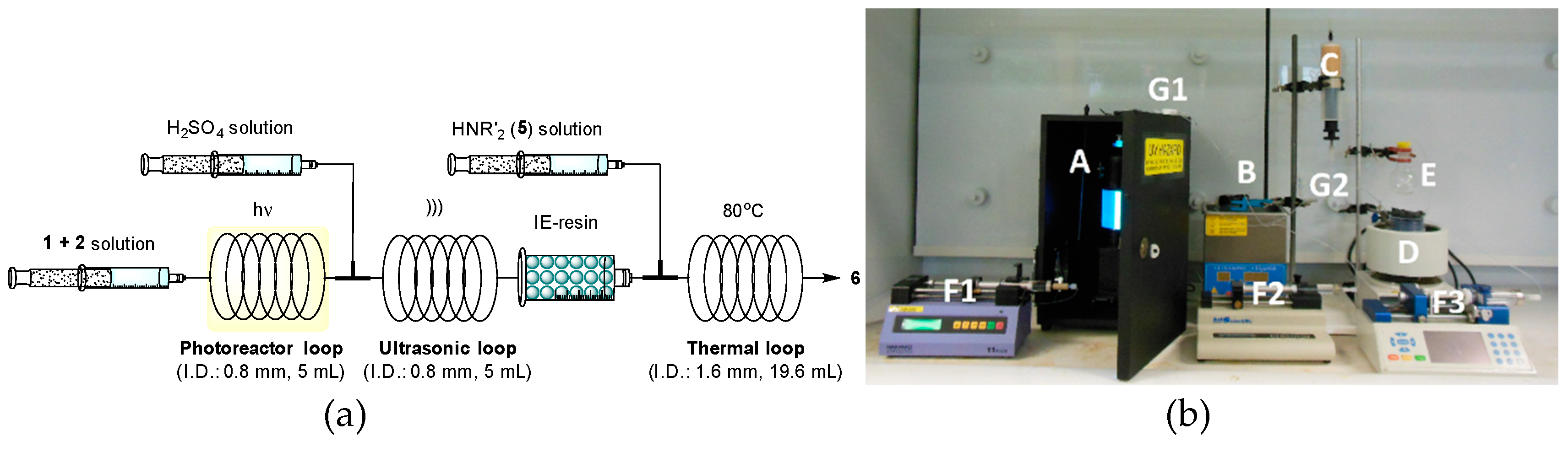

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Time [h] | Conversion [%] a |
|---|---|---|---|
| Photodecarboxylation | |||
| I | acetone b | 3 | 87 |
| II | acetonitrile b | 3 | 80 |
| III | DMF b | 3 | 45 |
| Dehydration | |||
| IV | acetone | 5 | 37 c |
| V | acetonitrile | 5 | 83 |
| VI | DMF | 5 | 80 |
| Amination | |||
| VII | acetone | 5 | 46 |
| VIII | acetonitrile | 5 | 53 |
| IX | DMF | 5 | 55 |
| Entry | n | R | Residence Time [min] | Yield of 3 [%] |
|---|---|---|---|---|
| pH 7 Buffer-Acetone | ||||
| I | 2 | H | 20 | 87 (3a) |
| II | 2 | 4-MeO | 20 | 85 (3b) |
| III | 3 | H | 20 | 95 (3c) |
| IV | 3 | 4-Me | 20 | 80 (3d) |
| V | 3 | 4-F | 20 | 89 (3e) |
| pH 7 Buffer-Acetonitrile | ||||
| VI | 2 | H | 30 | 83 (3a) |
| VII | 2 | 4-MeO | 30 | 85 (3b) |
| VIII | 3 | H | 30 | 91 (3c) |
| IX | 3 | 4-Me | 30 | 83 (3d) |
| X | 3 | 4-F | 30 | 90 (3e) |
| Entry | n | R | Residence Times [min] a | Yield of 4 [%] b |
|---|---|---|---|---|
| I | 2 | H | 30 + 15 | 91 (4a) |
| II | 2 | 4-MeO | 30 + 15 | 90 (4b) |
| III | 3 | H | 30 + 15 | 83 (4c) |
| IV | 3 | 4-Me | 30 + 15 | 86 (4d) |
| V | 3 | 4-F | 30 + 15 | 94 (4e) |
| Entry | n | R | R’ | Residence Times [min] a | Yield of 6 [%] b |
|---|---|---|---|---|---|
| I | 2 | H | Et | 30 + 15 + 33 | 75 (6a) |
| II | 2 | 4-MeO | Et | 30 + 15 + 33 | 73 (6b) |
| III | 3 | H | Me | 30 + 15 + 33 | 75 (6c) |
| IV | 3 | 4-Me | Et | 30 + 15 + 33 | 77 (6d) |
| V | 3 | 4-F | Et | 30 + 15 + 33 | 75 (6e) |
| Compound | Batch [7] | Flow |
|---|---|---|
| Reaction timea | 9 h | 78 min b |
| Overall yields of 6a–e [%] | 28–45 | 72–77 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mumtaz, S.; Robertson, M.J.; Oelgemöller, M. Continuous Flow Photochemical and Thermal Multi-Step Synthesis of Bioactive 3-Arylmethylene-2,3-Dihydro-1H-Isoindolin-1-Ones. Molecules 2019, 24, 4527. https://doi.org/10.3390/molecules24244527
Mumtaz S, Robertson MJ, Oelgemöller M. Continuous Flow Photochemical and Thermal Multi-Step Synthesis of Bioactive 3-Arylmethylene-2,3-Dihydro-1H-Isoindolin-1-Ones. Molecules. 2019; 24(24):4527. https://doi.org/10.3390/molecules24244527
Chicago/Turabian StyleMumtaz, Saira, Mark J. Robertson, and Michael Oelgemöller. 2019. "Continuous Flow Photochemical and Thermal Multi-Step Synthesis of Bioactive 3-Arylmethylene-2,3-Dihydro-1H-Isoindolin-1-Ones" Molecules 24, no. 24: 4527. https://doi.org/10.3390/molecules24244527
APA StyleMumtaz, S., Robertson, M. J., & Oelgemöller, M. (2019). Continuous Flow Photochemical and Thermal Multi-Step Synthesis of Bioactive 3-Arylmethylene-2,3-Dihydro-1H-Isoindolin-1-Ones. Molecules, 24(24), 4527. https://doi.org/10.3390/molecules24244527