Peptide/Peptoid Hybrid Oligomers: The Influence of Hydrophobicity and Relative Side-Chain Length on Antibacterial Activity and Cell Selectivity


Abstract
:1. Introduction
2. Results and Discussion
2.1. Selection and Synthesis of Peptidomimetics
2.2. Effects of Relative Side-Chain Length and Flexibility on Hydrophobicity
2.3. Effects of Relative Side-Chain Length and Flexibility on Antibacterial Activity
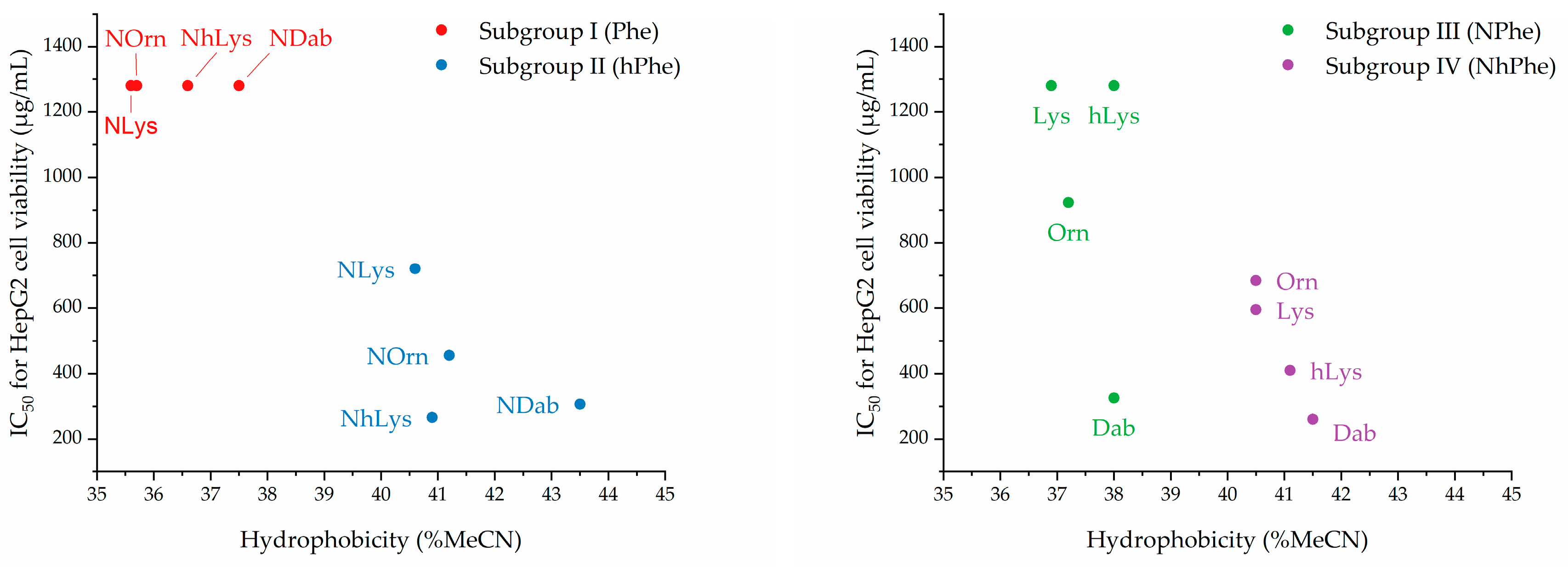
2.4. Effects of Relative Side-Chain Length and Flexibility on Mammalian Cell Viability
3. Materials and Methods
3.1. General Information
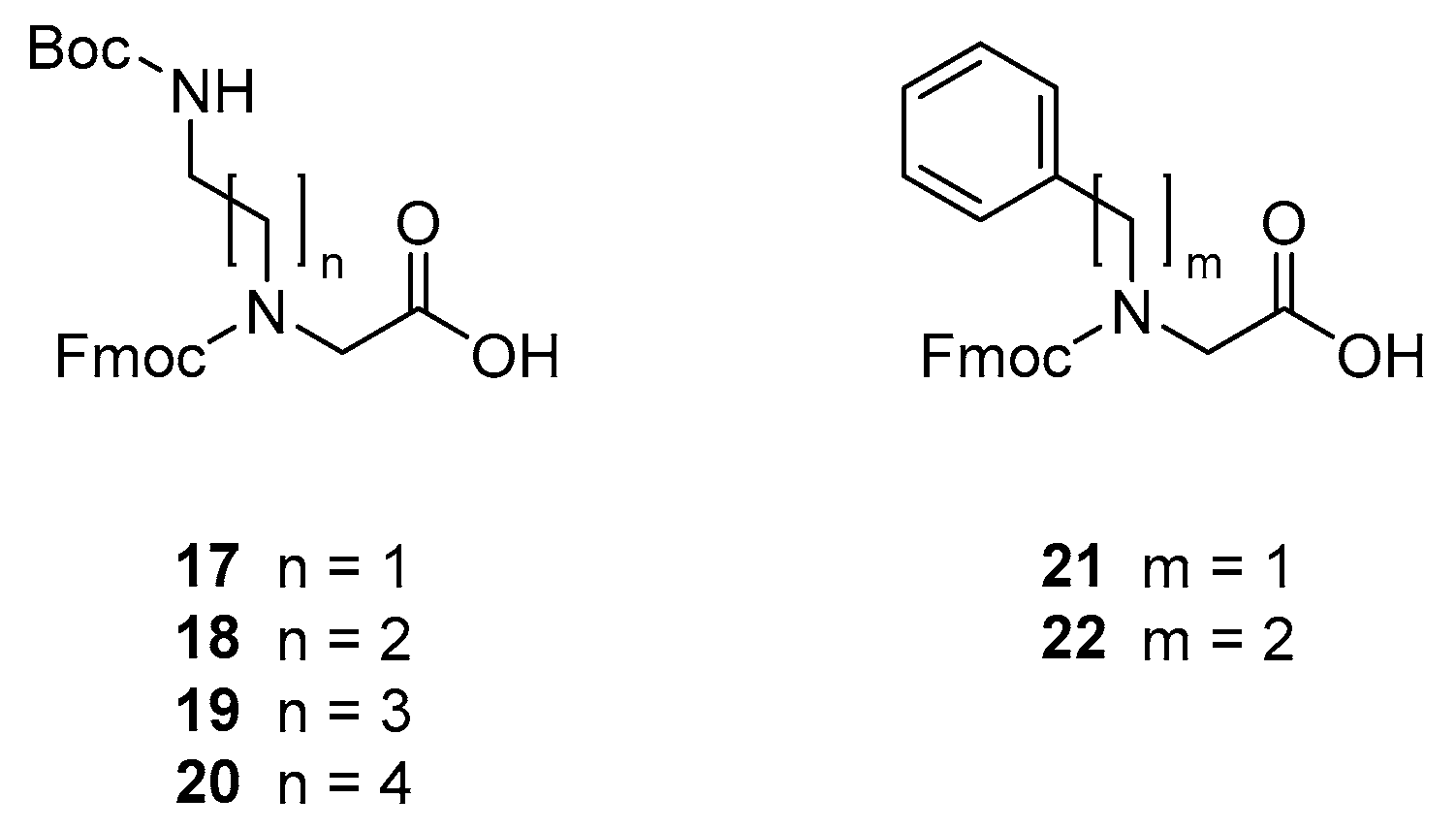
3.2. Building Block Synthesis
3.2.1. Synthesis of N-(((9H-fluoren-9-yl)methoxy)carbonyl)-N-(5-((tert-butoxycarbonyl)amino)ethyl)glycine [Fmoc-NDab(Boc)-OH, 17]
3.2.2. Synthesis of N-(((9H-fluoren-9-yl)methoxy)carbonyl)-N-(5-((tert-butoxycarbonyl)amino)propyl)glycine [Fmoc-NOrn(Boc)-OH, 18]
3.2.3. Synthesis of N-(((9H-fluoren-9-yl)methoxy)carbonyl)-N-(5-((tert-butoxycarbonyl)amino)butyl)glycine [Fmoc-NLys(Boc)-OH, 19]
3.2.4. Synthesis of N-(((9H-fluoren-9-yl)methoxy)carbonyl)-N-(5-((tert-butoxycarbonyl)amino)pentyl)glycine [Fmoc-NhLys(Boc)-OH, 20]
3.2.5. Synthesis of N-(((9H-fluoren-9-yl)methoxy)carbonyl)-N-benzylglycine [Fmoc-NPhe-OH, 21]
3.2.6. Synthesis of N-(((9H-fluoren-9-yl)methoxy)carbonyl)-N-phenethylglycine [Fmoc-NhPhe-OH, 22]
3.3. General Protocol for Manual Synthesis of Peptidomimetics
3.4. General Protocol for Microwave-Assisted Automated Synthesis of Peptidomimetics
3.5. Determination of Minimum Inhibitory Concentration
3.6. Determination of Hemolytic Activity
3.7. Determination of Antiproliferative Activity on HepG2 Cell Line
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morgan, D.J.; Okeke, I.N.; Laxminarayan, R.; Perencevich, E.N.; Weisenberg, S. Non-prescription antimicrobial use worldwide: A systematic review. Lancet Infect. Dis. 2011, 11, 692–701. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; The Review on Antimicrobial Resistance: London, UK, 2016. [Google Scholar]
- WHO Advisory Group on Integrated Surveillance of Antimicrobial Resistance. Critically Important Antimicrobials for Human Medicine; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- The 2019 Expert Committee on the Selection and Use of Essential Medicines. World Health Organization Model List of Essential Medicines; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Arzanlou, M.; Chai, W.C.; Venter, H. Intrinsic, adaptive and acquired antimicrobial resistance in Gram-negative bacteria. Essays Biochem. 2017, 61, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Boman, H.G. Peptide antibiotics and their role in innate immunity. Annu. Rev. Immunol. 1995, 13, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Hamill, P.; Hancock, R.E.W. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [Green Version]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Hamamoto, K.; Kida, Y.; Zhang, Y.; Shimizu, T.; Kuwano, K. Antimicrobial activity and stability to proteolysis of small linear cationic peptides with D-amino acid substitutions. Microbiol. Immunol. 2002, 46, 741–749. [Google Scholar] [CrossRef]
- Hong, S.Y.; Oh, J.E.; Lee, K.H. Effect of D-amino acid substitution on the stability, the secondary structure, and the activity of membrane-active peptide. Biochem. Pharmacol. 1999, 58, 1775–1780. [Google Scholar] [CrossRef]
- Simon, R.J.; Kania, R.S.; Zuckermann, R.N.; Huebner, V.D.; Jewell, D.A.; Banville, S.; Ng, S.; Wang, L.; Rosenberg, S.; Marlowe, C.K.; et al. Peptoids: A modular approach to drug discovery. Proc. Natl. Acad. Sci. USA 1992, 89, 9367–9371. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.K.; Lee, S.A.; Shin, S.; Lee, J.Y.; Jeong, K.W.; Nan, Y.H.; Park, Y.S.; Shin, S.Y.; Kim, Y. Structural flexibility and the positive charges are the key factors in bacterial cell selectivity and membrane penetration of peptoid-substituted analog of Piscidin 1. Biochim. Biophys. Acta 2010, 1798, 1913–1925. [Google Scholar] [CrossRef] [Green Version]
- Shuey, S.W.; Delaney, W.J.; Shah, M.C.; Scialdone, M.A. Antimicrobial β-peptoids by a block synthesis approach. Bioorg. Med. Chem. Lett. 2006, 16, 1245–1248. [Google Scholar] [CrossRef]
- Porter, E.A.; Weisblum, B.; Gellman, S.H. Mimicry of host-defense peptides by unnatural oligomers: Antimicrobial β-peptides. J. Am. Chem. Soc. 2002, 124, 7324–7330. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.A.; Weisblum, B.; Gellman, S.H. Interplay among folding, sequence, and lipophilicity in the antibacterial and hemolytic activities of α/β-peptides. J. Am. Chem. Soc. 2007, 129, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.A.; Bonke, G.; Vedel, L.; Adsersen, A.; Witt, M.; Franzyk, H.; Jaroszewski, J.W. α-peptide/β-peptoid chimeras. Org. Lett. 2007, 9, 1549–1552. [Google Scholar] [CrossRef] [PubMed]
- Padhee, S.; Hu, Y.; Niu, Y.; Bai, G.; Wu, H.; Costanza, F.; West, L.; Harrington, L.; Shaw, L.N.; Cao, C.; et al. Non-hemolytic α-AApeptides as antimicrobial peptidomimetics. Chem. Commun. (Camb.) 2011, 47, 9729–9731. [Google Scholar] [CrossRef]
- Niu, Y.; Padhee, S.; Wu, H.; Bai, G.; Harrington, L.; Burda, W.N.; Shaw, L.N.; Cao, C.; Cai, J. Identification of γ-AApeptides with potent and broad-spectrum antimicrobial activity. Chem. Commun. (Camb.) 2011, 47, 12197–12199. [Google Scholar] [CrossRef] [Green Version]
- Radzishevsky, I.S.; Rotem, S.; Bourdetsky, D.; Navon-Venezia, S.; Carmeli, Y.; Mor, A. Improved antimicrobial peptides based on acyl-lysine oligomers. Nat. Biotechnol. 2007, 25, 657–659. [Google Scholar] [CrossRef]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipathic, α-helical antimicrobial peptides. Biopolymers 2000, 55, 4–30. [Google Scholar] [CrossRef]
- Giangaspero, A.; Sandri, L.; Tossi, A. Amphipathic α-helical antimicrobial peptides. Eur. J. Biochem. 2001, 268, 5589–5600. [Google Scholar] [CrossRef]
- Zelezetsky, I.; Pacor, S.; Pag, U.; Papo, N.; Shai, Y.; Sahl, H.G.; Tossi, A. Controlled alteration of the shape and conformational stability of α-helical cell-lytic peptides: Effect on mode of action and cell specificity. Biochem. J. 2005, 390, 177–188. [Google Scholar] [CrossRef]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Rational design of α-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar] [CrossRef] [Green Version]
- Khandelia, H.; Kaznessis, Y.N. Molecular dynamics investigation of the influence of anionic and zwitterionic interfaces on antimicrobial peptides’ structure: Implications for peptide toxicity and activity. Peptides 2006, 27, 1192–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lu, H.; Lin, Y.; Cheng, J. Water-soluble polypeptides with elongated, charged side chains adopt ultra-stable helical conformations. Macromolecules 2011, 44, 6641–6644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangoni, M.L.; Carotenuto, A.; Auriemma, L.; Saviello, M.R.; Campiglia, P.; Gomez-Monterrey, I.; Malfi, S.; Marcellini, L.; Barra, D.; Novellino, E.; et al. Structure-activity relationship, conformational and biological studies of temporin L analogues. J. Med. Chem. 2011, 54, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, H.; Slaninova, J.; Bednarova, L.; Monincova, L.; Budesinsky, M.; Cerovsky, V. Effect of hydrocarbon stapling on the properties of α-helical antimicrobial peptides isolated from the venom of hymenoptera. Amino Acids 2012, 43, 2047–2058. [Google Scholar] [CrossRef] [PubMed]
- Bobone, S.; Bocchinfuso, G.; Park, Y.; Palleschi, A.; Hahm, K.S.; Stella, L. The importance of being kinked: Role of Pro residues in the selectivity of the helical antimicrobial peptide P5. J. Pept. Sci. 2013, 19, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Cherry, M.A.; Higgins, S.K.; Melroy, H.; Lee, H.S.; Pokorny, A. Peptides with the same composition, hydrophobicity, and hydrophobic moment bind to phospholipid bilayers with different affinities. J. Phys. Chem. B 2014, 118, 12462–12470. [Google Scholar] [CrossRef] [Green Version]
- Jahnsen, R.D.; Sandberg-Schaal, A.; Vissing, K.J.; Nielsen, H.M.; Frimodt-Møller, N.; Franzyk, H. Tailoring cytotoxicity of antimicrobial peptidomimetics with high activity against multidrug-resistant Escherichia coli. J. Med. Chem. 2014, 57, 2864–2873. [Google Scholar] [CrossRef]
- Jahnsen, R.D.; Frimodt-Møller, N.; Franzyk, H. Antimicrobial activity of peptidomimetics against multidrug-resistant Escherichia coli: A comparative study of different backbones. J. Med. Chem. 2012, 55, 7253–7261. [Google Scholar] [CrossRef]
- Lee, J.; Kang, D.; Choi, J.; Huang, W.; Wadman, M.; Barron, A.E.; Seo, J. Effect of side chain hydrophobicity and cationic charge on antimicrobial activity and cytotoxicity of helical peptoids. Bioorg. Med. Chem. Lett. 2018, 28, 170–173. [Google Scholar] [CrossRef]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J. Biol. Chem. 2012, 287, 7738–7745. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, Y.; Lev, N.; Shai, Y. Effect of the hydrophobicity to net positive charge ratio on antibacterial and anti-endotoxin activities of structurally similar antimicrobial peptides. Biochemistry 2010, 49, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Blondelle, S.E.; Houghten, R.A. Hemolytic and antimicrobial activities of the twenty-four individual omission analogues of melittin. Biochemistry 1991, 30, 4671–4678. [Google Scholar] [CrossRef] [PubMed]
- Kondejewski, L.H.; Jelokhani-Niaraki, M.; Farmer, S.W.; Lix, B.; Kay, C.M.; Sykes, B.D.; Hancock, R.E.W.; Hodges, R.S. Dissociation of antimicrobial and hemolytic activities in cyclic peptide diastereomers by systematic alterations in amphipathicity. J. Biol. Chem. 1999, 274, 13181–13192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachi, T.; Epand, R.F.; Epand, R.M.; Matsuzaki, K. Position-dependent hydrophobicity of the antimicrobial magainin peptide affects the mode of peptide-lipid interactions and selective toxicity. Biochemistry 2002, 41, 10723–10731. [Google Scholar] [CrossRef] [PubMed]
- Molchanova, N.; Hansen, P.R.; Damborg, P.; Nielsen, H.M.; Franzyk, H. Lysine-Based α-Peptide/β-Peptoid Peptidomimetics: Influence of Hydrophobicity, Fluorination, and Distribution of Cationic Charge on Antimicrobial Activity and Cytotoxicity. ChemMedChem 2017, 12, 312–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahnsen, R.O.; Sandberg-Schaal, A.; Frimodt-Møller, N.; Nielsen, H.M.; Franzyk, H. End group modification: Efficient tool for improving activity of antimicrobial peptide analogues towards Gram-positive bacteria. Eur. J. Pharm. Biopharm. 2015, 95, 40–46. [Google Scholar] [CrossRef]
- Tan, J.; Huang, J.; Huang, Y.; Chen, Y. Effects of single amino acid substitution on the biophysical properties and biological activities of an amphipathic α-helical antibacterial peptide against Gram-negative bacteria. Molecules 2014, 19, 10803–10817. [Google Scholar] [CrossRef] [Green Version]
- Glukhov, E.; Burrows, L.L.; Deber, C.M. Membrane interactions of designed cationic antimicrobial peptides: The two thresholds. Biopolymers 2008, 89, 360–371. [Google Scholar] [CrossRef]
- Molchanova, N.; Hansen, P.R.; Damborg, P.; Franzyk, H. Fluorinated antimicrobial lysine-based peptidomimetics with activity against methicillin-resistant Staphylococcus pseudintermedius. J. Pept. Sci. 2018, 24, e3098. [Google Scholar] [CrossRef]
- Chongsiriwatana, N.P.; Miller, T.M.; Wetzler, M.; Vakulenko, S.; Karlsson, A.J.; Palecek, S.P.; Mobashery, S.; Barron, A.E. Short alkylated peptoid mimics of antimicrobial lipopeptides. Antimicrob. Agents Chemother. 2011, 55, 417–420. [Google Scholar] [CrossRef] [Green Version]
- Chu-Kung, A.F.; Bozzelli, K.N.; Lockwood, N.A.; Haseman, J.R.; Mayo, K.H.; Tirrell, M.V. Promotion of peptide antimicrobial activity by fatty acid conjugation. Bioconjug. Chem. 2004, 15, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Radzishevsky, I.S.; Rotem, S.; Zaknoon, F.; Gaidukov, L.; Dagan, A.; Mor, A. Effects of acyl versus aminoacyl conjugation on the properties of antimicrobial peptides. Antimicrob. Agents Chemother. 2005, 49, 2412–2420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zweytick, D.; Pabst, G.; Abuja, P.M.; Jilek, A.; Blondelle, S.E.; Andra, J.; Jerala, R.; Monreal, D.; Martinez de Tejada, G.; Lohner, K. Influence of N-acylation of a peptide derived from human lactoferricin on membrane selectivity. Biochim. Biophys. Acta 2006, 1758, 1426–1435. [Google Scholar] [CrossRef] [Green Version]
- Bolt, H.L.; Eggimann, G.A.; Jahoda, C.A.B.; Zuckermann, R.N.; Sharples, G.J.; Cobb, S.L. Exploring the links between peptoid antibacterial activity and toxicity. MedChemComm 2017, 8, 886–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasupuleti, M.; Schmidtchen, A.; Chalupka, A.; Ringstad, L.; Malmsten, M. End-tagging of ultra-short antimicrobial peptides by W/F stretches to facilitate bacterial killing. PLoS ONE 2009, 4, e5285. [Google Scholar] [CrossRef] [Green Version]
- Gatto, E.; Mazzuca, C.; Stella, L.; Venanzi, M.; Toniolo, C.; Pispisa, B. Effect of peptide lipidation on membrane perturbing activity: A comparative study on two trichogin analogues. J. Phys. Chem. B 2006, 110, 22813–22818. [Google Scholar] [CrossRef]
- Schmidtchen, A.; Pasupuleti, M.; Morgelin, M.; Davoudi, M.; Alenfall, J.; Chalupka, A.; Malmsten, M. Boosting antimicrobial peptides by hydrophobic oligopeptide end tags. J. Biol. Chem. 2009, 284, 17584–17594. [Google Scholar] [CrossRef] [Green Version]
- Schmidtchen, A.; Ringstad, L.; Kasetty, G.; Mizuno, H.; Rutland, M.W.; Malmsten, M. Membrane selectivity by W-tagging of antimicrobial peptides. Biochim. Biophys. Acta 2011, 1808, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Schmidtchen, A.; Pasupuleti, M.; Malmsten, M. Effect of hydrophobic modifications in antimicrobial peptides. Adv. Colloid Interface Sci. 2014, 205, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Olsen, C.A.; Ziegler, H.L.; Nielsen, H.M.; Frimodt-Møller, N.; Jaroszewski, J.W.; Franzyk, H. Antimicrobial, hemolytic, and cytotoxic activities of β-peptoid-peptide hybrid oligomers: Improved properties compared to natural AMPs. ChemBioChem 2010, 11, 1356–1360. [Google Scholar] [CrossRef]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Molchanova, N.; Wang, H.; Hansen, P.R.; Hoiby, N.; Nielsen, H.M.; Franzyk, H. Antimicrobial ity of α-Peptide/β-Peptoid Lysine-Based Peptidomimetics Against Colistin-Resistant Pseudomonas aeruginosa Isolated From Cystic Fibrosis Patients. Front. Microbiol. 2019, 10, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruijtzer, J.A.W.; Hofmeyer, L.J.F.; Heerma, W.; Versluis, C.; Liskamp, R.M.J. Solid-Phase Syntheses of Peptoids using Fmoc-ProtectedN-Substituted Glycines: The Synthesis of (Retro)Peptoids of Leu-Enkephalin and Substance P. Chem. Eur. J. 1998, 4, 1570–1580. [Google Scholar] [CrossRef]
- Chen, C.X.; Hu, J.; Yang, C.; Zhang, Y.; Wang, F.; Mu, Q.M.; Pan, F.; Xu, H.; Lu, J.R. Amino acid side chains affect the bioactivity of designed short peptide amphiphiles. J. Mater. Chem. B 2016, 4, 2359–2368. [Google Scholar] [CrossRef]
- Hein-Kristensen, L.; Knapp, K.M.; Franzyk, H.; Gram, L. Bacterial membrane activity of α-peptide/β-peptoid chimeras: Influence of amino acid composition and chain length on the activity against different bacterial strains. BMC Microbiol. 2011, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Chongsiriwatana, N.P.; Patch, J.A.; Czyzewski, A.M.; Dohm, M.T.; Ivankin, A.; Gidalevitz, D.; Zuckermann, R.N.; Barron, A.E. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 2794–2799. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Teng, P.; Sang, P.; She, F.; Wei, L.; Cai, J. γ-AApeptides: Design, structure, and applications. Acc. Chem. Res. 2016, 49, 428–441. [Google Scholar] [CrossRef] [Green Version]
- Bolarinwa, O.; Nimmagadda, A.; Su, M.; Cai, J. Structure and function of AApeptides. Biochemistry 2017, 56, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.M.; Epand, R.F. Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim. Biophys. Acta 2009, 1788, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.M.; Epand, R.F. Bacterial membrane lipids in the action of antimicrobial agents. J. Pept. Sci. 2011, 17, 298–305. [Google Scholar] [CrossRef]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojsoska, B.; Zuckermann, R.N.; Jenssen, H. Structure-activity relationship study of novel peptoids that mimic the structure of antimicrobial peptides. Antimicrob. Agents Chemother. 2015, 59, 4112–4120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dathe, M.; Wieprecht, T.; Nikolenko, H.; Handel, L.; Maloy, W.L.; MacDonald, D.L.; Beyermann, M.; Bienert, M. Hydrophobicity, hydrophobic moment and angle subtended by charged residues modulate antibacterial and haemolytic activity of amphipathic helical peptides. FEBS Lett. 1997, 403, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Kondejewski, L.H.; Lee, D.L.; Jelokhani-Niaraki, M.; Farmer, S.W.; Hancock, R.E.W.; Hodges, R.S. Optimization of microbial specificity in cyclic peptides by modulation of hydrophobicity within a defined structural framework. J. Biol. Chem. 2002, 277, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, M.; Liu, L.P.; Deber, C.M. Cationic hydrophobic peptides with antimicrobial activity. Antimicrob. Agents Chemother. 2002, 46, 3585–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of α-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef] [Green Version]
- Uggerhøj, L.E.; Poulsen, T.J.; Munk, J.K.; Fredborg, M.; Søndergaard, T.E.; Frimodt-Møller, N.; Hansen, P.R.; Wimmer, R. Rational design of α-helical antimicrobial peptides: Do’s and don’ts. ChemBioChem 2015, 16, 242–253. [Google Scholar] [CrossRef]
- Modified MIC Method for Cationic Antimicrobial Peptides. Available online: http://cmdr.ubc.ca/bobh/method/modified-mic-method-for-cationic-antimicrobial-peptides/ (accessed on 3 October 2019).
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
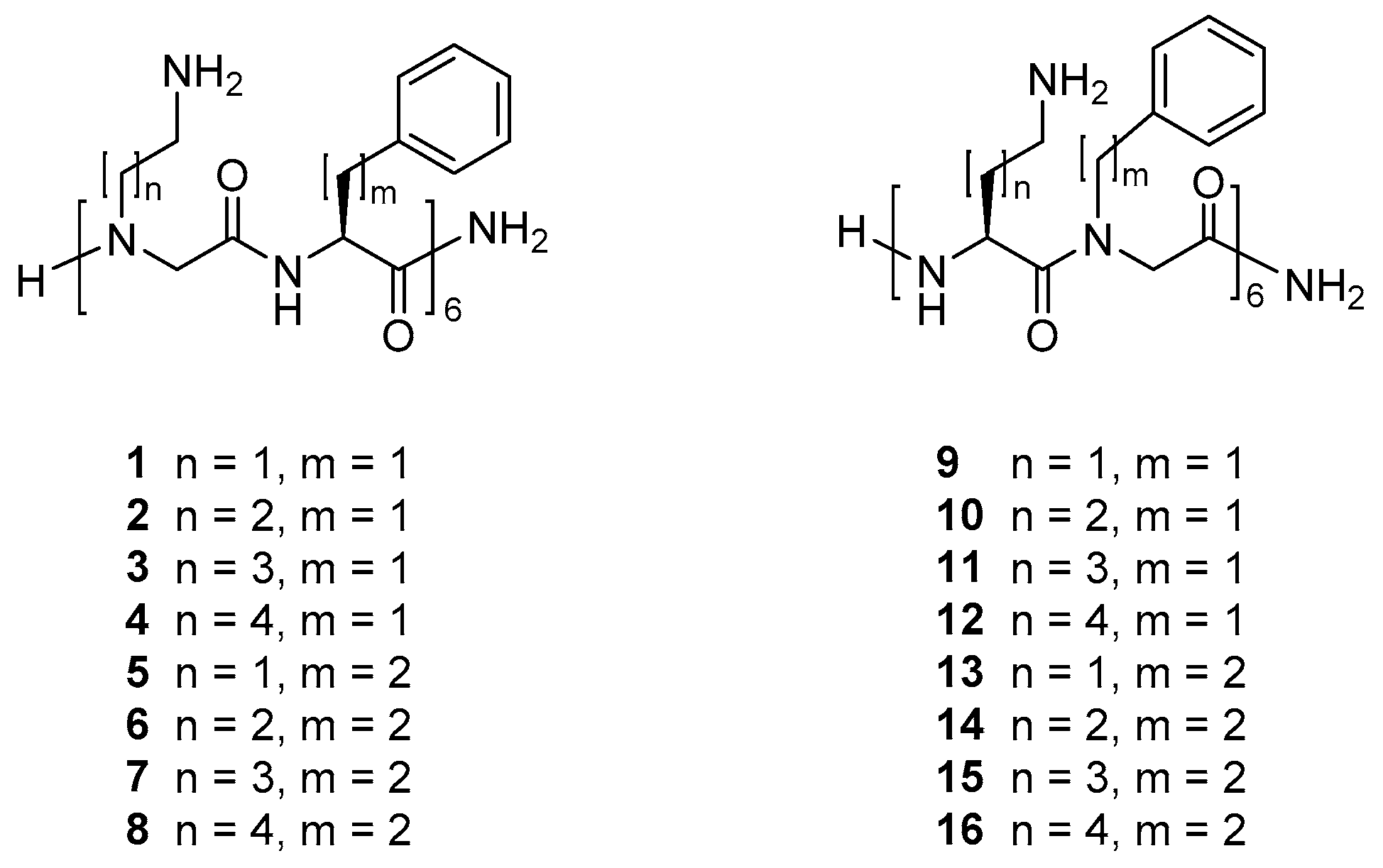

{kind=link}
{kind=link}
{kind=link}
| Subgroup | Cmpd | Hydrophobicity (% MeCN at elution a) | MIC (µg/mL) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gram-Negative | Gram-Positive | ||||||||
| E. coli ATCC 25922 | K. pneumoniae ATCC 13883 | P. aeruginosa ATCC 27853 | P. aeruginosa PAO1 | A. baumannii ATCC 19606 | S. aureus ATCC 29213 | E. faecalis ATCC 29212 | |||
| I | 1 | 37.5% | 4-8 | 64 | 64 | >64 | >64 | >64 | >64 |
| I | 2 | 35.7% | 8-16 | 64 | >64 | >64 | >64 | >64 | >64 |
| I | 3 | 35.6% | 8-16 | 32 | >64 | >64 | >64 | >64 | >64 |
| I | 4 | 36.6% | 4-8 | 64 | >64 | >64 | 64 | >64 | >64 |
| II | 5 | 43.5% | 4 | 64 | 4 | 16 | >64 | 8 | 32 |
| II | 6 | 41.2% | 4 | >64 | 8 | 16-32 | >64 | 16 | >64 |
| II | 7 | 40.6% | 2-4 | >64 | 32 | 32-64 | >64 | >64 | >64 |
| II | 8 | 40.9% | 2 | 64 | 64 | 64 | 4 | >64 | >64 |
| III | 9 | 38.0% | 2-4 | 32-64 | 1 | 2 | 16-32 | >64 | >64 |
| III | 10 | 37.2% | 4-8 | >64 | 2-4 | 4-8 | 64->64 | >64 | >64 |
| III | 11 | 36.9% | 4-8 | 64 | 4 | 8-16 | >64 | >64 | >64 |
| III | 12 | 38.0% | 2 | >64 | 64 | 64 | 16 | >64 | >64 |
| IV | 13 | 41.4% | 4 | 32 | 2 | 2-4 | >64 | >64 | >64 |
| IV | 14 | 40.5% | 4-8 | 32 | 8 | 8-16 | >64 | >64 | >64 |
| IV | 15 | 40.5% | 4 | >64 | 16 | 8-16 | 64 | >64 | >64 |
| IV | 16 | 41.1% | 4 | >64 | 16 | 32 | 8 | >64 | >64 |
| Colistin | 0.125-0.25 | 0.25 | 0.125-0.25 | 0.5 | 0.5 | >64 | >64 | ||
| Subgroup | Cmpd | Hydrophobicity (% MeCN at elution a) | IC50 (µg/mL) b | Viability at 1280 µg/mL c | Hemolytic Activity d | Cell Selectivity e |
|---|---|---|---|---|---|---|
| I | 1 | 37.5% | 1280 | 49% | 7% | 160-320 |
| I | 2 | 35.7% | >1280 | 62% | 5% | >80 |
| I | 3 | 35.6% | 1280 | 52% | 8% | 80-160 |
| I | 4 | 36.6% | 1280 | 49% | 9% | 160-320 |
| II | 5 | 43.5% | 307 | - | 43% | 77 |
| II | 6 | 41.2% | 456 | - | 8% | 114 |
| II | 7 | 40.6% | 720 | - | 6% | 180-360 |
| II | 8 | 40.9% | 266 | - | 9% | 133 |
| III | 9 | 38.0% | 326 | - | 79% | 82-163 |
| III | 10 | 37.2% | 923 | - | 10% | 115-231 |
| III | 11 | 36.9% | >1280 | 76% | 10% | >160 |
| III | 12 | 38.0% | 1280 | 47% | 9% | 640 |
| IV | 13 | 41.4% | 261 | - | 53% | 65 |
| IV | 14 | 40.5% | 684 | - | 11% | 86-171 |
| IV | 15 | 40.5% | 595 | - | 8% | 149 |
| IV | 16 | 41.1% | 410 | - | 9% | 103 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frederiksen, N.; Hansen, P.R.; Björkling, F.; Franzyk, H. Peptide/Peptoid Hybrid Oligomers: The Influence of Hydrophobicity and Relative Side-Chain Length on Antibacterial Activity and Cell Selectivity. Molecules 2019, 24, 4429. https://doi.org/10.3390/molecules24244429
Frederiksen N, Hansen PR, Björkling F, Franzyk H. Peptide/Peptoid Hybrid Oligomers: The Influence of Hydrophobicity and Relative Side-Chain Length on Antibacterial Activity and Cell Selectivity. Molecules. 2019; 24(24):4429. https://doi.org/10.3390/molecules24244429
Chicago/Turabian StyleFrederiksen, Nicki, Paul R. Hansen, Fredrik Björkling, and Henrik Franzyk. 2019. "Peptide/Peptoid Hybrid Oligomers: The Influence of Hydrophobicity and Relative Side-Chain Length on Antibacterial Activity and Cell Selectivity" Molecules 24, no. 24: 4429. https://doi.org/10.3390/molecules24244429